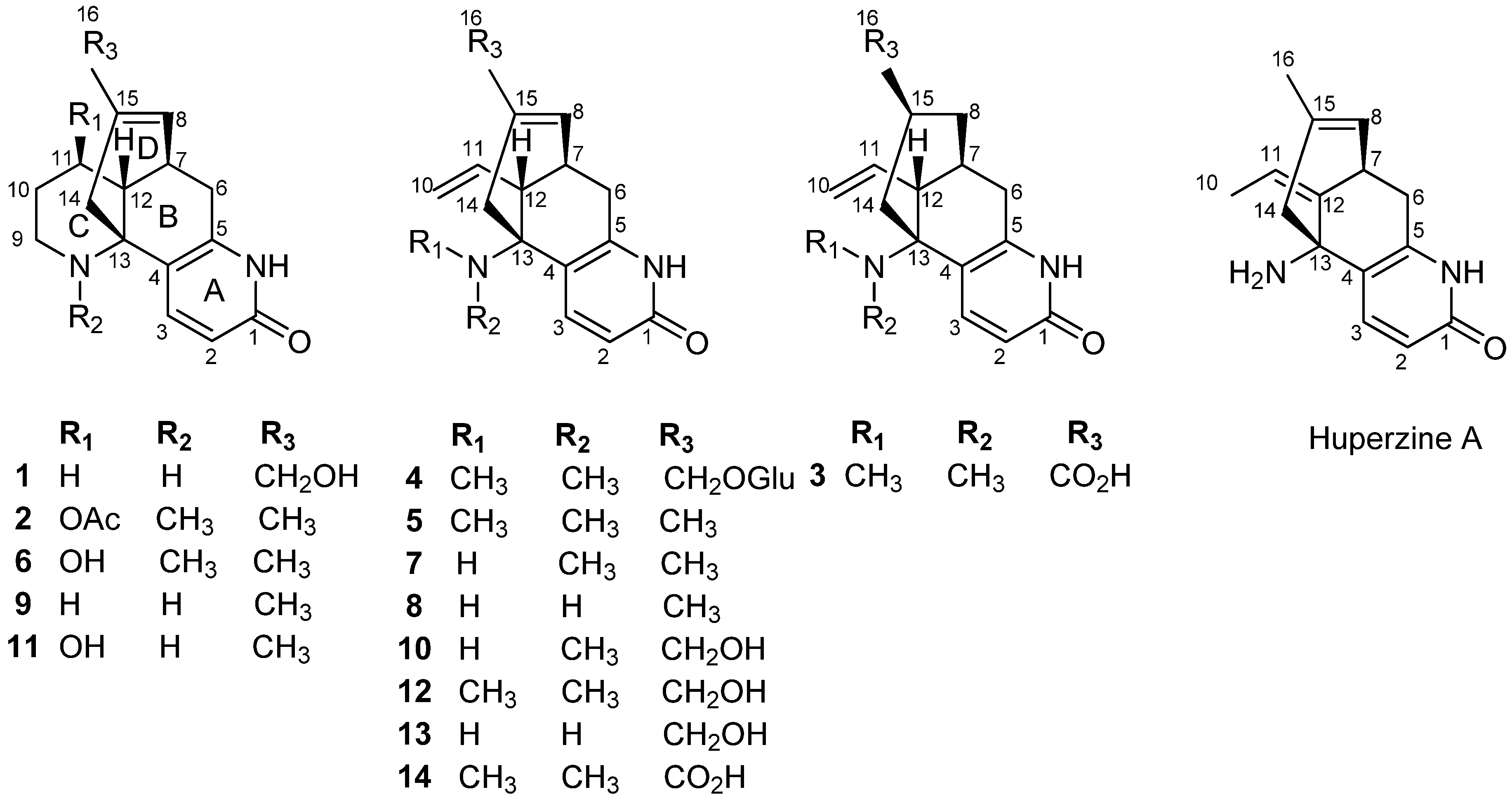
2. Results and Discussion
Compound
1, obtained as a colorless gum, showed an [M+H]
+ ion peak at
m/z 273.1600 in its HRESIMS, corresponding to the molecular formula of C
16H
20N
2O
2 (calcd. for C
16H
21N
2O
2, 273.1598), implying eight degrees of unsaturation. The IR absorption band (1,654 cm
−1) of compound
1 indicated the presence of an α,β-unsaturated carbonyl group. The existence of an
α-pyridone moiety was revealed by the absorption bands (227 and 306 nm) in its UV spectrum and two characteristic proton signals [
δH 7.79, 6.48 (each 1H, d,
J = 9.0 Hz)] in the low field region of the
1H-NMR spectrum [
13]. Additionally, a hydroxymethyl group [
δH 3.87, 3.83 (2H, ABq),
δC 65.9] and an olefinic proton [
δH 5.81 (br d,
J = 4.8 Hz)] were also displayed in the
1H-NMR spectrum (
Table 1). The
13C- and DEPT NMR spectra showed 16 carbon signals, including six methylenes (one oxygenated at
δC 65.9 and one nitrided at
δC 42.0), five methines and fivequaternary carbons (two nitrogenated at
δC 57.6 and 145.6, and one amide carbonyl carbon at
δC 165.5) (
Table 2). These spectroscopic data of compound 1 were similar to those of the known huperzine B (9) [
12] which co-occurs in this species [
9], indicating that compound 1 was a lycodine alkaloid possessing four connected six-membered rings.
