Dereplication Guided Discovery of Secondary Metabolites of Mixed Biosynthetic Origin from Aspergillus aculeatus

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion


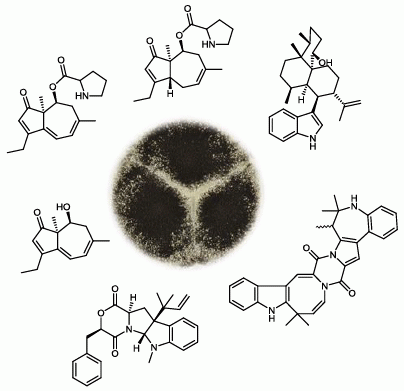
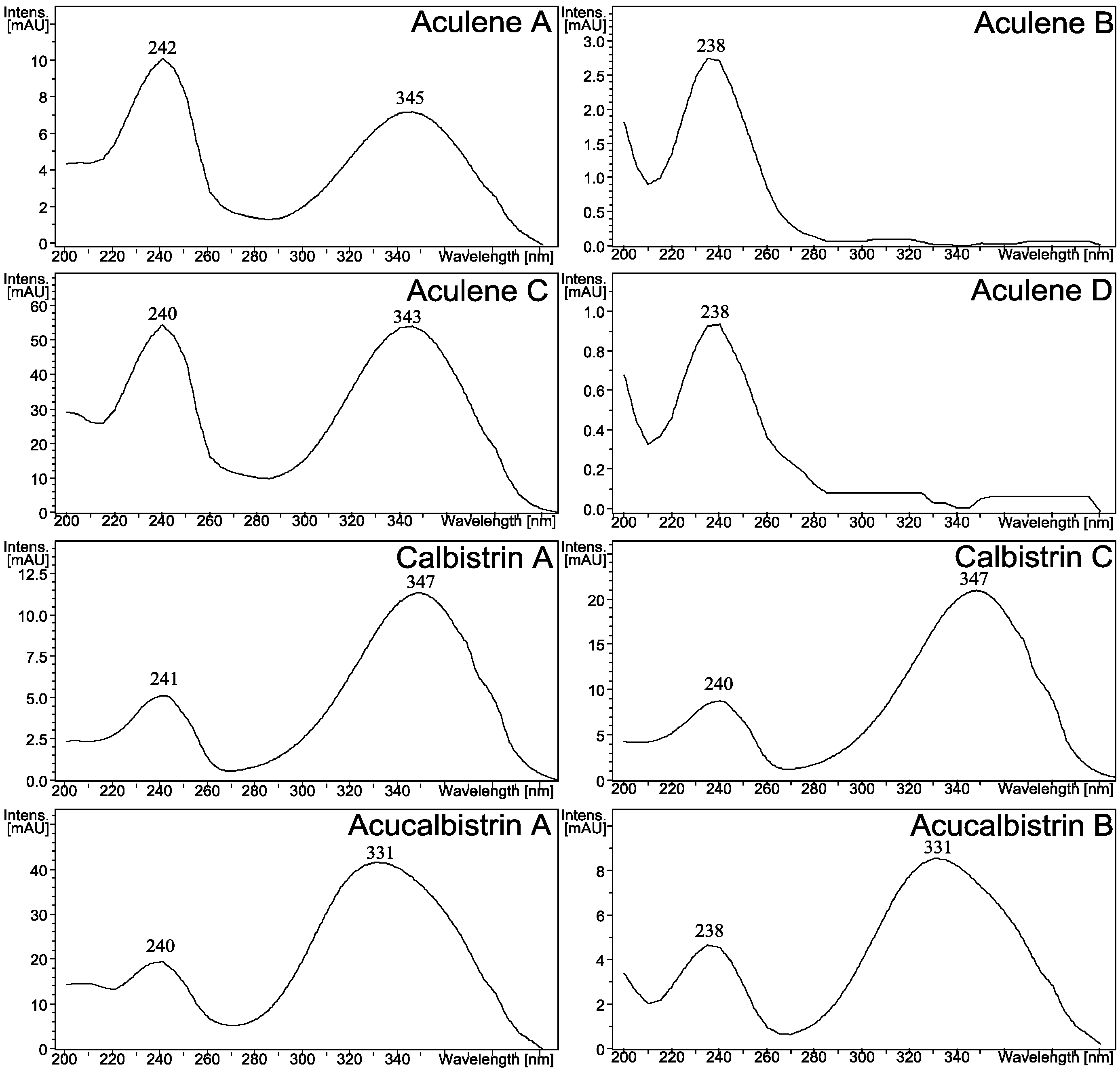
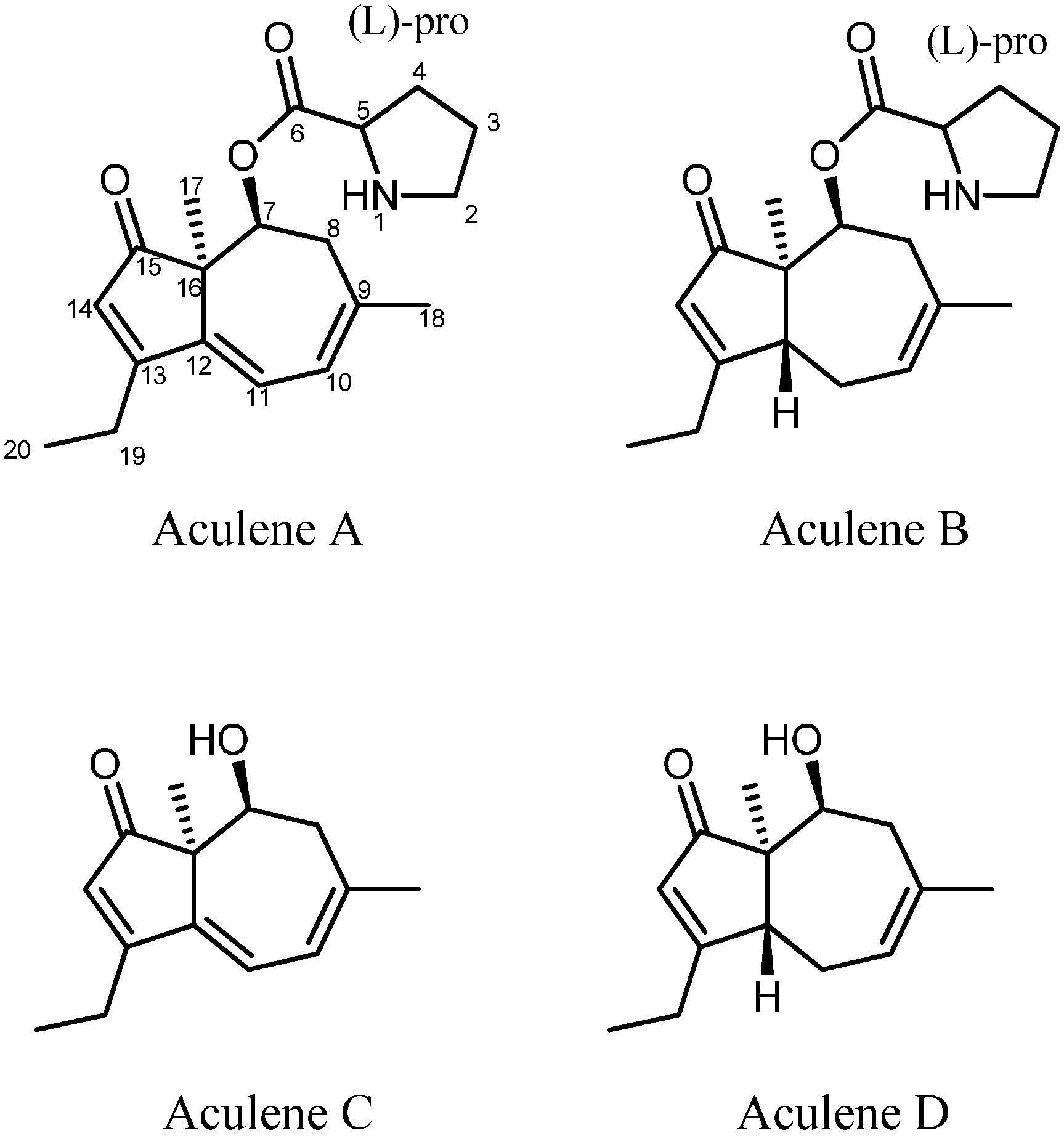
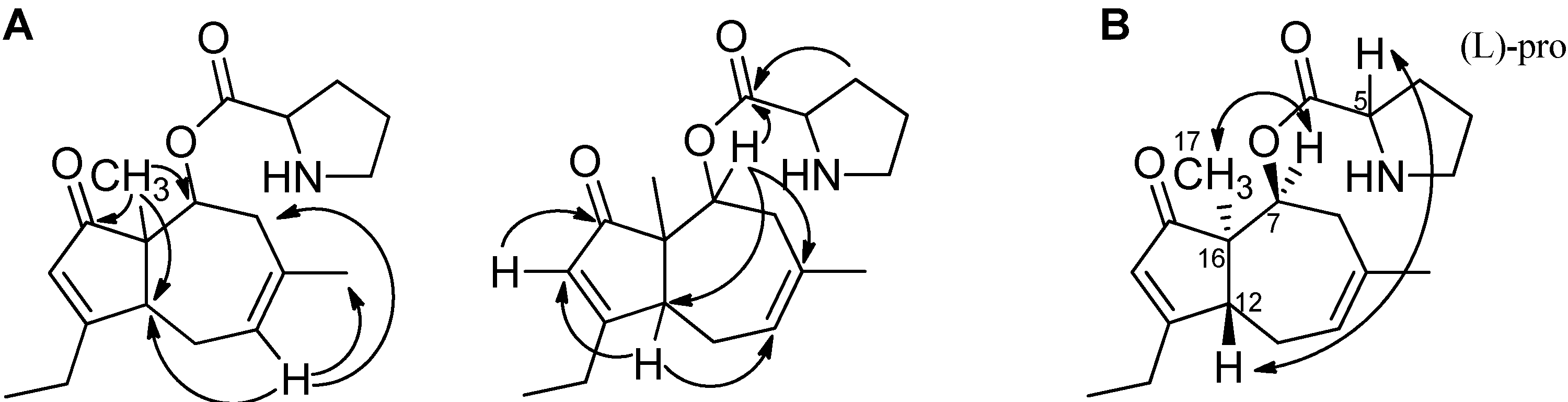
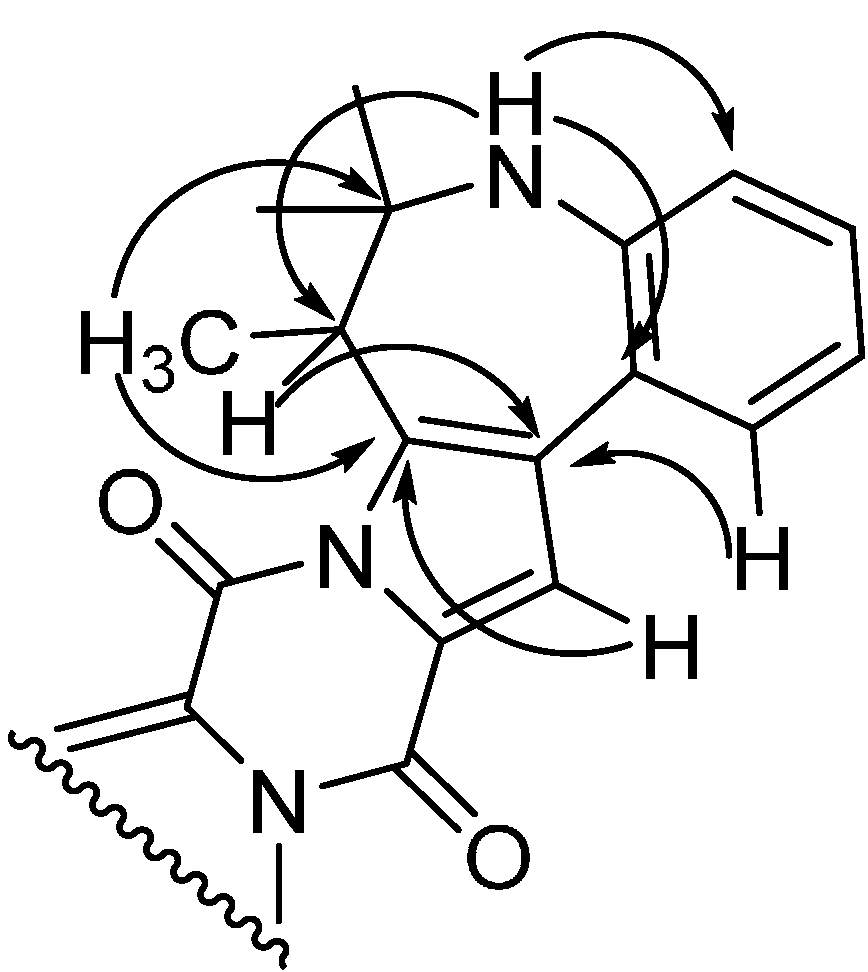
2.1. A Novel Class of Compounds, Named the aculenes, Has a Mixed Biosynthetic Origin




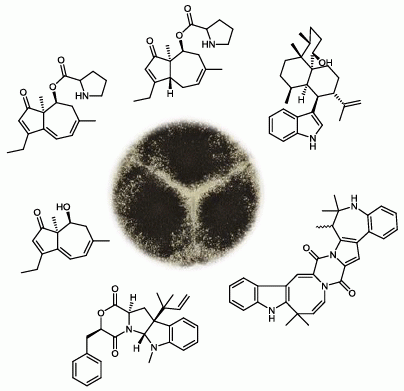{kind=link}
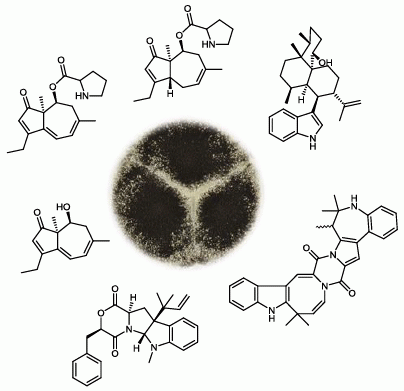{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
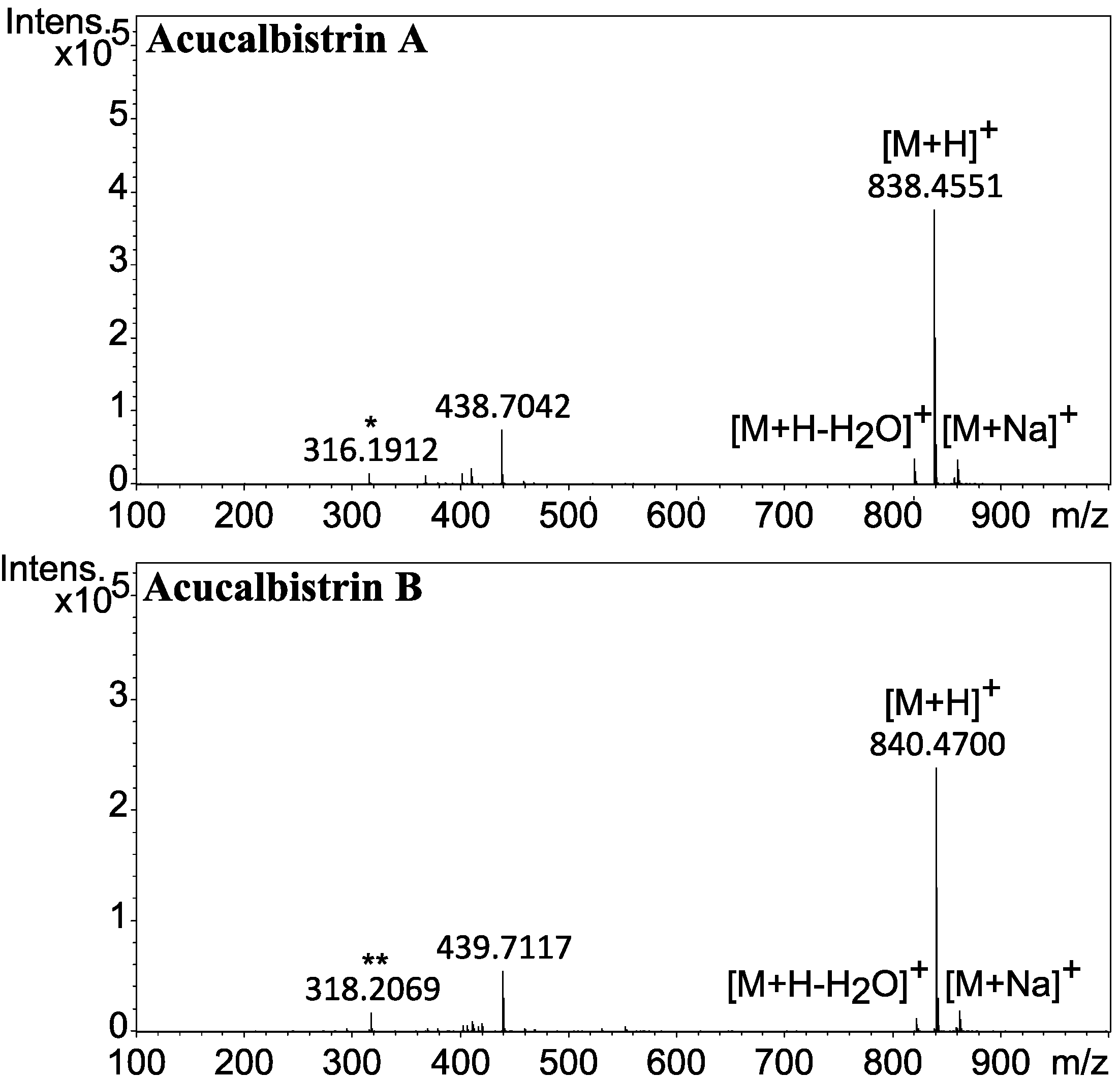{kind=link}
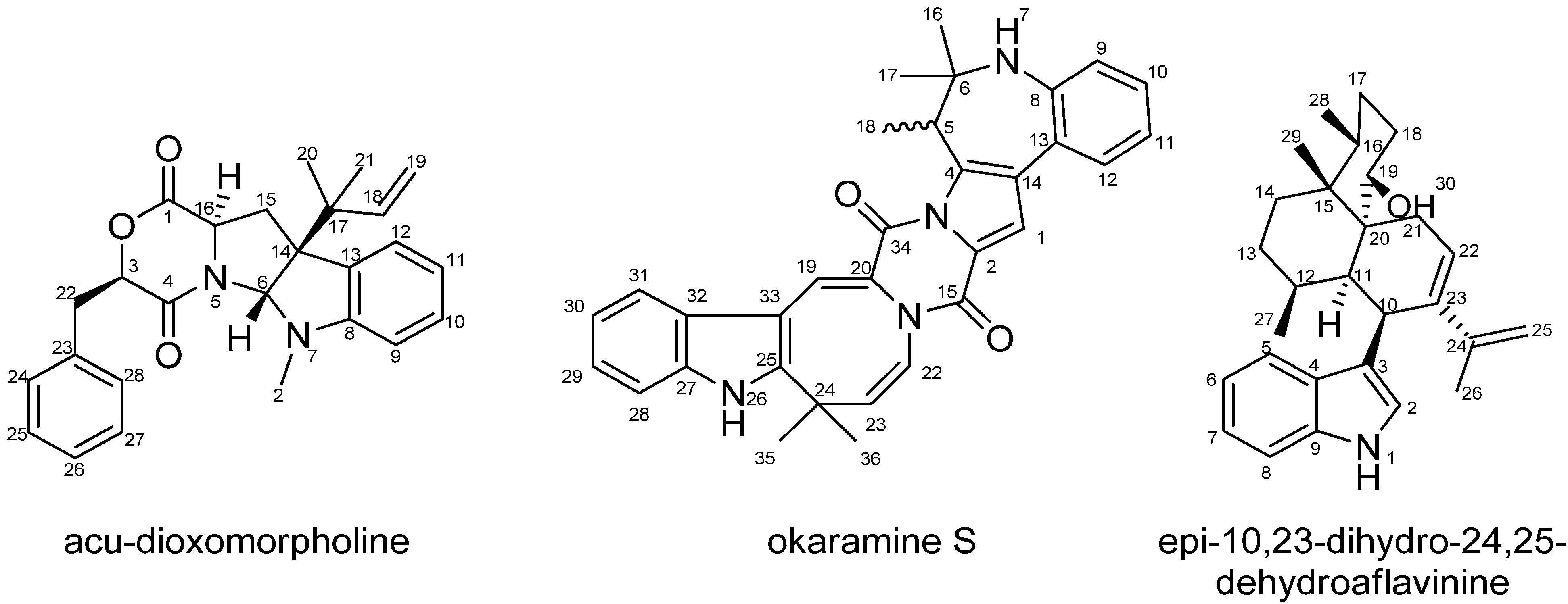{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aculene A | Aculene B | Aculene C | ||||||
|---|---|---|---|---|---|---|---|---|
| Position | δH (J in Hz) | HMBC | δH (J in Hz) | HMBC | δH (J in Hz) | HMBC | ||
| 1 | - | - | - | - | ||||
| 2 | 3.15 (m) | - | 3.21 (m) | 3,4,5 | ||||
| 2' | 3.09 (m) | - | 3.14 (m) | 3,4,5 | ||||
| 3 | 1.83 (m) | - | 1.84 (m) | 2,4,5 | ||||
| 3' | 1.67 (m) | - | 1.76 (m) | 2,4,5 | ||||
| 4 | 1.94 (m) | 2,6 | 2.05 (m) | 2,3,5,6 | ||||
| 4' | 1.46 (m) | 2,6 | 1.55 (m) | 2,6 | ||||
| 5 | 4.23 (t 7.7) | 6 | 4.31 (t 7.8) | 2,3,4,6 | ||||
| 6 | - | - | - | - | ||||
| 7 | 5.33 (dd 4.4, 2.4) | - | 5.28 (dd 5.3, 2.3) | 6,9,12,16 | 3.95 (t 3.5) | 9,13,15 | ||
| 8 | 2.82(br.d 20.8) | - | 2.67 (br. d 19.1) | 16 | 2.52 (br. d 20.1) | |||
| 8' | 2.55 (m) | 9,10,16 | 2.37 (m) | 7,9,10,16 | 2.31 (m) | 7,9,10,16 | ||
| 9 | - | - | - | - | - | - | ||
| 10 | 5.98 (d 7.4) | 8,12,18 | 5.56 (m) | 8,11,12,18 | 5.87 (d 7.3) | 8,12,18 | ||
| 11 | 6.24 (d 7.4) | 9,13,16 | 2.12 (m) | - | 6.08 (d 7.4) | 9,13,16 | ||
| 11' | - | - | 2.52 (m) | 18 | - | - | ||
| 12 | - | - | 3.31 (m) | 10,11,14 | - | - | ||
| 13 | - | - | - | - | - | - | ||
| 14 | 6.03 (s) | 12,13,15,16,19 | 5.81 (q 1.8) | 12,13,15,16,19 | 5.93 (s) | 12,13,15,16,19 | ||
| 15 | - | - | - | - | - | - | ||
| 16 | - | - | - | - | - | - | ||
| 17 | 0.96 (s) | 7,12,15,16 | 0.96 (s) | 7,12,15,16 | 0.79 (s) | 7,12,15,16 | ||
| 18 | 1.85 (s) | 8,9,10 | 1.68 (s) | 8,9,10 | 1.82 (s) | 8, 9,10 | ||
| 19 | 2.55 (m) | 13,20 | 2.39 (m) | 13,14,20 | 2.48 (m) | 12,13,14,20 | ||
| 20 | 1.16 (t 7.4) | 13,19 | 1.11 (t 7.3) | 13,19 | 1.14 (t 7.4) | 13,19 | ||
| -OH | - | - | - | - | 4.43 (s) | - | ||
| Position | Aculene A | Aculene B | Aculene C |
|---|---|---|---|
| δC | δC | δC | |
| 1 | - | - | - |
| 2 | 45.1 | 44.7 | - |
| 3 | 22.3 | 22.3 | - |
| 4 | 27.6 | 27.6 | - |
| 5 | 58.4 | 58.3 | - |
| 6 | 168.0 | 167.8 | - |
| 7 | 72.9 | 73.5 | 67.6 |
| 8 | 36.5 | 35.5 | 40.2 |
| 9 | 141.7 | 130.7 | 142.5 |
| 10 | 120.0 | 122.7 | 119.7 |
| 11 | 118.5 | 24.9 | 117.8 |
| 12 | 142.5 | 44.6 | 144.3 |
| 13 | 175.3 | 183.8 | 174.3 |
| 14 | 125.3 | 122.8 | 125.6 |
| 15 | 205.8 | 208.2 | 208.2 |
| 16 | 51.7 | 54.4 | 53.6 |
| 17 | 17.9 | 17.1 | 18.3 |
| 18 | 26.9 | 28.3 | 27.1 |
