2.1. Determination of the Theoretical Geometrical Parameters for the 1,2,13-Trioxane Ring of Artemisinin (Bond Length, Bond Angle, and Torsion Angle of Atoms in this Ring) in Different Methods and Basis Sets
We determined the geometrical parameters for the 1,2,13-trioxane ring of artemisinin (bond length, bond angle, and torsion angle of atoms in this ring), as shown in
Table 1.
Figure 1.
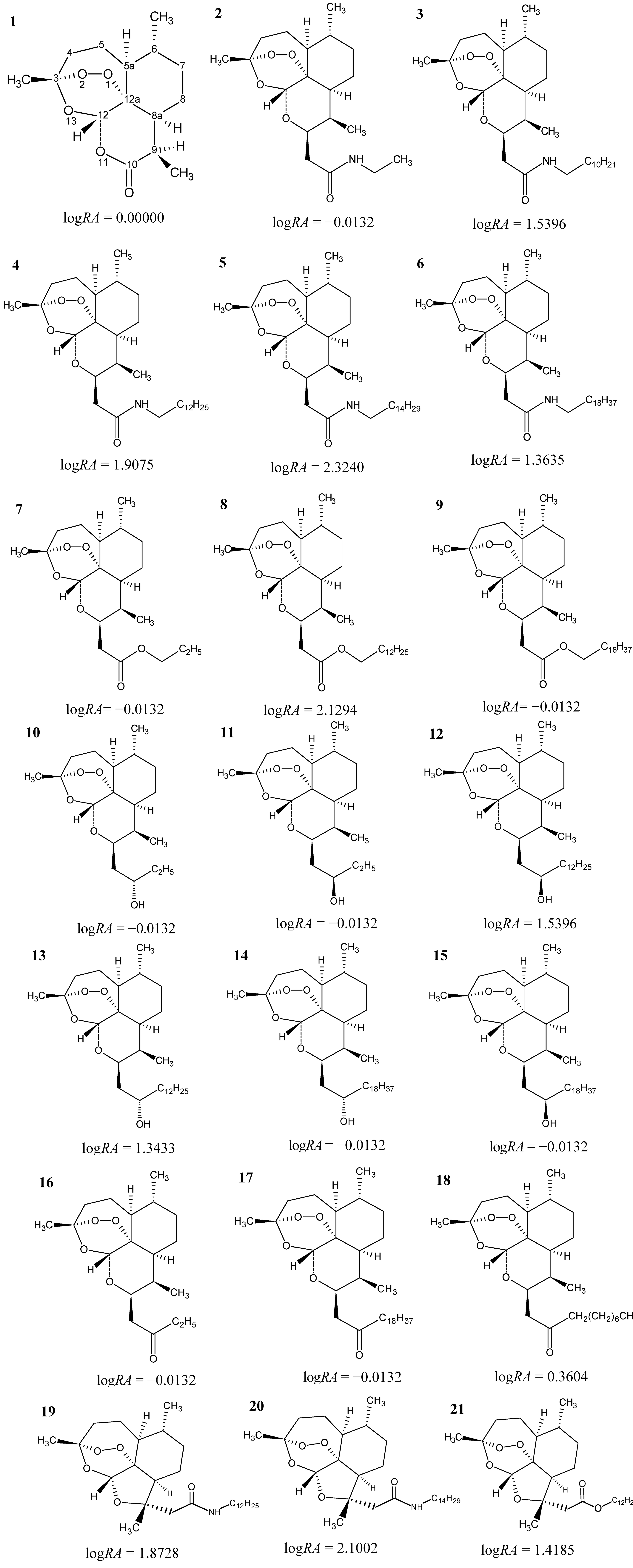
Structure and biological activity of artemisinin and derivatives with anticancer activity against human hepatocellular carcinoma HepG2 line.
Figure 1.
Structure and biological activity of artemisinin and derivatives with anticancer activity against human hepatocellular carcinoma HepG2 line.
Table 1.
Theoretical and experimental parameters for the 1,2,13-trioxane ring in artemisinin (compound 1).
Table 1.
Theoretical and experimental parameters for the 1,2,13-trioxane ring in artemisinin (compound 1).
| Parameters [a] | Semiempirical | Hartree-Fock/HF | DFT/B3LYP | EXP [29] |
|---|
| AM1 | PM3 | ZINDO | 6-31G | 6-31G* | 6-31G** | 3-21G | 3-21G* | 3-21G** | 6-311G | 6-31G | 6-31G* | 6-31G** [b] | 3-21G |
|---|
| Bond length (Å) |
| O1O2 | 1.288 | 1.544 | 1.237 | 1.447 | 1.391 | 1.390 | 1.461 | 1.461 | 1.462 | 1.429 | 1.524 | 1.459 | 1.459 | 1.524 | 1.469 |
| O2C3 | 1.447 | 1.403 | 1.400 | 1.435 | 1.393 | 1.396 | 1.440 | 1.440 | 1.439 | 1.432 | 1.452 | 1.413 | 1.414 | 1.455 | 1.416 |
| C3O13 | 1.427 | 1.428 | 1.396 | 1.435 | 1.388 | 1.408 | 1.436 | 1.435 | 1.435 | 1.434 | 1.473 | 1.441 | 1.441 | 1.473 | 1.445 |
| O13C12 | 1.416 | 1.403 | 1.392 | 1.403 | 1.400 | 1.376 | 1.407 | 1.407 | 1.407 | 1.401 | 1.425 | 1.395 | 1.396 | 1.430 | 1.379 |
| C12C12a | 1.537 | 1.555 | 1.513 | 1.533 | 1.533 | 1.532 | 1.529 | 1.529 | 1.530 | 1.530 | 1.438 | 1.539 | 1.539 | 1.535 | 1.523 |
| C12aO1 | 1.468 | 1.427 | 1.416 | 1.469 | 1.429 | 1.429 | 1.477 | 1.477 | 1.477 | 1.438 | 1.499 | 1.455 | 1.455 | 1.504 | 1.461 |
| Bond angle (°) |
| O1O2C3 | 112.530 | 110.340 | 114.310 | 108.800 | 106.100 | 109.460 | 107.100 | 107.080 | 107.060 | 109.210 | 107.300 | 108.280 | 108.280 | 105.590 | 108.100 |
| O2C3O13 | 103.600 | 104.810 | 105.370 | 106.760 | 110.800 | 107.800 | 107.270 | 107.285 | 107.300 | 106.670 | 107.730 | 108.490 | 108.490 | 108.220 | 106.600 |
| C3O13C12 | 115.480 | 116.010 | 115.843 | 117.300 | 112.800 | 115.300 | 115.670 | 115.680 | 115.710 | 116.960 | 114.990 | 114.080 | 114.060 | 113.200 | 114.200 |
| O13C12C12a | 113.510 | 115.200 | 113.270 | 112.280 | 108.700 | 112.300 | 112.080 | 112.080 | 112.030 | 112.360 | 113.640 | 113.250 | 113.240 | 113.300 | 114.500 |
| C12C12aO1 | 111.070 | 113.180 | 107.290 | 110.910 | 110.500 | 110.545 | 111.570 | 111.600 | 111.600 | 110.760 | 111.740 | 111.290 | 111.280 | 112.410 | 110.700 |
| C12aO1O2 | 113.740 | 112.290 | 118.380 | 113.240 | 112.700 | 112.700 | 111.290 | 111.290 | 111.290 | 113.360 | 111.400 | 111.600 | 111.590 | 109.620 | 111.200 |
| Torsion angle (°) |
| O1O2C3O13 | −77.800 | −73.310 | −70.403 | −71.840 | −73.369 | −73.400 | −74.670 | −74.700 | −74.690 | −71.940 | −73.460 | −73.900 | −73.910 | −76.610 | −75.500 |
| O2C3O13C12 | 42.070 | 52.700 | 36.370 | 33.390 | 31.034 | 31.100 | 32.300 | 32.360 | 32.180 | 33.010 | 34.970 | 32.800 | 32.780 | 33.750 | 36.000 |
| C3O13C12C12a | 11.400 | 2.811 | 17.420 | 25.320 | 27.432 | 27.400 | 28.290 | 28.190 | 28.330 | 25.380 | 26.260 | 27.460 | 25.500 | 29.060 | 25.300 |
| O13C12C12aO1 | −41.770 | −40.510 | −46.610 | −49.410 | −50.100 | −50.143 | −50.860 | −50.770 | −50.700 | −49.470 | −51.200 | −51.270 | −51.340 | −52.190 | −51.300 |
| C12C12aO1O2 | 12.050 | 19.940 | 18.110 | 12.510 | 10.900 | 10.924 | 9.989 | 9.940 | 9.750 | 12.480 | 12.740 | 11.730 | 11.780 | 9.060 | 12.700 |
| C12aO1O2C3 | 47.050 | 35.630 | 40.130 | 46.700 | 48.700 | 48.674 | 50.330 | 50.350 | 50.530 | 46.870 | 46.900 | 47.850 | 47.830 | 51.060 | 47.800 |
| Standard Deviation | 4.776 | 8.388 | 4.372 | 1.663 | 2.484 | 1.762 | 1.722 | 1.714 | 1.797 | 1.658 | 0.843 | 1.227 | 1.103 | 1.915 | ˗ |
Table 1 illustrates that for the DFT method, all four basis sets (B3LYP/6-31G, B3LYP/6-31G*, B3LYP/6-31G**, and B3LYP/3-21G) can accurately describe all of the structural parameters with respect to their magnitude and sign when compared with the experimental values.