Table 1.
1H-NMR data of compounds 1–4, δ in ppm and J in Hz.
Table 1.
1H-NMR data of compounds 1–4, δ in ppm and J in Hz.
| No. | δH (1) a | δH (2) b | δH (3) a | δH (4) b |
|---|
| 2 | 6.48, d (9.0) | 6.41, d (9.2) | 6.46, d (9.6) | 6.48, d (9.6) |
| 3 | 7.79, d (9.0) | 7.79, d (9.2) | 7.66, d (9.6) | 7.87, d (9.6) |
| 6a | 2.90, dd (18.6, 6) | 2.96, dd (17.6, 4.8) | 3.01, dd (18.6, 6.6) | 2.99, dd (17.6, 4.3) |
| 6b | 2.39, br d (18.6) | 2.39, br d (17.6) | 2.36, br d (18.6) | 2.38, br d (17.6) |
| 7 | 2.59, m | 2.68, m | 2.16, m | 2.60, m |
| 8a | 5.81, br d (4.8) | 5.42, br d (5.2) | 1.91, br d (13.8) | 5.82, br d (4.1) |
| 8b | | | 1.78, ddd (13.8, 13.2, 3.6) | |
| 9a | 3.06, br d (12.0) | 2.71, ddd (14.4, 14.2, 2.0) | | |
| 9b | 2.55, overlapped | 2.66, overlapped | | |
| 10a | 1.80, overlapped | 1.75, m | 5.33, dd (16.8, 1.8) | 5.42, dd (17.0, 2.0) |
| 10b | 1.80, overlapped | 1.64, m | 5.10, dd (10.2, 1.8) | 5.27, dd (10.2, 2.0) |
| 11a | 1.39, dddd (13.2, 12.6, 12.3, 4.6) | 4.76, ddd (11.2, 10.8, 5.2) | 6.02, ddd (16.8, 10.2, 10.2) | 6.09, ddd (17.0, 10.2, 10.1) |
| 11b | 1.69, m | | | |
| 12 | 2.07, br d (12.6) | 2.07, dd (10.8, 3.6) | 2.95, dd (10.8, 3.6) | 3.06, dd (10.3, 4.1) |
| 14a | 2.51, d (16.2) | 2.65, d (16.7) | 2.19, dd (12.6, 12.6) | 3.11, d (18.4) |
| 14b | 2.25, d (16.2) | 1.76, d (16.7) | 1.66, dd (12.6, 3) | 2.16, d (18.4) |
| 15 | | | 2.05, m | |
| 16 | 3.87, 3.83; ABq (13.8) | 1.56, br s | | 4.11, 4.02; ABq (12.2) |
| Glu | | | | |
| 1' | | | | 4.22, d (7.8) |
| 2' | | | | 3.17, dd (9.5, 7.8) |
| 3' | | | | 3.23, t (9.5) |
| 4' | | | | 3.25, t (9.5) |
| 5' | | | | 3.34, m |
| 6'a | | | | 3.64, dd (11.8, 5.4) |
| 6'b | | | | 3.85, dd (11.8, 1.3) |
| N-Me A | | 2.63, s | 2.58, s | 2.70, s |
| N-Me B | | | 2.58, s | 2.70, s |
| 11-OAc | | 2.04, s | | |
Table 2.
13C-NMR data of compounds 1−4.
Table 2.
13C-NMR data of compounds 1−4.
| No. | δC(1) a | δC(2) b | δC(3) a | δC(4) b |
|---|
| 1 | 165.5 | 165.0 | 165.0 | 165.5 |