| 19 | 20.1 | 23.1 | 19.8 |
| 20 | 12.0 | 10.8 | 11.8 |

2.2. Discovery of Novel Indole Terpenoids

| Position | δH (J in Hz) | δC | HMBC | Noesy |
|---|---|---|---|---|
| 1 | - | 168.0 | - | - |
| 2 | 2.96 (3H, s) | 32.7 | 6, 8 | 6,9 |
| 3 | 5.32 (1H, dd, 8.8, 3.4) | 78.1 | 4, 22, 23 | 16, 22, 22', 24/28 |
| 4 | - | 163.5 | - | - |
| 5 | - | - | - | - |
| 6 | 5.43 (1H, s) | 81.9 | 2, 4, 8, 13, 14, 15, 16, 17 | 12, 18, 19, 19', 20, 22 |
| 7 | - | - | - | - |
| 8 | - | 150.8 | - | - |
| 9 | 6.44 (1H, d, 7.6) | 105.6 | 11, 13 | 2, 10 |
| 10 | 7.09 (1H, td, 7.6, 0.8) | 128.5 | 8, 12 | 9, 11 |
| 11 | 6.64 (1H, t, 7.3) | 116.9 | 9, 12, 13 | 10, 12 |
| 12 | 7.17 (1H, d, 7.3) | 124.1 | 8, 10, 14 | 11, 15, 18, 20, 21 |
| 13 | - | 128.8 | - | - |
| 14 | - | 59.6 | - | - |
| 15 | 2.43 (1H, dd, 12.7, 6.6) | 36.6 | 1, 6, 13, 14, 16 | 12, 15', 16 |
| 15' | 2.21 ( 1H, dd, 12.7, 11.1) | 36.6 | 1, 13, 14, 16 | 15, 16, 18, 20, 21 |
| 16 | 4.17 (1H, dd, 11.1, 6.6) | 56.7 | 1, 15 | 3, 15, 15' |
| 17 | - | 40.1 | - | - |
| 18 | 5.87 (1H, dd, 17.4, 11.0) | 143.3 | 14, 17, 21 | 6, 12, 15', 19, 19', 20, 21 |
| 19 | 5.02 (1H, dd, 17.4, 1.1) | 113.7 | 14, 17, 18 | 6, 18, 19', 20, 21 |
| 19' | 5.06 (1H, dd, 11.0, 1.1) | 113.6 | 14, 17, 18 | 6, 18, 19, 20, 21 |
| 20 | 0.82 (3H, s) | 22.4 | 14, 17, 18, 21 | 6, 12, 15', 18, 19, 19', 21 |
| 21 | 0.97 (3H, s) | 21.7 | 14, 17, 18, 20 | 12, 15', 18, 19, 19', 20 |
| 22 | 2.98 (1H, dd, 14.8, 8.8) | 34.9 | 3, 4, 23, 28 | 3, 6, 22', 24/28 |
| 22' | 3.33 (1H, m) | 34.9 | 3, 4, 23, 28 | 3, 22, 24/28 |
| 23 | - | 136.3 | - | - |
| 24 | 7.26 (1H, m) | 129.2 | 22, 26, 28 | 3, 22, 22' |
| 25 | 7.28 (1H, m) | 127.7 | - | 26 |
| 26 | 7.22 (1H, tt, 7.2, 1.6) | 126.2 | 28 | 25 |
| 27 | 7.28 (1H, m) | 127.7 | 23, 25 | - |
| 28 | 7.26 (1H, m) | 129.2 | - | 3, 22, 22' |
| Position | δH (J in Hz) | δC | Hmbc | Noesy |
|---|---|---|---|---|
| 1 | 7.64 (1H, s) | 114.7 | 2, 4, 14, 15 | - |
| 2 | - | 123.8 | - | - |
| 4 | - | 137.6 | - | - |
| 5 | 4.26 (1H, q, 6.5) | 43.2 | 4, 14, 18 | 18 |
| 6 | - | 51.2 | - | - |
| 7 | 5.87 (1H, s) | - | 5, 9, 13, 17 | 9 |
| 8 | - | 144.5 | - | - |
| 9 | 6.90 (1H, d, 7.6) | 118.8 | 11, 13 | 10 |
| 10 | 6.99 (1H, t, 7.6) | 127.4 | 8, 12 | 9 |
| 11 | 6.65 (1H, t, 7.6) | 116.8 | 9, 13 | 12 |
| 12 | 7.74 (1H, d, 7.6) | 128.3 | 8, 10, 14 | 1,11 |
| 13 | - | 115.3 | - | - |
| 14 | - | 124.9 | - | - |
| 15 | - | 153.4 | - | - |
| 16 | 0.95 (3H, s) | 27.1 | 5, 6, 17 | 17 |
| 17 | 1.39 (3H, s) | 29.3 | 5, 6, 16 | 16 |
| 18 | 1.18 (3H, d, 7.2) | 18.1 | 4, 5, 6 | - |
| 19 | 7.76 (1H, s) | 117.5 | 20, 25, 32, 33, 34 | - |
| 20 | - | 125.2 | - | - |
| 22 | 5.90 (1H, d, 8.3) | 123.6 | 23, 24 | 23 |
| 23 | 6.15 (1H, d, 8.3) | 140.2 | 22, 24, 35/36 | 22 |
| 24 | - | 35.8 | - | - |
| 25 | - | 149.8 | - | - |
| 26 | 11.70 (1H, s) | - | 25, 32, 33 | - |
| 27 | - | 134.2 | - | - |
| 28 | 7.44 (1H, m) | 112.3 | - | 29/30 |
| 29 | 7.17 (1H, m) | 121.7 | 31 | 28, 31 |
| 30 | 7.17 (1H, m) | 121.7 | 31 | 28, 31 |