Meanwhile, in the semiempirical (AM1, PM3, and ZINDO) and Hartree-Fock (HF/6-31G, HF/6-31G*, HF/6-31G**, HF/3-21G, HF/3-21G*, HF/3-21G**, and HF/6-311G) methods there is not good agreement between the experimental and theoretical values for the torsion angles, especially the angle formed by atoms O2C3O13C12, with deviations −6.070° (AM1), −16.700° (PM3), −0.370° (ZINDO), +2.610° (HF/6-31G), +4.966° (HF/6-31G*), +4.900° (HF/6-31G**), +3.700° (HF/3-21G), +3.640° (HF/3-21G*), +3.820° (HF/3-21G**), +2.990° (HF/6-311G), +1.030° (B3LYP/6-31G), +3.200° (B3LYP/6-31G*), +3.220° (B3LYP/6-31G**) and +2.250° (B3LYP/3-21G) and exhibited standard deviations of 4.776 (AM1), 8.388 (PM3), 4.372 (ZINDO), 1.663 (HF/6-31G), 2.484 (HF/6-31G*), 1.762 (HF/6-31G**), 1.722 (HF/3-21G), 1.714 (HF/3-21G*), 1.797 (HF/3-21G**), 1.658 (HF/6-311G), 0.843 (B3LYP/6-31G), 1.227 (B3LYP/6-31G*), 1.103 (B3LYP/6-31G**) and 1.915 (B3LYP/3-21G), respectively.
Table 1 shows that for artemisinin (compound 1) the B3LYP/6-31G, B3LYP/6-31G*, B3LYP/6-31G** basis sets show excellent results for bond length, bond angle and torsion angle compared to the experimental data. The B3LYP/6-31G method described geometrical parameters well, with values close to the experimental results. However, the minimum base 6-31G has several deficiencies; thus, a polarization function was included to improve upon this base (
i.e.,
p orbitals represented by *). Thus, 6-31G* refers to basis set 6-31G with a polarization function for heavy atoms (
i.e., atoms other than hydrogen), and 6-31G** refers to the inclusion of a polarization function for hydrogen and helium atoms [
29,
30,
31,
32,
33,
34,
35].
When basis sets with polarization functions are used in calculations involving anions, good results are not obtained due to the electronic cloud of anionic systems, which tend to expand. Thus, appropriate diffuse functions must be included because they allow for a greater orbital occupancy in a given region of space. It then becomes necessary to include diffuse functions in the basis function associated with the configuration of a neutral metal atom to obtain a better description of the metal complex. The 6-31G** basis is particularly useful in the case of hydrogen bonds [
30,
31,
32,
33,
34,
35].
Cristino
et al. [
36] used the B3LYP/6-31G* method to model artemisinin and 19 10-substituted deoxoartemisinin derivatives, with different degrees of activity against the
Plasmodium falciparum D-6 strains of Sierra Leone. Chemometric methods (PCA, HCA, KNN, SIMCA, and SDA) were employed to reduce the dimensionality and to determine which subset of descriptors is responsible for the classification between more and less active agents.
Figueiredo
et al. [
37] conducted studies using the B3LYP/6-31G* method for antimalarial compounds against
Plasmodium falciparum K1. These studies led to multivariate models for artemisinin derivatives and series of dispiro-1,2,4-trioxolanes. The application of these models has enabled the prediction of activity for compounds designed without known biological activity. Moreover, a new series of antimalarial compounds is currently in the study phase.
Araújo
et al. [
38] used density functional theory (6-31G*) to verify the performance of a base set in reproducing experimental data, particularly geometrical parameters, and to calculate the interaction energies, electronic states, and geometrical arrangements for complexes composed of a heme group and artemisinin. The results demonstrated that the interaction between artemisinin and the heme group occurs at long distances through a complex in which the iron atom of the heme group retains its electronic characteristics, with the quintet state being the most stable. These results suggest that the interaction between artemisinin and heme is thermodynamically favorable.
Pereira
et al. [
39] studied four structures of artemisinin by reductive decomposition A, B1, B2, and B3 with 13 species (QHS, 1/2, 3, 4, 5, 5a, 6, 7, 18, 18a, 19, 20, and 21), and the structures of the studied species were analyzed in terms of geometrical parameters, Löwdin bond orders, atomic partial charges, spin densities, electronic energies, free energies, and entropy. These studies were carried out at the B3LYP/6-31G
** level.
Carvalho
et al. [
40] used the B3LYP/6-31G** method to study artemisinin and 31 analogues with antileishmanicidal activity against
Leishmania donovani. The authors proposed a set of 13 artemisinins, seven of which are less active and six of which that have not been tested; of these six, one is expected to be more active against
L. donovani.Barbosa
et al. [
41] performed molecular modeling and chemometric studies involving artemisinin and 28 derivatives exhibiting anticancer activity and the calculations of the compounds studied were performed at the B3LYP/6-31G** level.
By comparing these methods with the DFT method (see
Table 1), we find that all of the basis sets (B3LYP/6-31G, B3LYP/6-31G*, and B3LYP/6-31G**) have low standard deviations in relation to the semiempirical and Hartree-Fock methods at 0.843 (B3LYP/6-31G), 1.227 (B3LYP/6-31G*), and 1.103 (B3LYP/6-31G**). The variation was ±0.384 between B3LYP/6-31G and B3LYP/6-31G*, ±0.260 between B3LYP/6-31G and B3LYP/6-31G**, and ±0.124 between B3LYP/6-31G* and B3LYP/6-31G**. This study highlighted the B3LYP/6-31G** basis set, which is closer to the experimental results and shows good performance in the description when comparing the O2C3 and C3O13 bond length, O1O2C3 and C3O13C12 bond angles. The torsion angles or dihedral angle also showed good agreement with the experimental values reported in the literature, showing that with the 6-31G** basis set, the torsion angles O13C12C12aO1 and C12aO1O2C3 are closer to the artemisinin crystallographic data.
2.2. Principal Component Analysis (PCA) Results
The PCA results showed that the most important descriptors were the following: ALOGPS_logs, Mor29m, IC5 and GAP energy. They were chosen from the complete data set (1716 descriptors) and other variables were not selected because either they had a poor linear correlation with activity or they did not give a distinct separation between the more and less active.