| 2 | 119.6 | 118.1 | 118.8 | 118.9 |
| 3 | 140.6 | 140.8 | 142.4 | 143.1 |
| 4 | 115.3 | 119.8 | 117.7 | 117.5 |
| 5 | 145.6 | 142.6 | 146.4 | 144.9 |
| 6 | 29.8 | 29.3 | 30.1 | 29.4 |
| 7 | 34.3 | 29.7 | 38.7 | 39.7 |
| 8 | 127.1 | 124.0 | 37.5 | 128.8 |
| 9 | 42.0 | 48.6 | | |
| 10 | 24.8 | 25.3 | 117.3 | 119.1 |
| 11 | 25.0 | 71.2 | 141.4 | 139.8 |
| 12 | 39.3 | 37.3 | 48.1 | 46.2 |
| 13 | 57.6 | 58.1 | 64.2 | 64.3 |
| 14 | 41.6 | 43.2 | 41.9 | 40.6 |
| 15 | 136.6 | 132.6 | 41.4 | 135.6 |
| 16 | 65.9 | 23.0 | 180.8 | 72.8 |
| Glu | | | | |
| 1' | | | | 102.5 |
| 2' | | | | 75.0 |
| 3' | | | | 77.9 |
| 4' | | | | 71.7 |
| 5' | | | | 78.1 |
| 6' | | | | 62.8 |
| N-Me A | | 37.2 | 40.2 | 40.2 |
| N-Me B | | | 40.2 | 40.2 |
| 11-OAc | | 170.5 | | |
| 11-OAc | | 21.1 | | |
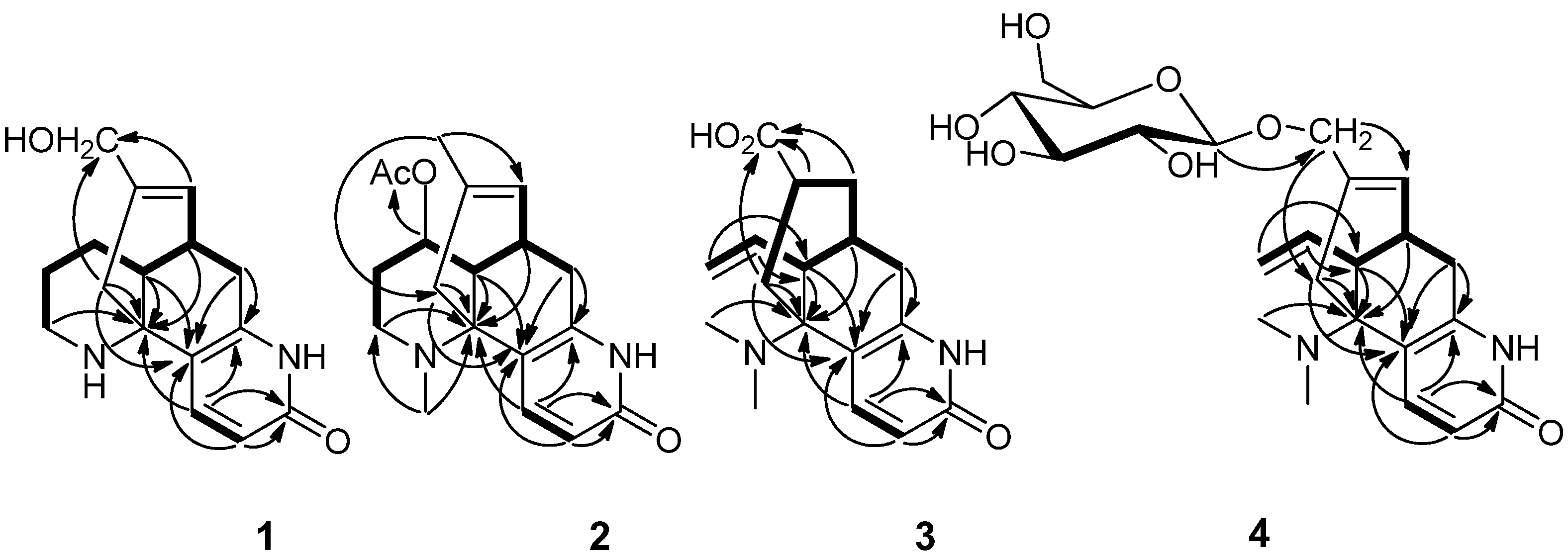
The most significant difference between two compounds was that the presence of an additional hydroxyl group in compound
1. The hydroxyl group might be localized to C-16, which was confirmed by HMBC correlations between the C-16 (
δC 65.9) with H-14, H-8 (
Figure 2).
Figure 2.
Key HMBC (H→C) correlations and 1H-1H COSY (▬) of compounds 1–4.
Figure 2.
Key HMBC (H→C) correlations and 1H-1H COSY (▬) of compounds 1–4.
The relative configuration of compound
1 was provided by the NOE difference spectra. In the biogenetic consideration of lycodine-type alkaloids derivatives isolated from Lycopodiaceae species, H-12 was assigned as the β-orientation. Irradiation of H-12 enhanced the signals of H-14a, thus, the H-7 was
α-oriented. The specific rotation of compound
1 was determined to be
![Molecules 19 09999 i001]()
−70 (
c 0.1, MeOH), which was similar to the value of
![Molecules 19 09999 i001]()
−54.2 (
c 0.2, MeOH) observed for huperzine B (
9) [
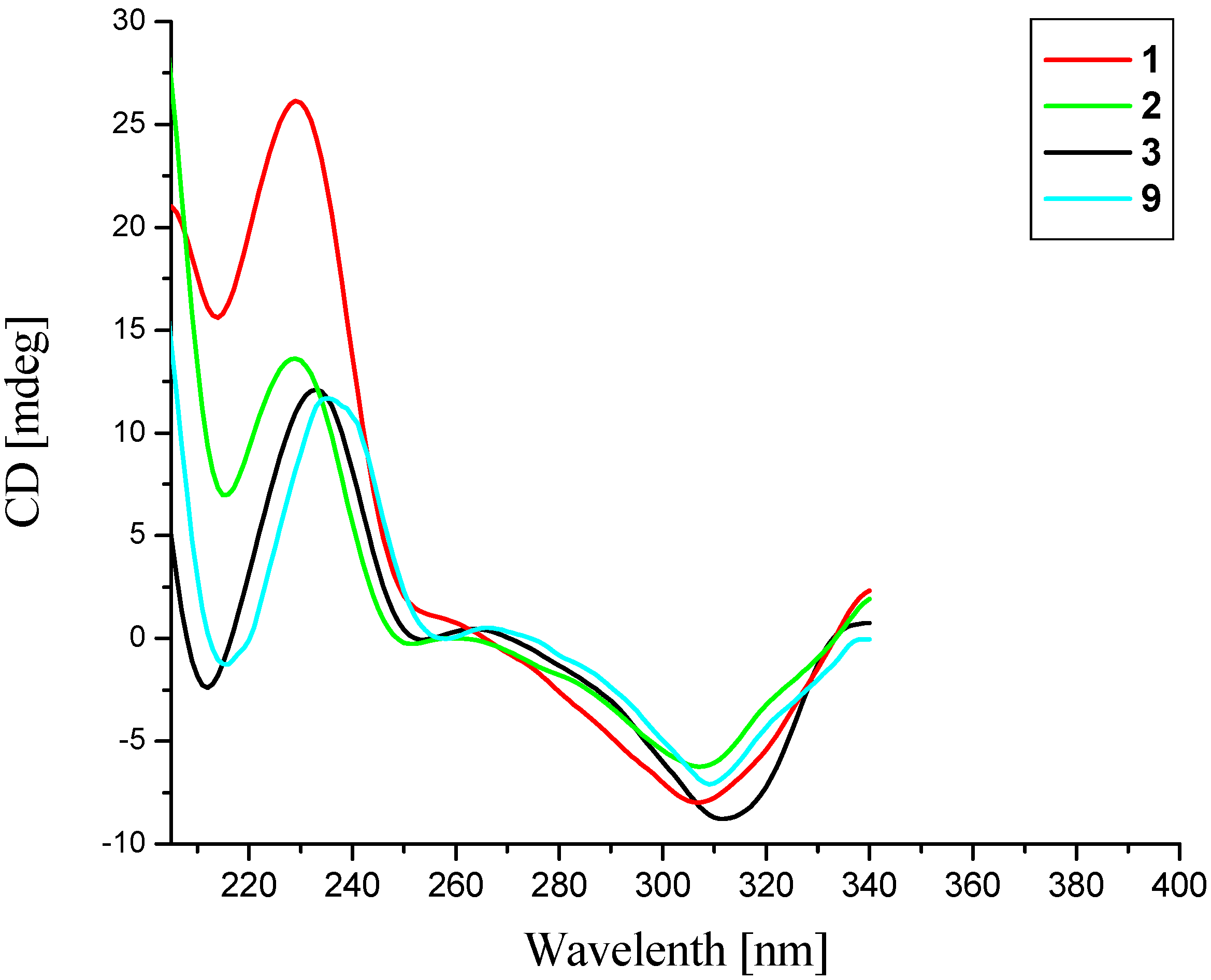
12]. Also, the CD spectrum of compound
1 (
Figure 3) showed a positive Cotton effect around 230 nm and a negative one near 307 nm, which was in agreement with that of huperzine B (