| 31 | 7.63 (1H, m) | 116.1 | 27, 29/30 | 12, 29/30 |
| 32 | - | 105.3 | - | - |
| 33 | - | 129.5 | - | - |
| 34 | - | 157.6 | - | - |
| 35 | 1.69 (3H, s) | 26.7 | 23, 24, 25, 35/36 | - |
| 36 | 1.69 (3H, s) | 26.7 | 23, 24, 25, 35/36 | - |
| Position | δH (J in Hz) | δC | HMBC | H2BC | NOESY |
|---|---|---|---|---|---|
| 1 | 10.66 (s) | - | 2, 3, 4, 9 | 2 | 2,8 |
| 2 | 7.07 (d 2.0) | 122.7 | 3, 4, 9 | - | 1, 23, 25, 27 |
| 3 | - | 114.4 | - | - | |
| 4 | - | 126.7 | - | - | |
| 5 | 7.38 (d 7.8) | 116.8 | 7, 9 | 6 | 10, 11, 18 |
| 6 | 6.96 (t 7.2) | 117.7 | 4, 8 | 5, 7 | |
| 7 | 7.02 (t 7.3) | 120.0 | 5, 9 | 6, 8 | |
| 8 | 7.29 (d 8.0) | 110.9 | 4, 6 | 7 | 1 |
| 9 | - | 135.5 | - | - | |
| 10 | 3.59 (dd 13.3, 5.0) | 33.4 | 2, 3, 11, 12, 23, 24 | 11, 23 | 5, 11, 19, 25, 26 |
| 11 | 2.47 (m) | 37.6 | 10, 12, 23 | 10, 12 | 5, 10, 12, 13, 16, 19 |
| 12 | 1.27 (m) | 28.9 | - | 17 | 11 |
| 13 | 1.53 (m) | 28.1 | 29 | - | 11, 13', 16 |
| 13' | 0.81 (d 12.9) | 28.1 | - | - | 13 |
| 14 | 1.46 (m) | 27.4 | - | - | |
| 14' | 1.08 (d 13.7) | 27.4 | 27 | - | 28 |
| 15 | - | 38.2 | - | - | |
| 16 | 2.04 (m) | 30.4 | - | 17, 28 | 11, 13, 17', 28 |
| 17 | 1.71 (m) | 24.8 | - | 18 | |
| 17' | 1.22 (m) | 24.8 | - | - | |
| 18 | 1.99 (d 11.3) | 29.6 | - | - | 5, 19 |
| 18' | 1.74 (m) | 29.6 | - | - | 19, 30 |
| 19 | 4.64 (s) | 65.6 | - | - | 10, 11, 18, 18', 22, 26 |
| 20 | - | 42.9 | - | - | |
| 21 | 2.07 (m) | 23.5 | - | - | 29, 30 |
| 21' | 1.65 (m) | 23.5 | 19, 23 | - | 27 |
| 22 | 1.85 (m) | 26.6 | 20, 24 | 21, 23 | 19, 30 |
| 22' | 1.54 (m) | 26.6 | - | - | |
| 23 | 3.13 (m) | 42.3 | 24, 25, 26 | 10, 22 | 2, 25, 27 |
| 24 | - | 149.8 | - | - | |
| 25 | 4.81 (d 1.8) | 110.4 | 23, 26 | 26 | 2, 10, 23, 25' |
| 25' | 4.58 (d 1.8) | 110.4 | 23, 26 | 26 | 25, 26 |
| 26 | 1.45 (s) | 17.7 | 23, 24, 25 | - | 10, 19, 25' |
| 27 | 1.21 (s) | 21.1 | 11, 12 | 12 | 2, 21', 23 |
| 28 | 0.71 (d 6.7) | 15.4 | 15, 16, 17 | 16 | 14', 16, 17' |
| 29 | 0.92 (s) | 17.7 | 13, 15, 16, 20 | - | 17, 21, 30 |
| 30 | 4.28 (d 4.3) | - | 19, 20 | 19 | 18', 21, 22, 29 |

2.3. Biological Testing of the Novel A. aculeatus Metabolites
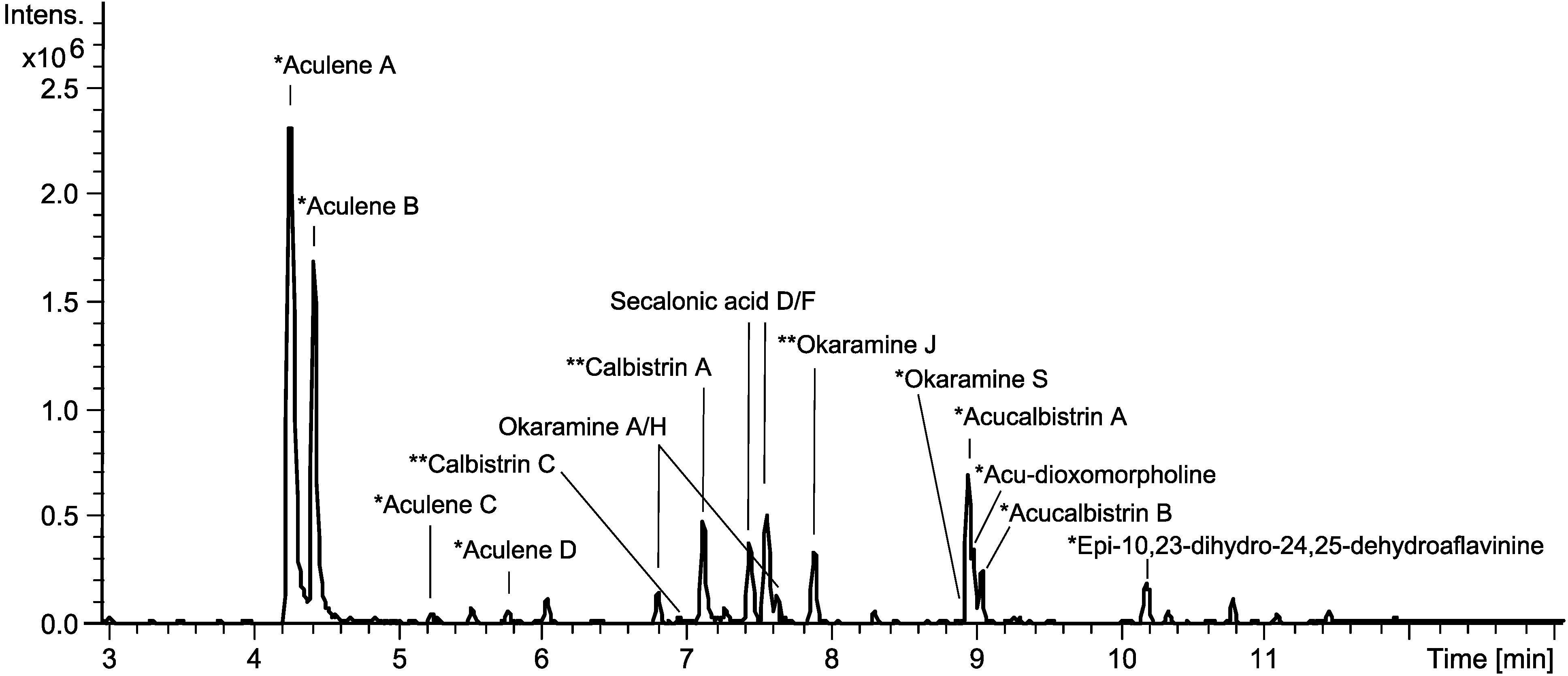
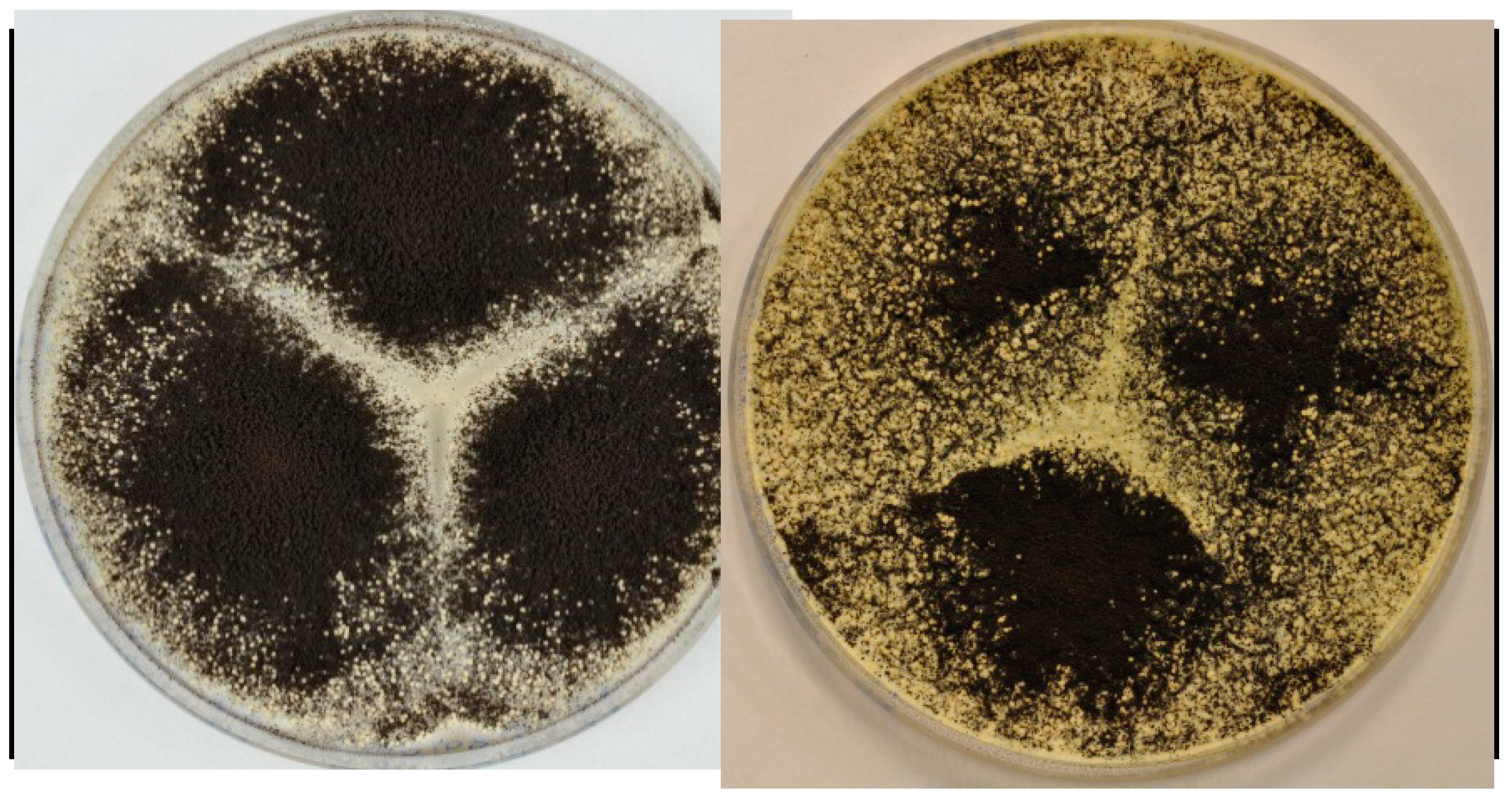
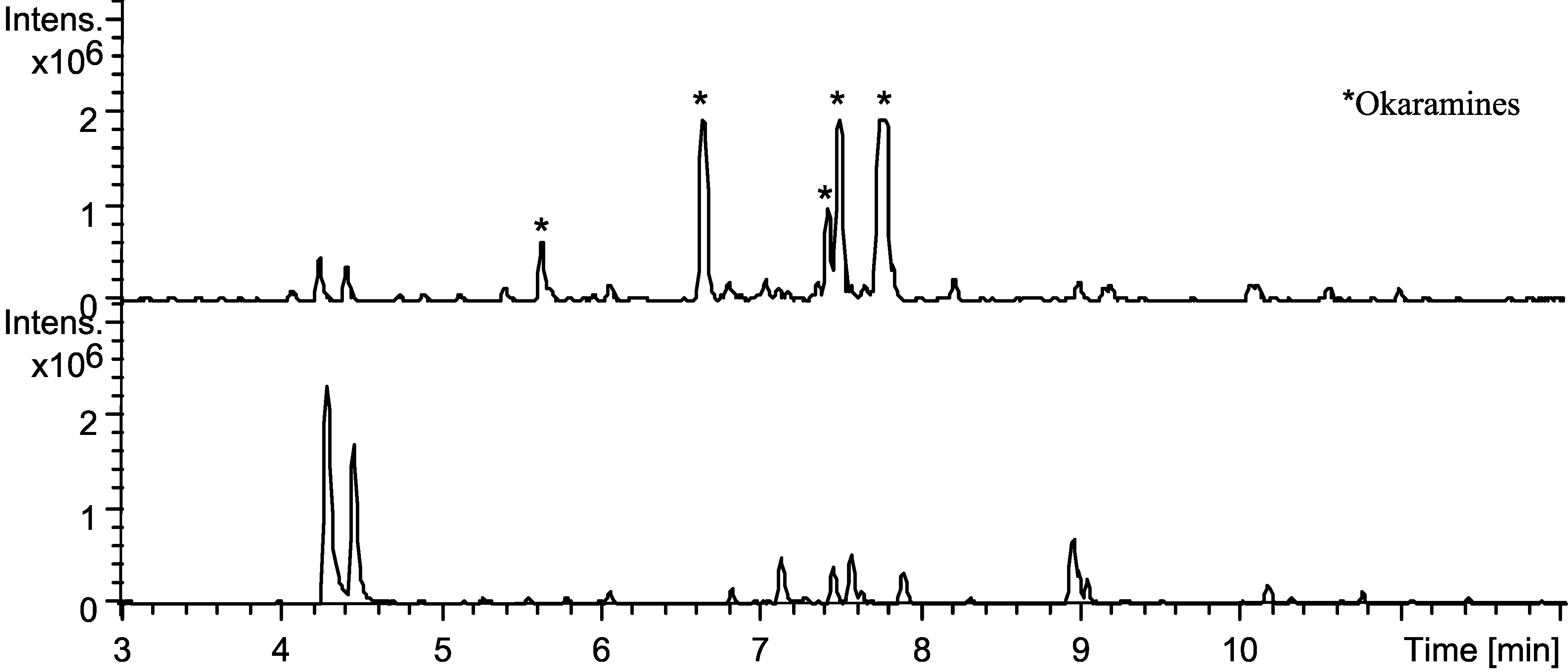
2.4. Production of Sclerotia Reveals a Highly Regulated Metabolic Profile


3. Experimental Section
3.1. Fungal Growth, Strains and Media
3.2. Large Scale Extraction
3.3. Plug Extraction
3.4. Sclerotium Extraction
3.5. UHPLC-DAD-HRMS Analysis
3.6. NMR
3.7. Marfey’s Reaction
3.8. Purification of Metabolites