The values of the important descriptors of each selected compound identified via PCA as well as the values of log
RA, relative activity (RA) and the
IC50 is the 50% inhibitory concentration are shown in
Table 2. The
Table 2 shows the Pearson correlation matrix between the descriptors and log
RA, and the correlation between pairs of descriptors is less than 0.2420, while the correlation between the descriptors and log
RA is less than 0.7459. The descriptors selected by PCA represent the characteristics necessary to separate between the more and less active with anticancer activity of these compounds against human hepatocellular carcinoma HepG2.
Table 2.
Physicochemical properties selected by PCA, experimental logRA values, IC50 and the correlation matrix.
Table 2.
Physicochemical properties selected by PCA, experimental logRA values, IC50 and the correlation matrix.
| Compounds | ALOGPS_log | Mor29m | IC5 | Gap Energy | logRA | RA | IC50/µΜ |
|---|
| 1- | −2.3500 | −0.3050 | 4.8620 | 0.2616 | 0.0000 | 1.0000 | 97 |
| 2- | −3.5200 | −0.3070 | 5.2530 | 0.2525 | −0.0132 | 0.9700 | 100 |
| 3+ | −6.3500 | −0.4550 | 5.6840 | 0.2521 | 1.5396 | 34.6417 | 2.8 |
| 4+ | −6.8400 | −0.5250 | 5.6240 | 0.2524 | 1.9075 | 80.8164 | 1.2 |
| 5+ | −7.1600 | −0.5140 | 5.5010 | 0.2527 | 2.3240 | 210.8628 | 0.46 |
| 6+ | −7.4900 | −0.5010 | 5.2250 | 0.2525 | 1.3635 | 23.0940 | 4.2 |
| 7- | −3.6400 | −0.2360 | 5.2170 | 0.2467 | −0.0132 | 0.9700 | 100 |
| 8+ | −7.0300 | −0.5260 | 5.5970 | 0.2462 | 2.1294 | 134.7100 | 0.72 |
| 9- | −7.6800 | −0.1790 | 5.1970 | 0.2462 | −0.0132 | 0.9700 | 100 |
| 10- | −3.6800 | −0.3650 | 5.2530 | 0.2367 | −0.0132 | 0.9700 | 100 |
| 11- | −3.6800 | −0.3050 | 5.2530 | 0.2359 | −0.0132 | 0.9700 | 100 |
| 12+ | −6.9700 | −0.3940 | 5.5080 | 0.2457 | 1.5396 | 34.6417 | 2.8 |
| 13+ | −6.9700 | −0.2910 | 5.5080 | 0.2552 | 1.3433 | 22.0444 | 4.4 |
| 14- | −7.4000 | −0.2280 | 5.1590 | 0.2217 | −0.0132 | 0.9700 | 100 |
| 15- | −7.4000 | −0.2280 | 5.1590 | 0.2287 | −0.0132 | 0.9700 | 100 |
| 16- | −3.7500 | −0.4430 | 5.1800 | 0.2194 | −0.0132 | 0.9700 | 100 |
| 17- | −7.6100 | −0.3330 | 5.1680 | 0.2177 | −0.0132 | 0.9700 | 100 |
| 18+ | −5.4900 | −0.3470 | 5.6380 | 0.2199 | 0.3604 | 2.2929 | 42.3 |
| 19+ | −6.7200 | −0.5520 | 5.5430 | 0.2491 | 1.8728 | 74.6105 | 1.3 |
| 20+ | −7.0600 | −0.5520 | 5.4190 | 0.2492 | 2.1002 | 125.9505 | 0.77 |
| 21+ | −6.8400 | −0.5150 | 5.5160 | 0.2449 | 1.4185 | 26.2119 | 3.7 |
| ALOGPS_
log | | 0.2420 | −0.4260 | 0.0497 | −0.5265 | - | - |
| Mor29m | | | −0.5892 | −0.2971 | −0.8249 | - | - |
| IC5 | | | | 0.1767 | 0.7459 | - | - |
| Gap energy | | | | | 0.5238 | - | - |
The results of the PCA model are presented in
Table 3. The model was constructed with three main components (3 PCs). The first principal component (PC1) describes 38.6537% of the total information, the second principal component (PC2) describes 21.5859%, and the third (PC3) 12.3501%. PC1 contains 48.3171% of the original data, and the combination of the first two components (PC1 + PC2) contains 75.2996%, and all three (PC1 + PC2 + PC3) explain 90.7373% of the total information, losing only 9.2627% of the original information. The descriptors ALOGPS_logs (0.4232), Mor29m (0.5937) and IC5 (−0.6223) contribute the most to PC1, while in PC2, the descriptor GAP energy (0.7746) is the primary contributor. The main components can be written as a linear combination of the selected descriptors. Mathematical expressions for PC1 (1) and PC2 (2) are shown below:
Table 3.
PCA and contribution of selected descriptors based on step multivariate analysis.
Table 3.
PCA and contribution of selected descriptors based on step multivariate analysis.
| Parameters | Main Component |
|---|
| PC1 | PC2 | PC3 |
|---|
| Variance (%) | 38.6537 | 21.5859 | 12.3501 |
| Cumulative variance (%) | 48.3171 | 75.2996 | 90.7373 |
| Molecular descriptors | | Contribution |
| | PC1 | PC2 |
| ALOGPS_log | | 0.4232 | 0.5936 |
| Mor29m | | 0.5937 | −0.1803 |
| IC5 | | −0.6223 | −0.1225 |
| Gap energy | | −0.2845 | 0.7746 |
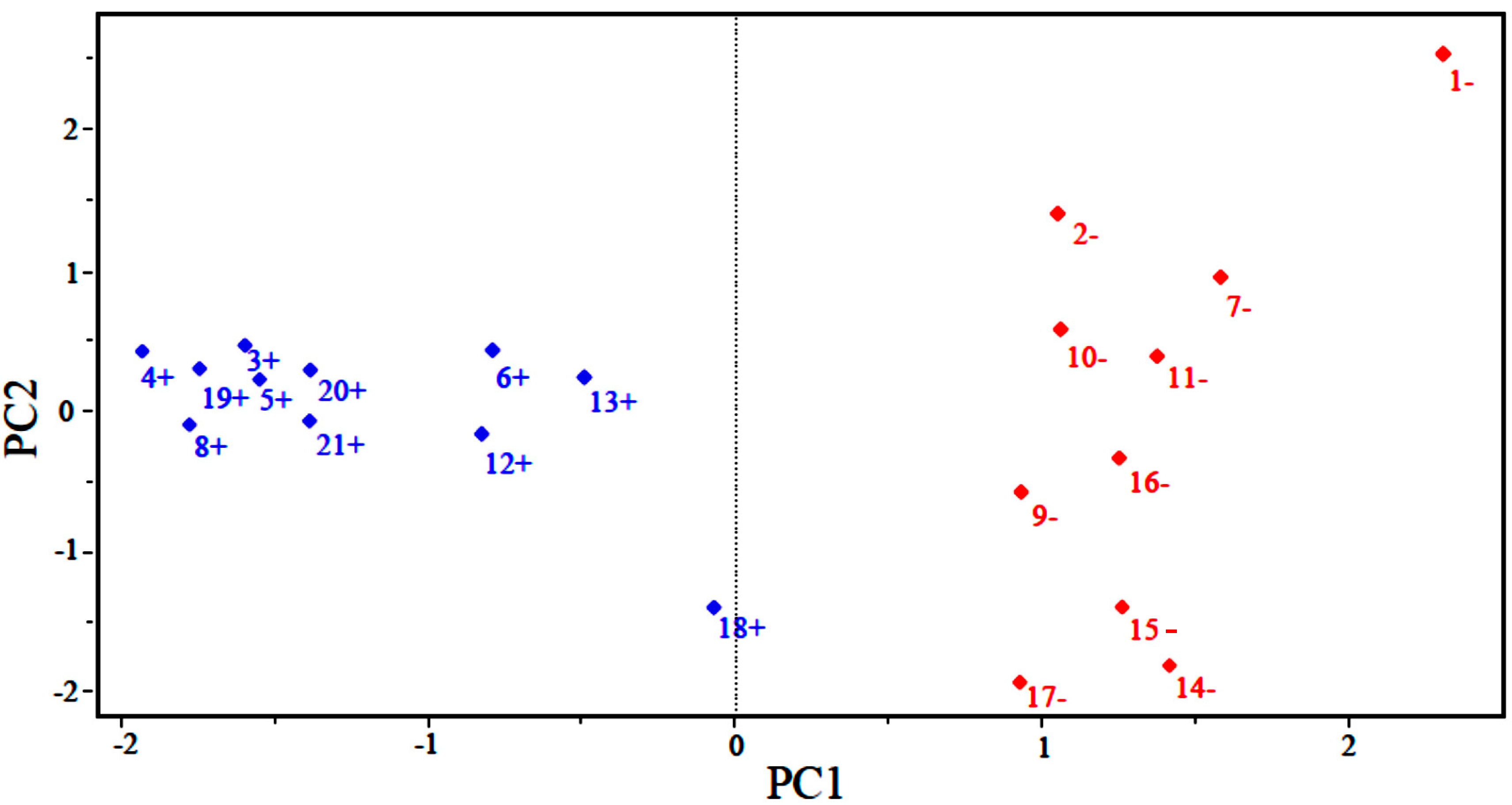
Figure 2 shows the scores for the 21 compounds studied. Based on the graph, PC1 distinguishes between compounds that are more potent and less potent. The most potent compounds are located at the left (3, 4, 5, 6, 8, 12, 13, 18, 19, 20 and 21), while the less potent compounds are located in the right side of the graph (1, 2, 7, 9, 10, 11, 14, 15, 16 and 17).