9) [
12], indicating a (13
R) configuration [
13]. Taken together, the absolute configuration of compound
1 was assigned to be 7
S/12
R/13
R. Finally, the structure of compound
1 was elucidated and named as 16-hydroxyhuperzine B.
Figure 3.
CD spectra of compounds 1–3 and 9.
Figure 3.
CD spectra of compounds 1–3 and 9.
Compound
2, obtained as a yellow powder, has an [M+H]
+ ion peak at
m/z 329.1863 in its HRESIMS, corresponding to the molecular formula of C
19H
24N
2O
3 (calcd. for C
19H
25N
2O
3, 329.1860), implying nine degrees of unsaturation. The NMR data of compound
2 were similar to those of huperzine B (
9) [
12], except for the presence of an additional acetoxy group [
δH 2.04 (3H, s),
δC 21.1, 170.5] and one methyl group [
δH 2.63 (3H, s),
δC 37.2]. The acetoxy group might be localized at C-11 in compound
2, which was supported by the shift of the C-11 signal in compound
2 (
δC 71.2) to lower field region relative to huperzine B (
9) (
δC 28.1), and was further confirmed by observed key HMBC correlations from H-11 (
δH 4.76) to the acetoxy carbonyl carbon at
δC 170.5 (
Figure 2). The methyl could be attached to the
N-atom, which was confirmed by HMBC correlations between the proton at
δH 2.63 with C-13 (
δC 58.1) and C-9 (
δC 48.6) (
Figure 2). The relative configuration of compound
2 was deduced by the NOE difference spectra experiment and the coupling constants. Like compound
1, the H-12 was assigned a β-orientation and the H-7 was
α-oriented. Furthermore, the large coupling constant between H-11 and H-12 (
J11, 12 = 10.8 Hz) indicated the vicinal protons H-11 (
δH 4.76) and H-12 (
δ 2.07) both took axial orientations. The CD spectrum of compound
2 showed a positive Cotton effect at 229 nm and a negative one at 307 nm (
Figure 3), which matched well with that of huperzine B (9) [
12]. Thus, the absolute configuration of compound
2 was assigned as 7
R/11
R/12
R/13
R.