 = +0.63° (MeOH). 1H- and 13C-NMR (see Table 1 and Table 2). = +6.96° (MeOH). 1H- and 13C-NMR (see Table 1 and Table 2). = 0.00° (MeOH). 1H- and 13C-NMR (see Table 1 and Table 2). = +46.1° (MeOH). 1H-NMR (499.87 MHz, DMSO-d6, 25 °C, 2.49 ppm): 0.77 (3H, d, J = 7.1 Hz), 0.97 (3H, d, J = 7.1 Hz), 1.13 (3H, s), 1.22 (3H, s), 1.32 (1H, m), 1.69 (3H, s), 1.98 (3H, s), 2.02 (1H, m), 2.30 (1H, dd, J = 13.8, 3.8 Hz), 2.37 (1H, m), 2.41 (1H, m), 2.69 (1H, dd, J = 13.8, 8.2 Hz), 2.80 (1H, m), 3.94 (1H, d, J = 9.4 Hz), 5.06 (1H, m, -OH), 5.10 (1H, m), 5.59 (1H, d, J = 9.6 Hz), 5.70 (1H, s), 5.86 (1H, m), 5.88 (1H, d, J = 14.9 Hz), 5.97 (1H, d, J = 9.8 Hz), 6.05 (1H, d, J = 11.2 Hz), 6.30 (1H, d, J = 12.0 Hz), 6.38 (1H, d, J = 15.1 Hz), 6.68 (1H, dd, J = 15.1, 11.2 Hz), 7.51 (1H, dd, J = 14.9, 12.0 Hz; 13C-NMR (125.70 MHz, DMSO-d6, 25 °C, 39.5 ppm): 10.9, 12.3, 13.0, 13.7, 20.6, 25.4, 26.0, 34.7, 39.8, 43.6, 43.7, 56.2, 56.3, 68.3, 78.3, 121.2, 126.4, 127.0, 127.2, 130.8, 132.7, 133.8, 135.7, 139.4, 140.9, 143.4, 173.9, 212.8. = +8.67° (MeOH); 1H-NMR (499.87 MHz, DMSO-d6, 25 °C, 2.49 ppm): 0.77 (3H, d, J = 7.1 Hz), 0.95 (3H, d, J = 7.1 Hz), 1.01 (3H, s), 1.21 (1H, m), 1.27 (3H, s), 1.71 (3H, s), 1.93 (1H, m), 1.98 (3H, s), 2.35 (1H, m), 2.38 (1H, qin, J = 8.4), 2.56 (1H, m), 2.92 (1H, dt, J = 17.5, 7.5 Hz), 2.99 (1H, m), 3.56 (2H, m), 3.95 (1H, dd, J = 9.4, 3.1 Hz), 5.09 (1H, br.d, J = 3.5 Hz, -OH), 5.05 (1H, s, -OH), 5.20 (1H, m), 5.37 (1H, d, J = 9.9 Hz), 5.59 (1H, m), 5.88 (1H, d, J = 14.9 Hz), 5.90 (1H, d, J = 9.9 Hz), 6.07 (1H, d, J = 11.1 Hz), 6.31 (1H, d, J = 12.0 Hz), 6.39 (1H, d, J = 15.1 Hz), 6.69 (1H, dd, J = 15.1, 11.1 Hz), 7.51 (1H, dd, J = 14.9, 12.0); 13C-NMR (125.70 MHz, DMSO-d6, 25 °C, 39.5 ppm): 10.9, 12.3, 13.0, 13.7, 20.6, 25.4, 26.0, 34.7, 39.8, 43.6, 43.7, 56.2, 56.3, 68.3, 78.3, 121.2, 126.4, 127.0, 127.2, 130.8, 132.7, 133.8, 135.7, 139.4, 140.9, 143.4, 173.9, 212.8. = −49.23° (MeOH). 1H- and 13C-NMR (see Table 3). = +63.58° (MeOH); 1H- and 13C-NMR (see Table 5). = −15.29° (MeOH); 1H- and 13C-NMR (see Table 4). = +15.38° (MeOH); 1H-NMR (499.87 MHz, DMSO-d6, 25 °C, 2.49 ppm): 1.52 (6H, s, H-17, H-18), 1.68 (3H, s, H-34), 1.73 (3H, s, H-35), 1.86 (1H, m, H-20), 2.42 (1H, dd, 13.2, 6.8, H-20'), 2.99 (1H, dd, 15.2, 9.4, H-1), 3.14 (1H, dd, 16.3, 7.1, H-31), 3.23 (1H, dd, 16.8, 7.5, H-31'), 3.59 (1H, dd, 15.2, 4.2, H-1'), 4.46 (1H, dd, 9.1, 4.4, H-2), 4.67 (1H, dd, 11.3, 6.4, H-19), 5.05 (1H, dd, 10.5, 1.1, H-4), 5.08 (1H, dd, 17.4, 1.1, H-4'), 5.26 (1H, t, 7.4, H-32), 5.33 (1H, d, 4.5, H-29), 6.01 (1H, s , H-36), 6.12 (1H, d, 4.4, H-28), 6.22 (1H, dd, 17.4, 10.5, H-5), 6.32 (1H, s, H-3), 6.67 (1H, t, 7.5, H-24), 6.88 (1H, d, 7.5, H-25), 6.96 (1H, t, 7.5, H-12), 6.96 (H-26), 7.05 (2H, H-11, H-23), 7.35 (1H, s, H-10), 7.53 (1H, s, H-13), 10.67 (1H, s, H-8); 13C-NMR (125.70 MHz, DMSO-d6, 25 °C, 39.5 ppm): 17.5 (C-34), 24.7 (C-1), 25.2 (C-35), 27.6 (C-17,C-18), 28.2 (C-31), 39.0 (C-6), 40.8 (C-20), 55.1 (C-2), 58.2 (C-19), 83.5 (C-29), 85.5 (C-21), 104.3 (C-15), 110.7 (C-10), 110.9 (C-4), 117.6 (C-13), 118.4 (C-12, C-24), 118.9 (C-23), 120.0 (C-11), 121.5 (C-32), 122.7 (C-22), 128.3 (C-14), 131.0 (C-26), 132.0 (C-33), 134.5 (C-9), 141.1 (C-7), 145.8 (C-27), 146.0 (C-5), 167.5 (C-16), 169.7 (C-30).