Figure 2.
Plot of PC1–PC2 scores for artemisinin and derivatives with anticancer activity against human hepatocellular carcinoma HepG2 line. Positive values indicate more potent analogs (in blue), and negative values indicate less potent analogs (in red).
Figure 2.
Plot of PC1–PC2 scores for artemisinin and derivatives with anticancer activity against human hepatocellular carcinoma HepG2 line. Positive values indicate more potent analogs (in blue), and negative values indicate less potent analogs (in red).
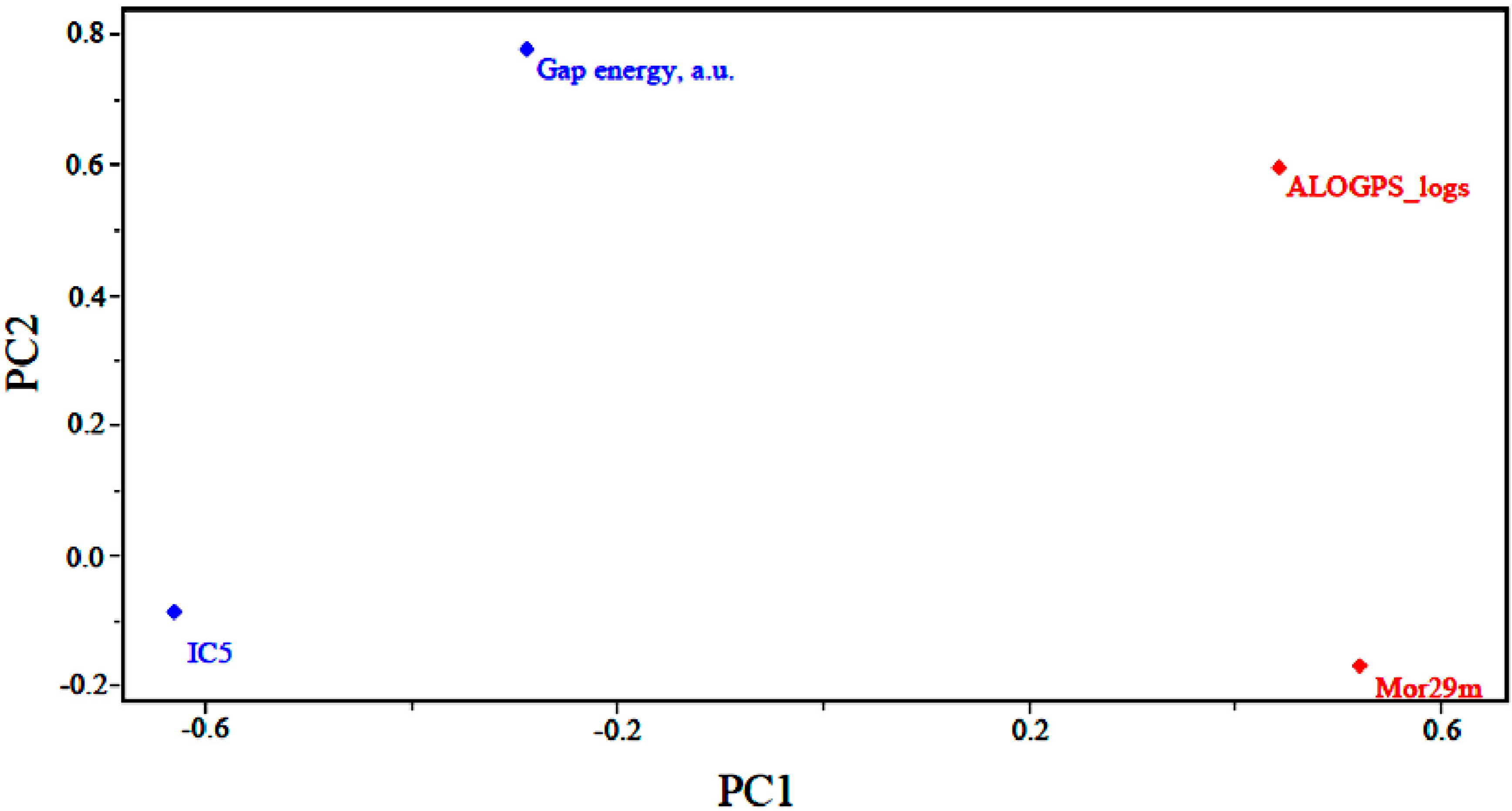
Figure 3 shows the loadings for the four (4) descriptors that are most important in the classification of compounds. Less potent compounds have high contributions from the descriptors ALOGPS_logs and Mor29m, while more potent compounds have a high contribution from the descriptor GAP energy and IC5. Thus, the descriptors GAP energy and IC5 are responsible for the location of more potent compounds at the left side of the graph. The descriptors ALOGPS_logs and Mor29m places less potent compounds in the right part of the graph.
Figure 3 also shows that the higher the contribution of the descriptors ALOGPS_logs and Mor29m in the first principal component,
i.e., the higher the value for a certain compound, the higher the score value will be, indicating that the compound is less potent than others. The other descriptors contribute to a lesser degree. For example, the descriptor GAP energy has negative weight in PC1, demonstrating that the most potent compounds generally have lower values of this descriptor.
Figure 3.
Plot of the PC1–PC2 loadings with the four descriptors selected to build the PLS and PCR models of artemisinin and derivatives with biological activity against human hepatocellular carcinoma HepG2 line.
Figure 3.
Plot of the PC1–PC2 loadings with the four descriptors selected to build the PLS and PCR models of artemisinin and derivatives with biological activity against human hepatocellular carcinoma HepG2 line.