Compound
3, a white powder, showed an [M+H]
+ ion peak at
m/z 303.1709 in its HRESIMS, corresponding to the molecular formula of C
17H
22N
2O
3 (calcd. for C
17H
23N
2O
3, 303.1703), implying eight degrees of unsaturation. In the
1H-NMR spectrum of compound
3, an ABX spin system for the exomethylene moiety (CH
2 = CH) resonating at
δH 6.02 (1H, ddd,
J = 16.8, 10.2, 10.2 Hz), 5.33 (1H, dd,
J = 16.8, 1.8 Hz) and 5.10 (1H, dd,
J = 10.2, 1.8 Hz) suggested that compound
3 was a piperidine ring (C ring) cleavage product. The NMR data of compound
3 were highly similar to those of lycoparin A (
14) [
8], except for the fact the Δ
8(15) double bond was saturated, which implied the structure of compound
3 to be 8,15-dihydrolycoparin A, which was supported by its 2D NMR experiments (
Figure 2). In particular, a long spin system [−CH
2−CH(X)−CH
2−CH−CH
2−(H
2-6/H-7(X)/H
2-8/H-15/H
2-14), X = −CH−CH−CH
2−(H-12/H-11/H
2-10)] was displayed in the
1H-
1H COSY spectrum of compound
3. The relative configuration of compound
3 was deduced by the NOE difference spectra experiment and the coupling constants. In the consideration of the biogenesis of the lycodine-type alkaloid derivatives isolated from Lycopodiaceae species, the H-12 was assigned as the β-orientation. Irradiation of H-12 enhanced the signals of H-8b and H-14a, indicating that these protons were on the same facial plane, the H-7 was therefore assigned as the
α-orientation. The large coupling constant between H-15 and H-14a (
J14a,
15 = 12.6 Hz) indicated that these two protons both took axial orientations, thus, H-15 was on the other side. Similarly, the CD curve of compound
3 showed a positive Cotton effect around 230 nm and a negative value near 310 nm (
Figure 3). Consequently, the absolute configuration of compound
3 was established as 7
S/12
R/13
R/15
R.
Compound
4, obtained as a colorless powder, showed a molecular ion peak at
m/z 449.2288 [M+H]
+ in HRESIMS, consistent with the molecular formula of C
23H
32N
2O
7 (calcd. for 449.2282, C
23H
33N
2O
7) requiring nine degrees of unsaturation. The strong IR absorptions at 3,385 and 1,658 cm
−1 indicated the presence of hydroxyl and carbonyl groups, respectively. The NMR data of compound
4 were analogous to those of huperzine D (
12) [
11] with a C-15 hydroxymethyl group except for a set of signals of one hexose group including the anomeric proton signal at
δH 4.22 (1H, d,
J = 7.8 Hz), the other 6 proton signals at
δH 3.17–3.85, the anomeric carbon signal at
δC 102.5 (C-1) and another 5 carbon signals at
δC 62–78. The hexose was suggested to be a D-glucose by the comparison of
13C-NMR data with those reported in the literature, and further confirmed by TLC comparison with an authentic sample and by its specific rotation value after acidic hydrolysis of compound
4 in HCl-methanol (9%) yielded huperzine D (
12) and glucose [
14]. The coupling constant of H-1' (
δH 4.22, d,
J = 7.8 Hz) indicated the D-glucose had a β-linkage. The C-16 of compound
4 should be glycosylated, which was confirmed by the observed key HMBC correlation from the anomeric proton at H-1' (
δH 4.22) to C-16 (
δC 72.8) (
Figure 2). Accordingly, the structure of compound
4 was assigned as (7
S, 12
S, 13
R)-huperzine D-16-O-β-D-glucopyranoside.
By comparison of our spectroscopic data with those reported in the literature, the remaining known compounds were identified as huperzinine (
5) [
11], casuarinine A (
6) [
13],
N-demethylhuperzinine (
7) [
10], huperzine C (
8) [
11], huperzine B (
9) [
12], casuarinine E (
10) [
13], carinatumin B (
11) [
15], huperzine D (
12) [
11], lycoparin C (
13) [
8], and lycoparin A (
14) [
8], respectively.
Lycodine-type alkaloids are a group of structurally unique secondary metabolites, characterized by four or three connected six-membered rings, including a pyridone or pyridine ring (ring A), a piperidine ring (ring C), and a bicyclo[3.3.1]nonane core formed by rings B and D. Many of them continued to be the hot spots of research interest because of their promising bioactivity [
16] and challenging total synthesis [
17,
18]. In our study, four new lycodine-type alkaloids and ten known analogues were isolated from
L.