= +0.63° (MeOH). 1H- and 13C-NMR (see Table 1 and Table 2). = +6.96° (MeOH). 1H- and 13C-NMR (see Table 1 and Table 2). = 0.00° (MeOH). 1H- and 13C-NMR (see Table 1 and Table 2). = +46.1° (MeOH). 1H-NMR (499.87 MHz, DMSO-d6, 25 °C, 2.49 ppm): 0.77 (3H, d, J = 7.1 Hz), 0.97 (3H, d, J = 7.1 Hz), 1.13 (3H, s), 1.22 (3H, s), 1.32 (1H, m), 1.69 (3H, s), 1.98 (3H, s), 2.02 (1H, m), 2.30 (1H, dd, J = 13.8, 3.8 Hz), 2.37 (1H, m), 2.41 (1H, m), 2.69 (1H, dd, J = 13.8, 8.2 Hz), 2.80 (1H, m), 3.94 (1H, d, J = 9.4 Hz), 5.06 (1H, m, -OH), 5.10 (1H, m), 5.59 (1H, d, J = 9.6 Hz), 5.70 (1H, s), 5.86 (1H, m), 5.88 (1H, d, J = 14.9 Hz), 5.97 (1H, d, J = 9.8 Hz), 6.05 (1H, d, J = 11.2 Hz), 6.30 (1H, d, J = 12.0 Hz), 6.38 (1H, d, J = 15.1 Hz), 6.68 (1H, dd, J = 15.1, 11.2 Hz), 7.51 (1H, dd, J = 14.9, 12.0 Hz; 13C-NMR (125.70 MHz, DMSO-d6, 25 °C, 39.5 ppm): 10.9, 12.3, 13.0, 13.7, 20.6, 25.4, 26.0, 34.7, 39.8, 43.6, 43.7, 56.2, 56.3, 68.3, 78.3, 121.2, 126.4, 127.0, 127.2, 130.8, 132.7, 133.8, 135.7, 139.4, 140.9, 143.4, 173.9, 212.8. = +8.67° (MeOH); 1H-NMR (499.87 MHz, DMSO-d6, 25 °C, 2.49 ppm): 0.77 (3H, d, J = 7.1 Hz), 0.95 (3H, d, J = 7.1 Hz), 1.01 (3H, s), 1.21 (1H, m), 1.27 (3H, s), 1.71 (3H, s), 1.93 (1H, m), 1.98 (3H, s), 2.35 (1H, m), 2.38 (1H, qin, J = 8.4), 2.56 (1H, m), 2.92 (1H, dt, J = 17.5, 7.5 Hz), 2.99 (1H, m), 3.56 (2H, m), 3.95 (1H, dd, J = 9.4, 3.1 Hz), 5.09 (1H, br.d, J = 3.5 Hz, -OH), 5.05 (1H, s, -OH), 5.20 (1H, m), 5.37 (1H, d, J = 9.9 Hz), 5.59 (1H, m), 5.88 (1H, d, J = 14.9 Hz), 5.90 (1H, d, J = 9.9 Hz), 6.07 (1H, d, J = 11.1 Hz), 6.31 (1H, d, J = 12.0 Hz), 6.39 (1H, d, J = 15.1 Hz), 6.69 (1H, dd, J = 15.1, 11.1 Hz), 7.51 (1H, dd, J = 14.9, 12.0); 13C-NMR (125.70 MHz, DMSO-d6, 25 °C, 39.5 ppm): 10.9, 12.3, 13.0, 13.7, 20.6, 25.4, 26.0, 34.7, 39.8, 43.6, 43.7, 56.2, 56.3, 68.3, 78.3, 121.2, 126.4, 127.0, 127.2, 130.8, 132.7, 133.8, 135.7, 139.4, 140.9, 143.4, 173.9, 212.8. = −49.23° (MeOH). 1H- and 13C-NMR (see Table 3). = +63.58° (MeOH); 1H- and 13C-NMR (see Table 5). = −15.29° (MeOH); 1H- and 13C-NMR (see Table 4). = +15.38° (MeOH); 1H-NMR (499.87 MHz, DMSO-d6, 25 °C, 2.49 ppm): 1.52 (6H, s, H-17, H-18), 1.68 (3H, s, H-34), 1.73 (3H, s, H-35), 1.86 (1H, m, H-20), 2.42 (1H, dd, 13.2, 6.8, H-20'), 2.99 (1H, dd, 15.2, 9.4, H-1), 3.14 (1H, dd, 16.3, 7.1, H-31), 3.23 (1H, dd, 16.8, 7.5, H-31'), 3.59 (1H, dd, 15.2, 4.2, H-1'), 4.46 (1H, dd, 9.1, 4.4, H-2), 4.67 (1H, dd, 11.3, 6.4, H-19), 5.05 (1H, dd, 10.5, 1.1, H-4), 5.08 (1H, dd, 17.4, 1.1, H-4'), 5.26 (1H, t, 7.4, H-32), 5.33 (1H, d, 4.5, H-29), 6.01 (1H, s , H-36), 6.12 (1H, d, 4.4, H-28), 6.22 (1H, dd, 17.4, 10.5, H-5), 6.32 (1H, s, H-3), 6.67 (1H, t, 7.5, H-24), 6.88 (1H, d, 7.5, H-25), 6.96 (1H, t, 7.5, H-12), 6.96 (H-26), 7.05 (2H, H-11, H-23), 7.35 (1H, s, H-10), 7.53 (1H, s, H-13), 10.67 (1H, s, H-8); 13C-NMR (125.70 MHz, DMSO-d6, 25 °C, 39.5 ppm): 17.5 (C-34), 24.7 (C-1), 25.2 (C-35), 27.6 (C-17,C-18), 28.2 (C-31), 39.0 (C-6), 40.8 (C-20), 55.1 (C-2), 58.2 (C-19), 83.5 (C-29), 85.5 (C-21), 104.3 (C-15), 110.7 (C-10), 110.9 (C-4), 117.6 (C-13), 118.4 (C-12, C-24), 118.9 (C-23), 120.0 (C-11), 121.5 (C-32), 122.7 (C-22), 128.3 (C-14), 131.0 (C-26), 132.0 (C-33), 134.5 (C-9), 141.1 (C-7), 145.8 (C-27), 146.0 (C-5), 167.5 (C-16), 169.7 (C-30).3.9. Antifungal Susceptibility Testing