2.3. Hierarchical Cluster Analysis (HCA) Results
The HCA method classified the compounds into two classes (more active and less active) and was based on the Euclidean distance and the incremental method [
42]. In the incremental linkage, the distance between two clusters is the maximum distance between a variable in one cluster and a variable in the other cluster. The descriptors employed to perform HCA were the same as those used for PCA,
i.e., ALOGPS_logs, Mor29m, IC5 and GAP energy.
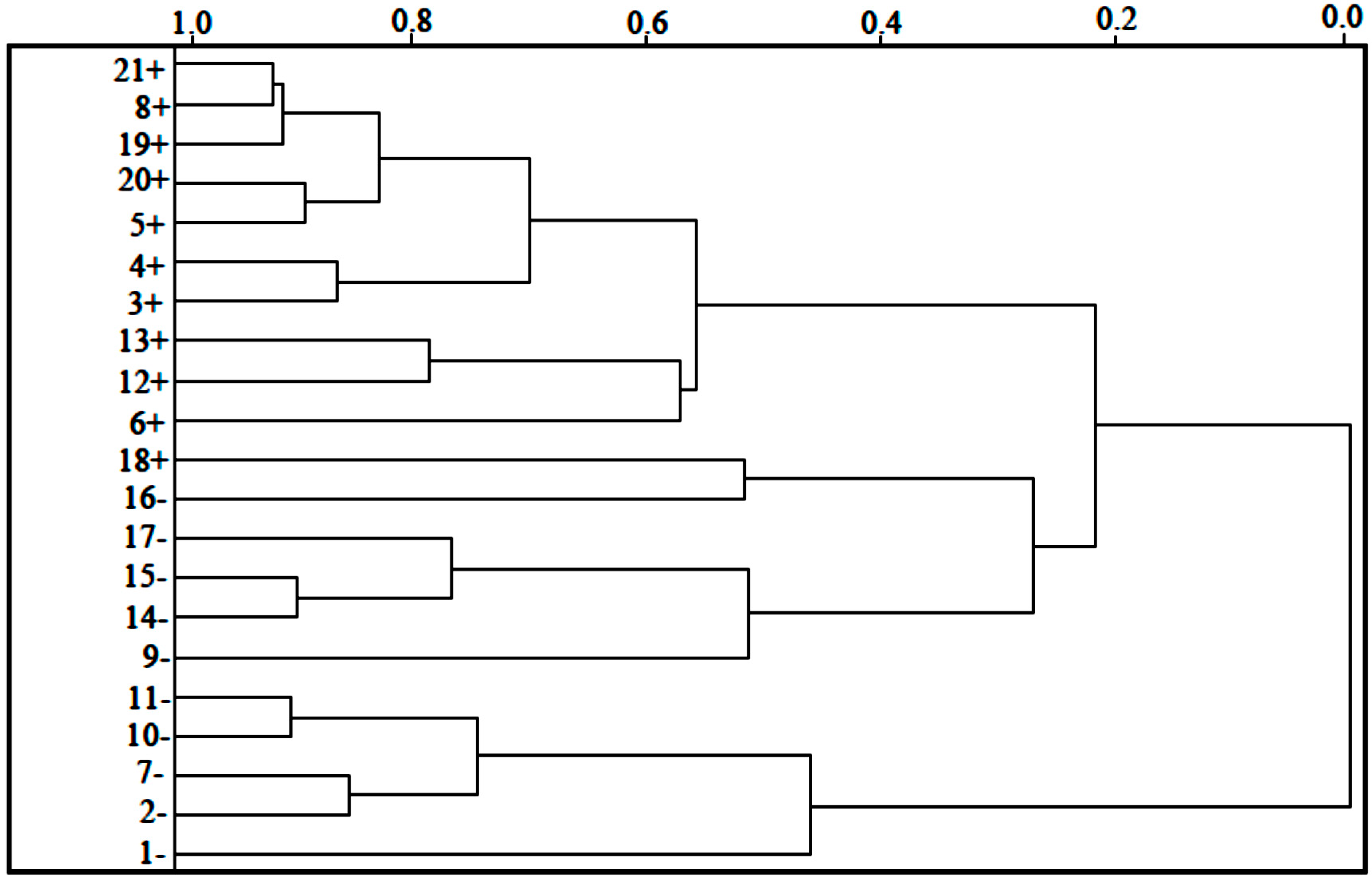
In the HCA technique, the distances between pairs of samples are computed and compared. Small distances imply that compounds are similar, while dissimilar samples will be separated by relatively large distances. The dendrogram in
Figure 4 shows the HCA graphic as well as the compounds separated into two main classes. The scale of similarity varies from 0 for samples with no similarity to 1 for samples with identical similarity. By analyzing the dendrogram, some conclusions can be drawn even though the compounds present some structural diversity.
HCA showed results similar to those obtained with PCA. The compounds are grouped according to their biological activities. The most potent compounds are 3, 4, 5, 6, 8, 12, 13, 18, 19, 20 and 21. The less potent compounds are 1, 2, 7, 9, 10, 11, 14, 15, 16 and 17. Compound 18 has the lowest value of logRA = 0.3604, among the compounds classified as most potent of the series studied. Whereas, the compound 5 has the highest value of logRA = 2.3240, whereas the variation between the activities of the compounds 5 and 18 is ±1.9636 between them.
Figure 4.
HCA dendrogram for artemisinin and derivatives with anticancer activity against human hepatocellular carcinoma HepG2. Positive values indicate more potent analogs, and negative values indicate less active compounds.
Figure 4.
HCA dendrogram for artemisinin and derivatives with anticancer activity against human hepatocellular carcinoma HepG2. Positive values indicate more potent analogs, and negative values indicate less active compounds.
2.4. Partial Least Squares (PLS) and Principal Component Regression (PCR) Results
The statistical quality [
43] of the PLS and PCR models was gauged by parameters such as correlation coefficient or squared correlation coefficient (R
2), explained variance (R
2ajust,
i.e., adjusted R
2), standard deviation (s), variance ratio (F—a statistic of assessing the overall significance), cross-validated correlation coefficient (
Q2), standard error of validation (
SEV), predicted residual error sum of squares (
PRESS) and standard deviation of cross-validation (
SPRESS) [
44,
45,
46]. The best regression models were selected based on high values of R
2, R
2ajust, Q
2 and F and low values of s,
SEV,
PRESS and
Spress.
The calculated properties and the experimental activity values for the compounds studied were used to build the PLS and PCR regression models (see
Table 4). The models built using the PLS and PCR were based on three latent variables and 21 compounds.
The regression equations obtained for PLS (Equation (3)) and PCR (Equation (4)) models that relate the descriptors and anticancer activity are the following:
n = 21, R
2 = 0.9473, R
2ajust = 0.9381, s = 0.2280, F
(4,17) = 71.9013,
Q2 = 0.9151,
SEV = 0.2620,
PRESS = 0.8937,
SPRESS = 0.0590.
n = 21, R
2 = 0.9367, R
2ajust = 0.9256, s = 0.2514, F
(4,17) = 59.1211,
Q2 = 0.9063,
SEV = 0.2752,
PRESS = 1.0745,
SPRESS = 0.0647.
The results obtained with the PLS and PCR models were very close, with variation between PLS and PCR of R
2 = ±0.0106, R
2ajust = ±0.0125, s = ±0.0234, F
(4,11) = ±12.7802, Q
2 = ±0.0088, SEV = ±0.0132, PRESS = ±0.4808 and S
PRESS = ±0.0057. The quality of the PLS and PCR models can be demonstrated by comparing the measured and the predicted activities. The validation errors obtained by the leave-one-out cross-validation method are shown in
Table 4. For the PLS model, only six compounds (1, 3, 5, 18, 20 and 21) had high validation errors, and the PCR model yielded seven compounds (1, 3, 4, 5, 17, 18 and 20) with high residual values.
Table 4.
Predicted PLS and PCR results and validation errors for logRA (experimental).
Table 4.