casuarinoides. As far as we know, compound
4 is the first reported example of a lycopodium alkaloidal glycoside. It may be worthwhile to point out that we initially doubted the origin of compound
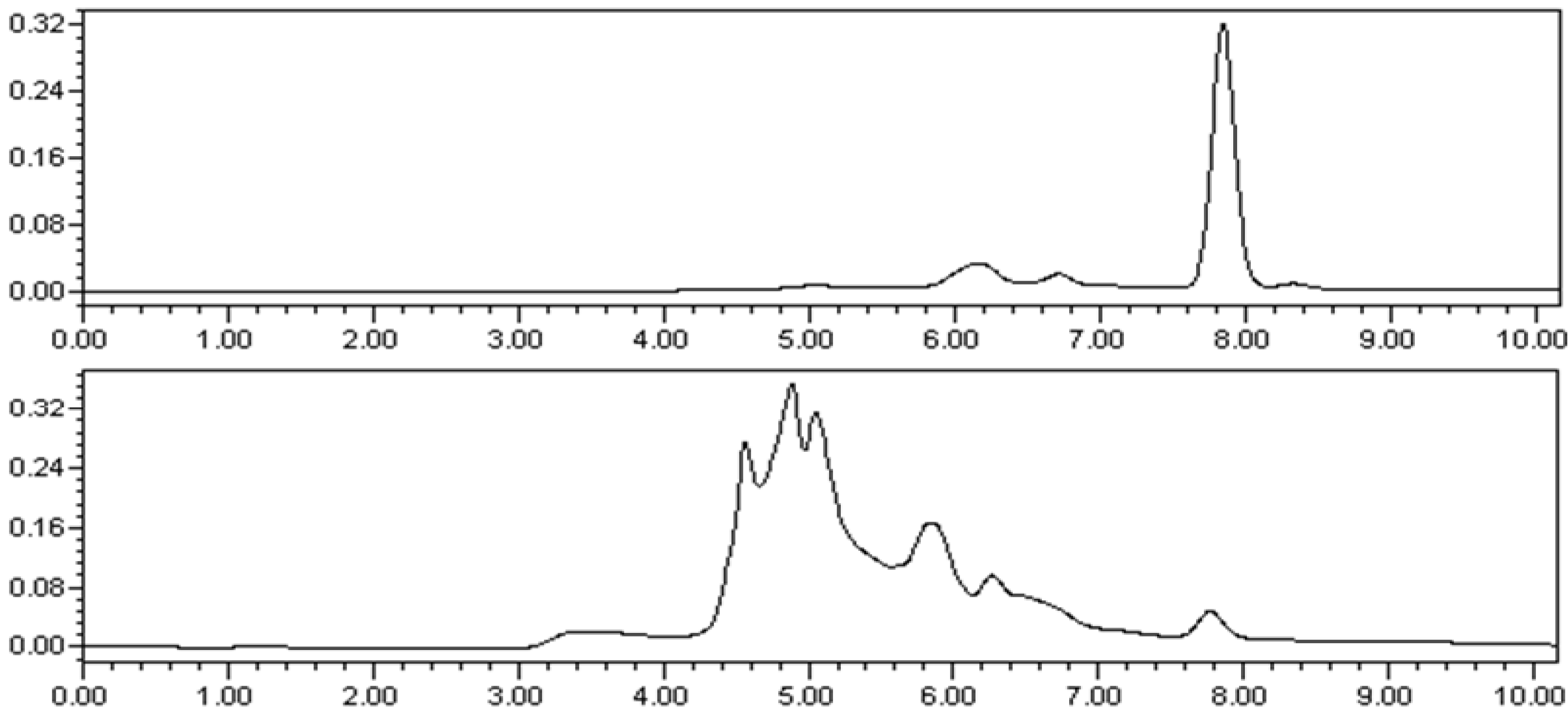
2 because it could be an artifact produced from the present extraction and isolation procedure, which employed EtOAc. In order to rule out that possibility, the MeOH extract of the plant was re-examined by the HPLC with pure compound
2 as reference (
Figure 4). The unequivocal detection of the same compound in the original extract with an identical HPLC t
R value as that of the reference compound proved that compound
2 was a natural product and not an artifact.
Figure 4.
(A) HPLC analysis of compound 2 (retention time: 7.848 min); (B): HPLC analysis of the MeOH extracts of L. casuarinoides (retention time: 7.821 min). HPLC analyses were performed on Waters 1525-2998 series HPLC system (C-18 column, Sun Fire, 5 μm, 4.6 mm × 150 mm); mobile phase, CH3CN/H2O = 7/3; flow rate, 1.0 mL/min; injection volume, 10 μL).
Figure 4.
(A) HPLC analysis of compound 2 (retention time: 7.848 min); (B): HPLC analysis of the MeOH extracts of L. casuarinoides (retention time: 7.821 min). HPLC analyses were performed on Waters 1525-2998 series HPLC system (C-18 column, Sun Fire, 5 μm, 4.6 mm × 150 mm); mobile phase, CH3CN/H2O = 7/3; flow rate, 1.0 mL/min; injection volume, 10 μL).
The isolated lycodine-type alkaloids
1–
14 were evaluated for their AChE inhibiting activity by Ellman’s method in 96-well microplates [
19,
20]. The results are listed in
Table 3. Huperzine A with an IC
50 value of 74.3 nM was used as a reference compound, and it showed good agreement with literature data (IC
50 = 72.4 nM) [
21]. Huperzine C (
8) possessed the most potent inhibition against AChE, with an IC
50 value of 0.6 μM. Also,
N-demethylhuperzinine (
7), huperzine B (
9) and lycoparin C (
13) showed significant AChE inhibitory activity with IC
50 values of 1.9, 20.2 and 23.9 μM, respectively, and the new compound
1 exhibited moderate inhibitory activity with IC
50 value of 87.3 µM. Interestingly,
N-demethylhuperzinine (
7), huperzine C (
8), huperzine B (
9) and lycoparin C (
13) have been previously reported to show inhibition of AChE activity with IC
50 values of 15.0, 0.489, 19.3 and 25.0 µM, repectively [
8,
13,
21]. Our reports were found to be close to the literature data. In fact, the inhibiton of huperzine C (
8) with an amino group at C-13 was 3-fold higher than that of
N-demethyl-huperzinine (
7), and huperzinine (
5) show very weak activity, suggested that the
N-methyl group on the position 13 might cause the sharp decrease in AChE inhibition. In other words, the amino group on the positions 13 may be a structural requirement for the anti-AChE activity of lycodine-type alkaloids, which was also supported by comparing the structure-activity relationships between casuarinine E (
10) (IC
50 > 250 μM) and lycoparin C (
13). Morever, the activity of huperzine C (
8) with a methyl at C-15 was 40-fold higher than those of lycoparin C (
13) with a hydroxymethyl at C-15, indicated the methyl of the three-carbon bridge ring was important for AChE inhibition in this kind of alkaloids. Interestingly, huperzine A is very similar to huperzine C (
8), except for the position of double bond, but the activity of the former was 8-fold higher than that of the latter, implying the exocyclic double bond was required for high anti-AChE activity. The findings mentioned above were consistent with previous observations [
22].