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nielsen, K.F.; Mogensen, J.M.; Johansen, M.; Larsen, T.O.; Frisvad, J.C. Review of secondary metabolites and mycotoxins from the Aspergillus niger group. Anal. Bioanal. Chem. 2009, 395, 1225–1242. [Google Scholar] [CrossRef]
- Song, Y.C.; Li, H.; Ye, Y.H.; Shan, C.Y.; Yang, Y.M.; Tan, R.X. Endophytic naphthopyrone metabolites are co-inhibitors of xanthine oxidase, SW1116 cell and some microbial growths. FEMS Microbiol. Lett. 2004, 241, 67–72. [Google Scholar] [CrossRef]
- Koyama, K.; Ominato, K.; Natori, S.; Tashiro, T.; Tsuruo, T. Cytotoxicity and antitumor activities of fungal bis(naphtho-gamma-pyrone) derivatives. J. Pharmacobio-Dyn. 1988, 11, 630–635. [Google Scholar] [CrossRef]
- Abarca, M.; Bragulat, M. Ochratoxin A production by strains of Aspergillus niger var. niger. Appl. Environ. Microbiol. 1994, 60, 2650–2652. [Google Scholar]
- Frisvad, J.C.; Smedsgaard, J.; Samson, R.A.; Larsen, T.O.; Thrane, U. Fumonisin B2 Production by Aspergillus niger. J. Agric. Food Chem. 2007, 55, 9727–9732. [Google Scholar] [CrossRef]
- Varga, J.; Frisvad, J.C.; Kocsubé, S.; Brankovics, B.; Tóth, B.; Szigeti, G.; Samson, R.A. New and revisited species in Aspergillus section Nigri. Stud. Mycol. 2011, 69, 1–17. [Google Scholar] [CrossRef]
- Jurjević, Z.; Peterson, S.W.; Stea, G.; Solfrizzo, M.; Varga, J.; Hubka, V.; Perrone, G. Two novel species of Aspergillus section Nigri from indoor air. IMA Fungus 2012, 3, 159–173. [Google Scholar] [CrossRef]
- Mizuno, K.; Yagi, A.; Satoi, S.; Takada, M.; Hayashi, M.; Asano, K.; Matsuda, T. Studies on aculeacin. I. Isolation and characterization of aculeacin A. J. Antibiot. 1977, 4, 297–302. [Google Scholar]
- Satoi, S.; Yagi, A.; Asano, K.; Misuno, K.; Watanabe, T. Studies on aculeacin. II. Isolation and characterization of aculeacins B, C, D, E, F and G. J. Antibiot. 1977, 30, 303–307. [Google Scholar]
- Watanabe, S.; Hirai, H.; Ishiguro, M.; Kambara, T.; Kojima, Y.; Matsunaga, T.; Nishida, H.; Suzuki, Y.; Harwood, H.J.; Huang, L.H.; et al. CJ-15,183, a new inhibitor of squalene synthase produced by a fungus, Aspergillus aculeatus. J. Antibiot. 2001, 54, 904–910. [Google Scholar]
- Andersen, R.; Buechi, G.; Kobbe, B.; Demain, A.L. Secalonic acids D and F are toxic metabolites of Aspergillus aculeatus. J. Org. Chem. 1977, 42, 352–353. [Google Scholar]
- Hayashi, H.; Furutsuka, K.; Shiono, Y. Okaramines H and I, new Okaramine Congeners, from Aspergillus aculeatus. J. Nat. Prod. 1999, 62, 315–317. [Google Scholar] [CrossRef]
- Ingavat, N.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Asperaculin A, a sesquiterpenoid from a marine-derived fungus, Aspergillus aculeatus. J. Nat. Prod. 2011, 74, 1650–1652. [Google Scholar] [CrossRef]