Predicted PLS and PCR results and validation errors for logRA (experimental).
| Compounds | Predicted | Validation Error | Experimental |
|---|
| PLS | PCR | PLS | PCR | logRA |
|---|
| 1− | −0.4002 | −0.3420 | −0.4002 | −0.3420 | 0.0000 |
| 2− | 0.3129 | 0.2298 | 0.3161 | 0.2166 | −0.0132 |
| 3+ | 1.9110 | 1.8824 | 0.3714 | 0.3428 | 1.5396 |
| 4+ | 2.0905 | 2.0404 | 0.1830 | 1.1329 | 1.9075 |
| 5+ | 1.8148 | 1.7574 | −0.5092 | −0.5666 | 2.3240 |
| 6+ | 1.4038 | 1.3075 | 0.0403 | −0.0560 | 1.3635 |
| 7− | −0.1312 | −0.1548 | −0.1444 | −0.1680 | −0.0132 |
| 8+ | 1.9071 | 1.9093 | −0.2223 | −0.2201 | 2.1294 |
| 9− | 0.2824 | 0.2716 | 0.2692 | 0.2584 | −0.0132 |
| 10− | 0.1883 | 0.1772 | 0.1751 | 0.1640 | −0.0132 |
| 11− | −0.0429 | −0.0270 | −0.0561 | −0.0402 | −0.0132 |
| 12+ | 1.3212 | 1.3357 | −0.2184 | −0.2039 | 1.5396 |
| 13+ | 1.1437 | 1.1276 | −0.1996 | −0.2157 | 1.3433 |
| 14− | −0.1448 | 0.0796 | −0.1580 | 0.0664 | −0.0132 |
| 15− | 0.0023 | 0.1410 | −0.0109 | 0.1278 | −0.0132 |
| 16− | 0.0131 | 0.1077 | 0.0001 | 0.0945 | −0.0132 |
| 17− | 0.1968 | 0.3439 | 0.1836 | 0.3307 | −0.0132 |
| 18+ | 0.7639 | 0.8522 | 0.4035 | 0.4918 | 0.3604 |
| 19+ | 1.9530 | 1.9139 | 0.0802 | 0.0411 | 1.8728 |
| 20+ | 1.7459 | 1.6991 | −0.3543 | −0.4011 | 2.1002 |
| 21+ | 1.7443 | 1.7392 | 0.3258 | 0.3207 | 1.4185 |
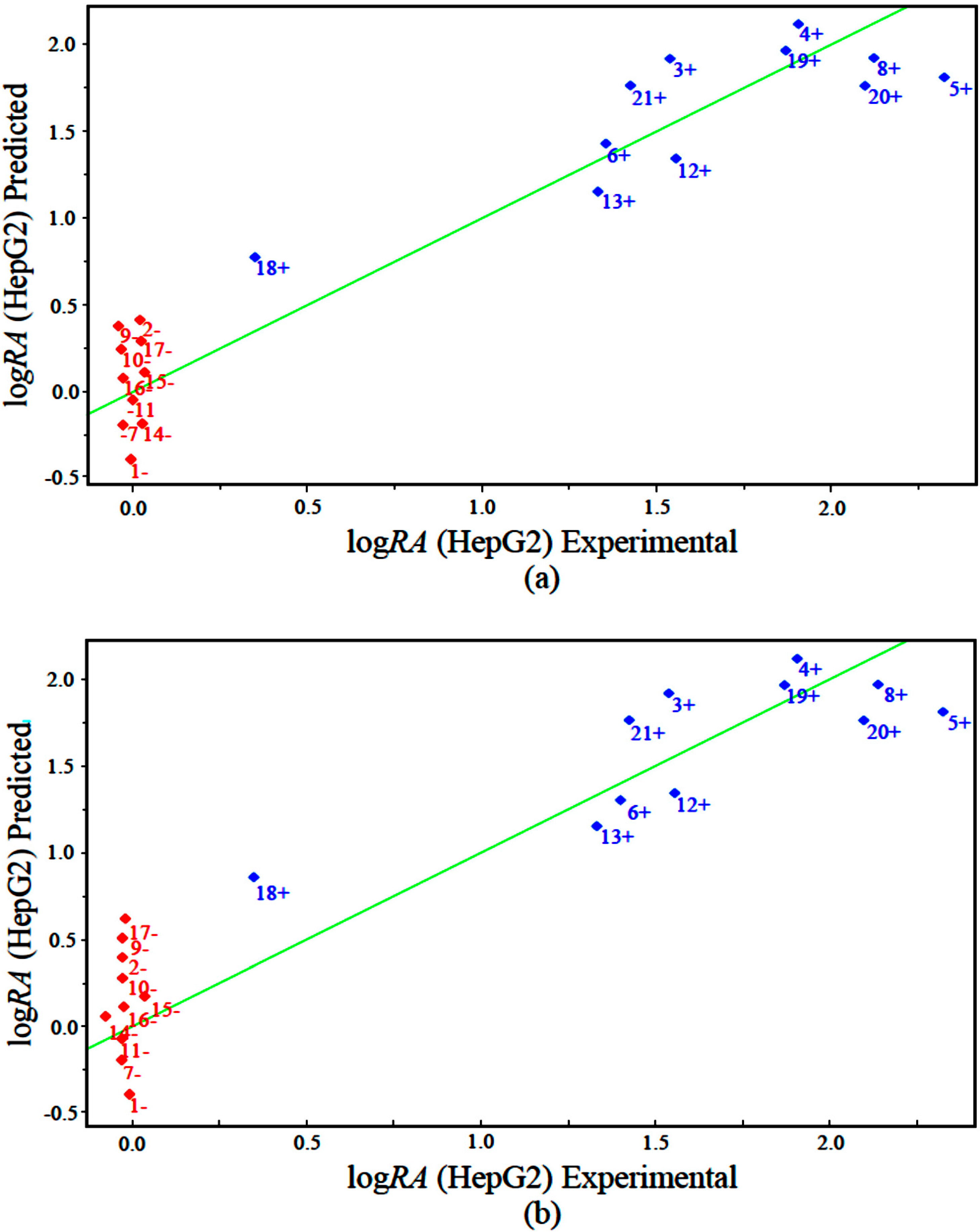
The measured versus predicted values using our PLS and PCR models are presented in
Figure 5a,b, respectively. The PLS and PCR plots identify compounds with higher activity (blue) and compounds with lower activity (red). According to the PLS and PCR models, the four variables present different magnitudes of regression coefficients (in absolute value). The models reveal that compounds with high biological potency against human hepatocellular carcinoma HepG2 have a combination of higher values of IC5 and GAP energy and lower values of ALOGPS_logs and Mor29m for the PLS and PCR models.
Figure 5.
Plot of experimental versus predicted values for logRA modeled by (a) PLS and (b) PCR.
Figure 5.
Plot of experimental versus predicted values for logRA modeled by (a) PLS and (b) PCR.