Table 3.
AChE inhibiting activity of compounds 1, 7, 8, 9 and 13.
Table 3.
AChE inhibiting activity of compounds 1, 7, 8, 9 and 13.
| Compounds | IC50 (μM) |
|---|
| Compound 1 | 87.3 ± 1.9 |
| N-Demethylhuperzinine (7) | 1.9 ± 0.2 |
| Huperzine C (8) | 0.6 ± 0.1 |
| Huperzine B (9) | 20.2 ± 1.3 |
| Lycoparin C (13) | 23.9 ± 2.2 |
| Huperzine A a | (74.3 ± 2.8) ×10−3 |
3. Experimental
3.1. General
Optical rotations were measured on a Perkin-Elmer 341 polarimeter. IR spectra were recorded on a 170SX FT-IR instrument using KBr discs over the range of 400–4,000 cm−1. UV spectra were measured using a Shimadzu UV-260 spectrophotometer. CD spectra were obtained on an Olis DSM 1000 spectrometer. NMR spectra were recorded on a Bruker AM-400 and a Varian Mercury-600BB NMR (600 MHz) spectrometer using TMS as an internal standard. High-resolution electrospray ionization mass spectra (HRESIMS) were measured on a Bruker Daltonics APEX II 47e spectrometer. Column chromatography was performed on silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, People’s Republic of China), Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) and RP-C18 (100–200 µm, Waters). Semipreparative HPLC was performed on a Waters 1525 binary pump system with a Waters 2489 detector (210 nm) using a YMC-Pack ODS-A (250 × 10 mm, S-5 μm) column. Fractions were monitored by TLC, which were visualized by heating the silica gel plates after being sprayed with 5% H2SO4 in EtOH.
3.2. Plant Material
The whole L. casuarinoides were collected in Tunchang County of Hainan Province, China (19°36′N; 110°12′E; elevation 328 m), in July 2009, and identified by Qiongxin Zhong of Hainan Normal University. The voucher specimen (No. 2009020) was deposited in the State Key Laboratory of Applied Organic Chemistry, Lanzhou University, China.
3.3. Extraction and Isolation
An air-dried and powdered sample (4.7 kg) was extracted with 95% MeOH three times (each time 30 L for 7 days) at room temperature. Evaporation of the solvent gave a residue (430 g), which was partitioned between EtOAc (3 × 2 L) and 2% HCl solution (2 L). The acidic water-soluble materials, adjusted to pH 9–10 with 10% ammonia solution, were extracted with CHCl3 (4 × 2.5 L) and BuOH (4 × 2.5 L). The CHCl3 extract (4.3 g) was subjected to silica gel (Φ 4 × 40, 200–300 mesh, 400 g) column eluting with a CHCl3/MeOH (80:0, 80:1, 40:1, 20:1, 10:1, 5:1, 2:1, 1:1, 0:1, each 1.5 L) gradient system to give fractions 1–9. Fraction 3 (0.15 g) was chromatographed on silica gel column (Φ 1 × 10, 200–300 mesh, 8 g) eluting with PE/EtOAc/Et2NH (1:1:0.002, 0.1 L) to give compound 2 (6.0 mg). Huperzinine (5, 900 mg) was recrystallized in MeOH from fraction 4 (1.4 g). Fraction 5 (0.3 g) was subjected to silica gel column chromatography (Φ 1.5 × 15, 200–300 mesh, 30 g) eluting with CHCl3/EtOAc/MeOH/Et2NH (7:7:1:0.015, 0.15 L) to yield casuarinine A (6) (20.6 mg), N-demethyl-huperzinine (7) (15.6 mg) and huperzine C (8) (10.3 mg). Fraction 6 (0.3 g) was rechromatographed on silica gel (Φ 1.5 × 15, 200–300 mesh, 30 g) eluting with EtOAc/MeOH/Et2NH (10:1:0.01, 0.13 L) to give huperzine B (9) (15.7 mg). Fraction 7 (0.5 g) was chromatographed on a silica gel column (Φ 2 × 10, 200–300 mesh, 35 g) eluted with CHCl3/EtOAc/MeOH, (5:5:1) to afford three fractions (7A–C). Then fraction 7B was purified by reversed-phase preparative HPLC (MeOH/H2O, 28:72, v/v; flow rate, 2.0 mL/min) to yield compound 3 (1.2 mg, tR = 16.9 min), casuarinine E (10) (2.8 mg, tR = 20.9 min), carinatumin B (11) (18 mg, tR = 18.6 min) and huperzine D (12) (19.2 mg, tR = 35.3 min). The BuOH extract (2.3 g) was subjected to an HP-20 column (Φ 5 × 20, 0.8 kg) eluting with a H2O/MeOH (10:0, 9:1, 8:2, 7:3, 6:4, 5:5, each 2 L) gradient system to give fractions 1–6. Fraction 2 (0.15 g) was chromatographed on a reversed-phase column (Φ 1 × 15, 30 g) using ODS and was eluted with 20% MeOH (fractions 2A–B). Then fraction 2B was purified by reversed-phase preparative HPLC (MeOH/H2O, 16:84, v/v; flow rate, 2.0 mL/min) to yield lycoparin C (13) (3.1 mg, tR = 8.9 min), lycoparin A (14) (3.8 mg, tR = 11.2 min), compound 1 (1.8 mg, tR=19.3 min) and compound 4 (6.3 mg, tR = 12.6 min).