- Ingavat, N.; Dobereiner, J.; Wiyakrutta, S.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Aspergillusol A, an alpha-glucosidase inhibitor from the marine-derived fungus Aspergillus aculeatus. J. Nat. Prod. 2009, 72, 2049–2052. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Q.-Q.; Zhang, W.-W.; Liu, Q.-Y.; Zheng, Q.-H.; Zhong, P.; Hu, X.; Fang, Z.-X. Aculeatusquinones A-D, novel metabolites from the marine-derived fungus Aspergillus aculeatus. Heterocycles 2013, 87, 861–868. [Google Scholar] [CrossRef]
- Goldberg, I.; Rokem, J.; Pines, O. Organic acids: Old metabolites, new themes. J. Chem. Technol. Biotechnol. 2006, 81, 1601–1611. [Google Scholar] [CrossRef]
- Pel, H.J.; de Winde, J.H.; Archer, D.B.; Dyer, P.S.; Hofmann, G.; Schaap, P.J.; Turner, G.; de Vries, R.P.; Albang, R.; Albermann, K.; et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat. Biotechnol. 2007, 25, 221–231. [Google Scholar] [CrossRef]
- Perrone, G.; Susca, A.; Cozzi, G.; Ehrlich, K.C.; Varga, J.; Frisvad, J.C.; Meijer, M.; Noonim, P.; Mahakarnchanakul, W.; Samson, R.A. Biodiversity of Aspergillus species in some important agricultural products. Stud. Mycol. 2007, 59, 53–66. [Google Scholar] [CrossRef]
- Adisa, V.A. Pectic enzymes of Aspergillus aculeatus associated with post-harvest deterioration of citrus sinensis fruit. J. Food Biochem. 1989, 13, 243–252. [Google Scholar] [CrossRef]
- Bhat, M.K. Cellulases and related enzymes in biotechnology. Biotechnol. Adv. 2000, 18, 355–383. [Google Scholar] [CrossRef]
- Fujimoto, H.; Ooi, T.; Wang, S.; Takizawa, T. Purification and properties of three xylanases from Aspergillus aculeatus. Biosci. Biotechnol. Biochem. 1995, 59, 538–540. [Google Scholar] [CrossRef]
- Polizeli, M.L.T.M.; Rizzatti, A.C.S.; Monti, R.; Terenzi, H.F.; Jorge, J.A.; Amorim, D.S. Xylanases from fungi: Properties and industrial applications. Appl. Microbiol. Biotechnol. 2005, 67, 577–591. [Google Scholar] [CrossRef]
- Olutiola, P.O.; Nwaogwugwu, R.I. Growth, sporulation and production of maltase and proteolytic enzymes in Aspergillus aculeatus. Trans. Br. Mycol. Soc. 1982, 78, 105–113. [Google Scholar] [CrossRef]
- Christophersen, C.; Crescente, O.; Frisvad, J.C.; Gram, L.; Nielsen, J.; Nielsen, P.H.; Rahbaek, L. Antibacterial activity of marine-derived fungi. Mycopathologia 1999, 143, 135–138. [Google Scholar]
- Samson, R.A.; Houbraken, J.; Thrane, U.; Frisvad, J.C.; Andersen, B. Food and Indoor Fungi; CBS-KNAW Fungal Biodiversity Centre: Utrecht, The Netherlands, 2010. [Google Scholar]
- Smedsgaard, J. Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. J. Chromatogr. A 1997, 760, 264–270. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Ma, M.; Rank, C.; Frisvad, J.C.; Larsen, T.O. Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 2011, 74, 2338–2348. [Google Scholar] [CrossRef]
- Laatsch, H. Antibase 2012. Available online: http://www.wiley-vch.de/stmdata/antibase.php (accessed on 1 February 2014).