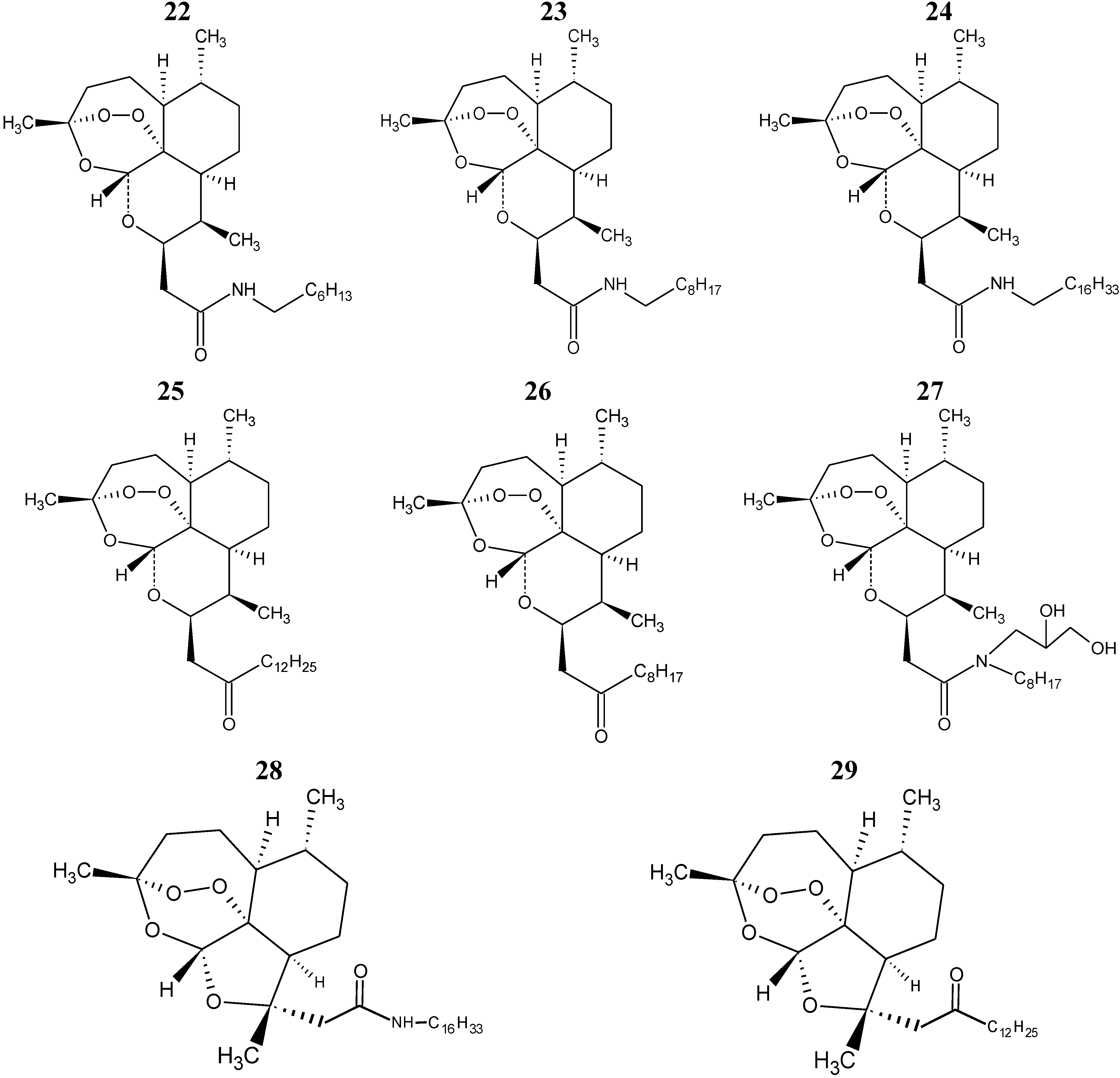
The eight compounds of the test set (22–29) were molded from the most stable structure of artemisinin, compound 1 of
Figure 1, and constructed using GaussView 5.0 program, carrying the complete optimization of the geometry of each compound with the basis set of separated valence B3LYP/6-31G** using the DFT method as implemented in Gaussian 03 program. After obtain the most stable geometry of each compound was determined only selected descriptors in PCA and used in the construction of the QSAR models, namely ALOGPS_logs, Mor29m, IC5 and GAP energy, shown in
Table 5.
Table 5.
Molecular properties selected by analysis of main components of test set with anticancer activity unknown.
Table 5.
Molecular properties selected by analysis of main components of test set with anticancer activity unknown.
| Test Set | ALOGPS_log | Mor29m | IC5 | Gap energy, a.u. |
|---|
| 22 | −5.030000 | −0.412000 | 5.514000 | 0.252200 |
| 23 | −5.760000 | −0.443000 | 5.628000 | 0.252200 |
| 24 | −7.390000 | −0.515000 | 5.364000 | 0.252400 |
| 25 | −7.140100 | −0.305100 | 5.571100 | 0.219700 |
| 26 | −6.030000 | −0.311000 | 5.572000 | 0.252400 |
| 27 | −4.820000 | −0.518000 | 5.856000 | 0.251700 |
| 28 | −7.350000 | −0.601000 | 5.280000 | 0.227600 |
| 29 | −7.010000 | −0.543000 | 5.488000 | 0.232300 |
The QSAR models (PLS and PCR) were built used to predict the unknown anticancer activity of eight new artemisinin derivatives shown in
Figure 6, compounds 22–29.
Table 6 shows the results of the log
RA by PCR and PLS models. According to
Table 6 the PLS and PCR models showed that all the compounds of the test set are predicted to be more active, they had values of log
RA greater than zero (log
RA > 0) in both models (PLS and PCR) with residues of prediction ranging from 0.0650 to −0.0560, suggesting that these new compounds in the two models (PLS and PCR) are more potent than artemisinin may be synthesized and tested for anticancer activity.
Figure 6.
Compounds of the test set artemisinin derivatives with unknown anticancer activity against human hepatocellular carcinoma HepG2.
Figure 6.
Compounds of the test set artemisinin derivatives with unknown anticancer activity against human hepatocellular carcinoma HepG2.
Table 6.
Anticancer activity predicted (logRA) by PCR and PLS models for the test set compounds and residues of prediction between models.
Table 6.
Anticancer activity predicted (logRA) by PCR and PLS models for the test set compounds and residues of prediction between models.
Test Set
Compounds | Predicted (logRA) | Residues of Prediction
(PLS-PCR) |
|---|
| PLS | PCR |
|---|
| 22 | 1.2458 | 1.2048 | 0.0410 |
| 23 | 1.6431 | 1.6210 | 0.0221 |
| 24 | 1.6804 | 1.6154 | 0.0650 |
| 25 | 0.6841 | 0.8649 | −0.1808 |
| 26 | 1.1631 | 1.1564 | 0.0067 |
| 27 | 2.1201 | 2.1163 | 0.0038 |
| 28 | 1.3444 | 1.3850 | −0.0406 |
| 29 | 1.5410 | 1.5970 | −0.0560 |
2.5. Pharmacokinetic and Toxicological Results
The prediction of Absorption, Distribution, Metabolism and Excretion (ADME) proprieties for artemisinin and its derivatives of the test set (compounds 22–29) classified by PLS and PCR models as more potent are shown in
Table 7 and
Table 8. In
Table 7, one can observe the absorption values (HIA, PCaCO2 and PMDCK) predicted for the compounds. The prediction of human intestinal absorption is a major objective in the optimization and selection of candidates for the development of oral medications. The focus on the discovery of modern drugs is not simply in the pharmacological activity, but also in search of more favorable pharmacokinetic properties [
47]. The results of human intestinal absorption are the sum of absorption and bioavailability, evaluated from the proportion of excretion or cumulative excretion in urine, bile and feces [
48,
49].
The test compounds showed good human intestinal absorption, having values of HIA > 90%, being close to that of artemisinin (compound 1). Compound 27 showed the lowest absorption equal to 94.2039%, whereas compound 26 showed the highest value of HIA equal to 98.1189%, as shown in
Table 7.
The P
Caco2 (nm/s) and P
MDCK (nm/s) cell models have been used as a reliable
in vitro model for the prediction of oral drug absorption, being the Caco-2 cells derived from human colon adenocarcinoma and have various routes of drug transport through the intestinal epithelium [
49]. The results of the compounds shown in
Table 7 showed an average permeability of 45.4351, as proposed by Yazdanian [
50]. The values obtained of P
CaCO2 (nm/s) were higher than 30.3276 nm/s (compound 1, artemisinin). The compounds 25 and 26 showed higher values of cell permeability of 51.2476 and 51.5452 nm/s, respectively.
Table 7.