3.4. Spectral Data
16-Hydroxyhuperzine B (
1). Colorless gum;
![Molecules 19 09999 i001]()
= −70 (
c 0.1, MeOH); IR (KBr)
νmax: 3340, 2919, 2850, 1654, 1611, 1464, 1115, 752 cm
−1; UV (MeOH) λ
max (log
ε): 227 (3.90), 306 (3.81) nm;
1H-NMR (CD
3OD, 600 MHz) and
13C-NMR (CD
3OD, 150 MHz) data: see
Table 1 and
Table 2; HRESIMS
m/z 273.1600 [M+H]
+ (273.1598 calcd. for C
16H
21N
2O
2).
N-Methyl-11-acetoxyhuperzine B (
2). Yellow powder;
![Molecules 19 09999 i001]()
= −50 (
c 0.1, MeOH); IR (KBr)
νmax: 3373, 2919, 2850, 1658, 1598, 1456, 1095, 761 cm
−1; UV (MeOH) λ
max (log
ε): 226 (3.86), 306 (3.79) nm;
1H-NMR CDCl
3, 400 MHz) and
13C-NMR (CDCl
3, 100 MHz) data: see
Table 1 and
Table 2; HRESIMS
m/z 329.1863 [M+H]
+ (329.1860 calcd. for C
19H
25N
2O
3).
8,15-Dihydrolycoparin A (
3). White powder;
![Molecules 19 09999 i001]()
= −120 (
c 0.1, MeOH); IR (KBr)
νmax: 3384, 2919, 2851, 1658, 1609, 1462, 1119, 796 cm
−1; UV (MeOH) λ
max (log
ε): 227 (3.85), 306 (3.75) nm;
1H-NMR (CD
3OD, 600 MHz) and
13C-NMR (CD
3OD, 150 MHz) data: see
Table 1 and
Table 2; HRESIMS
m/z 303.1709 [M+H]
+ (303.1703 calcd. for C
17H
23N
2O
3).
(7S,12S,13R)-HuperzineD-16-O-β-d-glucopyranoside (
4). Colorless powder;
![Molecules 19 09999 i001]()
= −70 (
c 0.2, MeOH); IR (KBr)
νmax: 3385, 2919, 2851, 1658, 1602, 1459, 1093, 761 cm
−1; UV (MeOH) λ
max (log
ε): 227 (3.81), 305 (3.69) nm;
1H-NMR (CD
3OD, 400 MHz) and
13C-NMR (CD
3OD, 100 MHz) data: see
Table 1 and
Table 2; HRESIMS
m/z 449.2288 [M+H]
+ (449.2282 calcd. for C
23H
33N
2O
7).
3.5. Hydrolysis of Compound 4
Compound 4 (5 mg) was dissolved in 9% dry HCl-Methanol (2 mL) at 80 ºC for 3 h. After neutralization with NaHCO3, the mixture was evaporated. The residue was resuspended in H2O and then filtered to yield compound 1 (1.2 mg). The sugar components in the filtrate were identified by TLC (on silica gel, developed with CHCl3/MeOH = 5:1) as D-(+)-glucose (Rf 0.40) by comparison with an authentic sample.
3.6. Biological Material
AChE (EC3.1.1.7, Sigma product no. C2888), acetylthiocholine iodide (ATCI), 5,5'-dithiobis [2-nitrobenzoic acid] (DTNB) and huperzine A (purity 98%) were purchased from Sigma (St. Louis, MO, USA).
3.7. Assay of AChE Inhibition
The fourteen compounds were tested for AChE inhibitory activities by the modified Ellman’s method in 96-well microplates [
19,
20]. Briefly, 0.1 M sodium phosphate buffer (140 µL, pH = 8.0), sample solution (20 µL) and enzyme solution (15 µL) were mixed and incubated at 4 °C for 20 min. Ten µL of 0.01 M DTNB was added and the reaction was then started by adding 0.075 M ATCI (10 µL). After incubating the reaction solution at 37 °C for 20 min, the optical densities were measured in a 96-well plate reader at 405 nm immediately. A blank positive control was set up by adding 20 µL huperinze A (0.100 µg/mL in phosphate buffered saline) instead of 20 µL sample solution. Blanks were set up by adding 20 µL buffer solutions instead of 20 µL sample solution. Experiment control was set up by adding 15 µL buffer solutions instead of 15 µL enzyme solution in order to deduct the sample background. All reactions were carried out thrice. The inhibition rate (%) was calculated by the following equation:
The concentration of test samples that inhibited the hydrolysis of acetylthiocholine by 50% (IC50) was determined by monitoring the effect of increasing concentrations of these samples in assays on the inhibition values. Huperzine A was chosen as the positive control, with an IC50 value of 74 nmol.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

 −70 (c 0.1, MeOH), which was similar to the value of
−70 (c 0.1, MeOH), which was similar to the value of 