- Shiono, Y.; Akiyama, K.; Hayashi, H. New okaramine congeners, okaramines J, K, L, M and related compounds, from Penicillium Simplicissimum ATCC90288. Biosci. Biotechnol. Biochem. 1999, 63, 1910–1920. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Smedsgaard, J. Fungal metabolite screening: Database of 474 mycotoxins and fungal metabolites for dereplication by standardised liquid chromatography-UV-mass spectrometry methodology. J. Chromatogr. A 2003, 1002, 111–136. [Google Scholar] [CrossRef]
- Jackson, M.; Karwowski, J.; Humphrey, P. Calbistrins, novel antifungal agents produced by Penicillium restrictum. I: Production, taxonomy of the producing organism and biological activity. J. Antibiot. 1993, 46, 34–38. [Google Scholar] [CrossRef]
- Brill, G.; Chen, R.; Rasmussen, R. Calbistrins, novel antifungal agents produced by Penicillium restrictum. II: Isolation and elucidation of structure. J. Antibiot. 1993, 46, 39–47. [Google Scholar] [CrossRef]
- Sørensen, A.; Lübeck, P.S.; Lübeck, M.; Nielsen, K.F.; Ahring, B.K.; Teller, P.J.; Frisvad, J.C. Aspergillus saccharolyticus sp. nov., a black Aspergillus species isolated in Denmark. Int. J. Syst. Evol. Microbiol. 2011, 61, 3077–3083. [Google Scholar] [CrossRef]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-di-fluoro-2,4-dinitrobenzen. Carlsb. Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef]
- Rank, C.; Klejnstrup, M.L.; Petersen, L.M.; Kildgaard, S.; Frisvad, J.C.; Gotfredsen, C.H.; Larsen, T.O. Comparative chemistry of Aspergillus oryzae (RIB40) and A. flavus (NRRL 3357). Metabolites 2012, 2, 39–56. [Google Scholar] [CrossRef]
- Boyes-Korkis, J.; Gurney, K.; Penn, J. Anacine, a new benzodiazepine metabolite of Penicillium aurantiogriseum produced with other alkaloids in submerged fermentation. J. Nat. Prod. 1993, 56, 1707–1717. [Google Scholar] [CrossRef]
- Wang, H.J.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. Mollenines A and B: new dioxomorpholines from the ascostromata of Eupenicillium molle. J. Nat. Prod. 1998, 61, 804–807. [Google Scholar] [CrossRef]
- TePaske, M.R.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. Three new aflavinines from the sclerotia of Aspergillus tubingensis. Tetrahedron 1989, 45, 4961–4968. [Google Scholar] [CrossRef]
- Samson, R.A.; Noonim, P.; Meijer, M.; Houbraken, J.; Frisvad, J.C.; Varga, J. Diagnostic tools to identify black aspergilli. Stud. Mycol. 2007, 59, 129–145. [Google Scholar] [CrossRef]
- Butts, C.P.; Jones, C.R.; Towers, E.C.; Flynn, J.L.; Appleby, L.; Barron, N.J. Interproton distance determinations by NOE--surprising accuracy and precision in a rigid organic molecule. Org. Biomol. Chem. 2011, 9, 177–184. [Google Scholar]
- Frisvad, J.C.; Petersen, L.M.; Lyhne, E.K.; Larsen, T.O. Formation of sclerotia and production of indoloterpenes by Aspergillus niger and other species in section Nigri. PLoS One 2014, 9, e94857. [Google Scholar]
- Murao, S.; Hayashi, H.; Takiuchi, K.; Arai, M. Okaramine A, a novel indole alkaloid with insecticidal activity, from Penicillium simplicissimum AK-40. Agric. Biol. Chem. 1988, 52, 885–886. [Google Scholar] [CrossRef]
- Hayashi, H.; Takiuchi, K.; Murao, S.; Arai, M. Okaramine B, an insecticidal indole alkaloid, produced by Penicillium simplicissimum AK-40. Agric. Biol. Chem. 1988, 52, 2131–2133. [Google Scholar] [CrossRef]
- Hayashi, H.; Asabu, Y.; Murao, S.; Arai, M. New Okaramine Congeners, Okaramins D, E, and F, from Penicillium simplicissimum ATCC 90288. Jpn. Soc. Biosci. Biotechnol. Agrochem. 1995, 59, 246–250. [Google Scholar] [CrossRef]
- Wicklow, D.T. Metabolites in the coevolution of fungal chemical defence systems. In Coevolution of Fungi with Plant and Animals; Pirozynski, K., Hawksworth, D., Eds.; Academic Press: London, UK, 1988; pp. 173–201. [Google Scholar]
- Macura, S.; Farmer, B., II; Brown, L. An improved method for the determination of cross-relaxation rates from NOE data. J. Magn. Reson. 1986, 70, 493–499. [Google Scholar]
- Hu, H.; Krishnamurthy, K. Revisiting the initial rate approximation in kinetic NOE measurements. J. Magn. Reson. 2006, 182, 173–177. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G.; de Leeuw, F.A.A.M.; Altona, C. The relationship between proton-proton NMR coupling constants and substituents electronegativitis-I. Tetrahedron 1979, 36, 2783–2792. [Google Scholar] [CrossRef]
- Bally, T.; Rablen, P.R. Quantum-chemical simulation of 1H-NMR spectra. 2. Comparison of DFT-based procedures for computing proton-proton coupling constants in organic molecules. J. Org. Chem. 2011, 76, 4818–4830. [Google Scholar] [CrossRef]
- Bochevarov, A.; Harder, E.; Hughes, T.; Greenwood, J.; Braden, D.A.; Philipp, D.; Rinaldo, D.; Halls, M.; Zhang, J.; Friesner, R. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Menucci, B.; Petersson, G.A. Gaussian 09, Revision B.01. Available online: http://www.gaussian.com/ (accessed on 1 February 2014).
- Reference methods for broth dilution antifungal susceptibility testing of yeasts. In Fourth International Supplement; CLSI Document M27-S4; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012.
- Holm, D.K.; Petersen, L.M.; Klitgaard, A.; Knudsen, P.B.; Jarczynska, Z.D.; Nielsen, K.F.; Gotfredsen, C.H.; Larsen, T.O.; Mortensen, U.H. Molecular and chemical characterization of the biosynthesis of the 6-MSA derived meroterpenoid yanuthone D in Aspergillus niger. Chem. Biol. 2014, 21, 519–529. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Petersen, L.M.; Hoeck, C.; Frisvad, J.C.; Gotfredsen, C.H.; Larsen, T.O. Dereplication Guided Discovery of Secondary Metabolites of Mixed Biosynthetic Origin from Aspergillus aculeatus. Molecules 2014, 19, 10898-10921. https://doi.org/10.3390/molecules190810898
Petersen LM, Hoeck C, Frisvad JC, Gotfredsen CH, Larsen TO. Dereplication Guided Discovery of Secondary Metabolites of Mixed Biosynthetic Origin from Aspergillus aculeatus. Molecules. 2014; 19(8):10898-10921. https://doi.org/10.3390/molecules190810898
Chicago/Turabian StylePetersen, Lene M., Casper Hoeck, Jens C. Frisvad, Charlotte H. Gotfredsen, and Thomas O. Larsen. 2014. "Dereplication Guided Discovery of Secondary Metabolites of Mixed Biosynthetic Origin from Aspergillus aculeatus" Molecules 19, no. 8: 10898-10921. https://doi.org/10.3390/molecules190810898
APA StylePetersen, L. M., Hoeck, C., Frisvad, J. C., Gotfredsen, C. H., & Larsen, T. O. (2014). Dereplication Guided Discovery of Secondary Metabolites of Mixed Biosynthetic Origin from Aspergillus aculeatus. Molecules, 19(8), 10898-10921. https://doi.org/10.3390/molecules190810898