Absorption properties for artemisinin (compound 1) and compounds of the test set.
Table 7.
Absorption properties for artemisinin (compound 1) and compounds of the test set.
| Compounds | Absorption |
|---|
| HIA(%) [a] | PCaCO2(nm/s) [b] | PMDCK(nm/s) [c] | Pskin [d] |
|---|
| 1 | 96.3143 | 30.3276 | 72.4627 | −3.00248 |
| 22 | 95.9522 | 48.074 | 0.2820 | −2.78573 |
| 23 | 96.0180 | 49.0102 | 2.7481 | −2.38535 |
| 24 | 96.1170 | 50.8969 | 64.4258 | −1.10239 |
| 25 | 97.6636 | 51.2473 | 54.1962 | −1.00477 |
| 26 | 98.1189 | 51.5452 | 13.6801 | −1.4846 |
| 27 | 94.2039 | 35.0362 | 0.0437 | −2.66011 |
| 28 | 96.1170 | 46.0453 | 64.766 | −0.792156 |
| 29 | 97.6636 | 46.7337 | 55.4025 | −0.768943 |
Table 8.
Distribution properties in percentages of PPB and penetration of the blood brain barrier for artemisinin (compound 1) and compounds of the test set.
Table 8.
Distribution properties in percentages of PPB and penetration of the blood brain barrier for artemisinin (compound 1) and compounds of the test set.
| Compounds | Distribution |
|---|
| PPB(%) [a] | CBrain/CBlood [b] |
|---|
| 1 | 93.368123 | 1.30488 |
| 22 | 90.481620 | 3.1575 |
| 23 | 91.279366 | 5.35648 |
| 24 | 93.306402 | 11.0801 |
| 25 | 96.696312 | 8.39023 |
| 26 | 95.399268 | 2.65831 |
| 27 | 90.056670 | 1.91129 |
| 28 | 93.838777 | 10.9862 |
| 29 | 97.347576 | 8.08563 |
In accordance with Irvine
et al. [
51], P
MDCK (nm/s) system cells can be used as tool for rapid screening permeability. The test compounds (22, 23, 26 and 27) were those that presented low permeability in the P
MDCK (<25) cell system. In the studied set, compounds 22 and 27 showed the lowest permeability values P
MDCK equal to 0.2820 and 0.0437 nm/s, respectively. Compounds 24, 25, 28 and 29 showed the highest permeability values varying in the range from 54.1962 to 64.7660 nm/s, close to the permeability value of artemisinin.
In the pharmaceutical, cosmetic and agrochemical industries, predicting the rate of skin permeability is a crucial parameter for transdermal administration of medications and for the risk assessment of chemical products that come into contact with the skin accidentally [
52]. The test set compounds showed negative values of skin permeability,
i.e, it is not important to be administered for transdermal use, and also not present any risk accordance results described in
Table 7.
The distribution of a drug depends on its plasma protein binding (PPB) and partition in adipose tissue and other tissues. In plasma the drug may be in unbound or bound form, which depends on the affinity that the drug presents by the plasmatic protein (drug target). If the protein binding is reversible, then a chemical equilibrium will exist between bound and unbound states. The proteins binding can influence in the biological half-life in the body. The bound portion may act as a reservoir or deposit to which the drug is slowly released in the unbound form. As the non-bound form being metabolized and/or excreted from the body, fraction bound to will be released in order to that maintain balance [
53,
54]. In
Table 8 shows the results of the distribution properties (PPB% and C
Brain/C
Blood) for artemisinin and classified as most potent compounds of test set. Compounds 22–29 showed strong plasma protein binding with PPB > 90.0566%, being close to the value of PPB of artemisinin which was equal to 93.3681%. Compounds 25, 26 and 29 showed higher strength in plasma protein binding equal to 96.6963%, 95.3992% and 97.3475%, respectively.
The penetration of the blood brain barrier is critical in the pharmaceutical field, because compounds that act on the central nervous system (CNS) should go through it, and inactive compounds in CNS should not go in order to avoid collateral effects of CNS [
55]. In the test set, all compounds showed absorption values to the CNS higher than 1, and in accordance with the classification proposed by Ma
et al. [
56], compounds that have values greater than 1 (C
Brain/C
Blood > 1) are classified as active in the CNS may cause collateral effects, and compounds that have values below 1 (C
Brain/C
Blood < 1) are classified as inactive in the CNS. Therefore, compounds 22–29 had a variation of C
Brain/C
Blood in relation to the artemisinin of 1.8526, 4.0516, 9.7752, 7.0853, 1.3534, 0.6064 and 9.6813, respectively. Since the compound 27 showed the value of penetration of the blood brain barrier (C
Brain/C
Blood) closest to of artemisinin (C
Brain/C
Blood = 1.304) having the smallest variation between test compounds studied (C
Brain/C
Blood[compound 27] − C
Brain/C
Blood[artemisinin]), showing value equal to 0.6064.
Table 9 shows the results of the toxicological properties of mutagenicity (Ames Test) and carcinogenicity (Mouse and rat) for artemisinin and its derivatives of the test set (22–29) classified by PLS and PCR models as more potent with anticancer activity against human hepatocellular carcinoma HepG2. One of the important reasons for the discovery of new drugs is the evaluation of the toxicity of drug candidates. This means that the conception of drugs with consideration of its toxicity is very important, as well as predicts the mutagenicity and carcinogenicity of new compounds that may be toxic.
Table 9.
Toxicological properties of mutagenicity (Ames Test) and carcinogenicity (mouse and rat) for artemisinin and its derivatives of the test set (22–29).
Table 9.
Toxicological properties of mutagenicity (Ames Test) and carcinogenicity (mouse and rat) for artemisinin and its derivatives of the test set (22–29).
| Compounds | Ames Test | Carcinogenicity |
|---|
| Mutagenicity | Mouse | Rat |
|---|
| 1 | Mutagenic | Negative | Positive |
| 22 | Non-mutagenic | Negative | Positive |
| 23 | Non-mutagenic | Negative | Positive |
| 24 | Non-mutagenic | Negative | Positive |
| 25 | Non-mutagenic | Positive | Positive |
| 26 | Non-mutagenic | Positive | Positive |
| 27 | Non-mutagenic | Negative | Negative |
| 28 | Non-mutagenic | Negative | Positive |
| 29 | Non-mutagenic | Negative | Positive |
The Ames test is a simple method to test mutagenicity of a compound, suggested by Ames, where various strains of
Salmonella typhimurium bacterium with mutations in the genes involved in histidine synthesis, so they require histidine for growth, are used. The variable being tested is the ability of the mutagenic agent to provoke the reversal of the growth in histidine-exempt medium [
57]. In this method, compound 1 (artemisinin) presented positive prediction, which means that this compound was predicted as a mutagen. The other compounds (22–29) showed a negative prediction, ie, were predicted as non-mutagenic, as shown in
Table 9.
Carcinogenicity is the ability that a substance has to induce alterations that lead to cancer. The carcinogenicity assays require a long time (>2 years). The principal methodologies use “in vivo” assays, using mice or rats by exposing them to a chemical compound, where the observed variable is the existence of cancer. In this study, PreADMET server was used to predict the result which is constructed from the data of the NTP (National Toxicology Program) and the USA/FDA, which are the results of in vivo tests for carcinogenicity in mice and rats for 2 years.
In the prediction of carcinogenicity in mouse, compounds 25 and 26 showed positive prediction, ie, no evidence of carcinogenic activity. The others compounds were predicted as negative, which means that there is evidence of carcinogenic activities in mouse, for such compounds (1, 22–24 and 27–29). In the prediction of carcinogenicity in rat, the following compounds 1, 22–26, 28 and 29 had positive prediction, demonstrating that show no carcinogenic activity. Whereas compound 27 showed negative prediction, meaning that this compound may exhibit carcinogenic activity.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









