Dissociative Electron Transfer to Diphenyl-Substituted Bicyclic Endoperoxides: The Effect of Molecular Structure on the Reactivity of Distonic Radical Anions and Determination of Thermochemical Parameters

Abstract
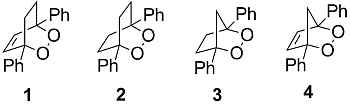
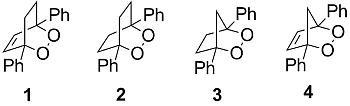
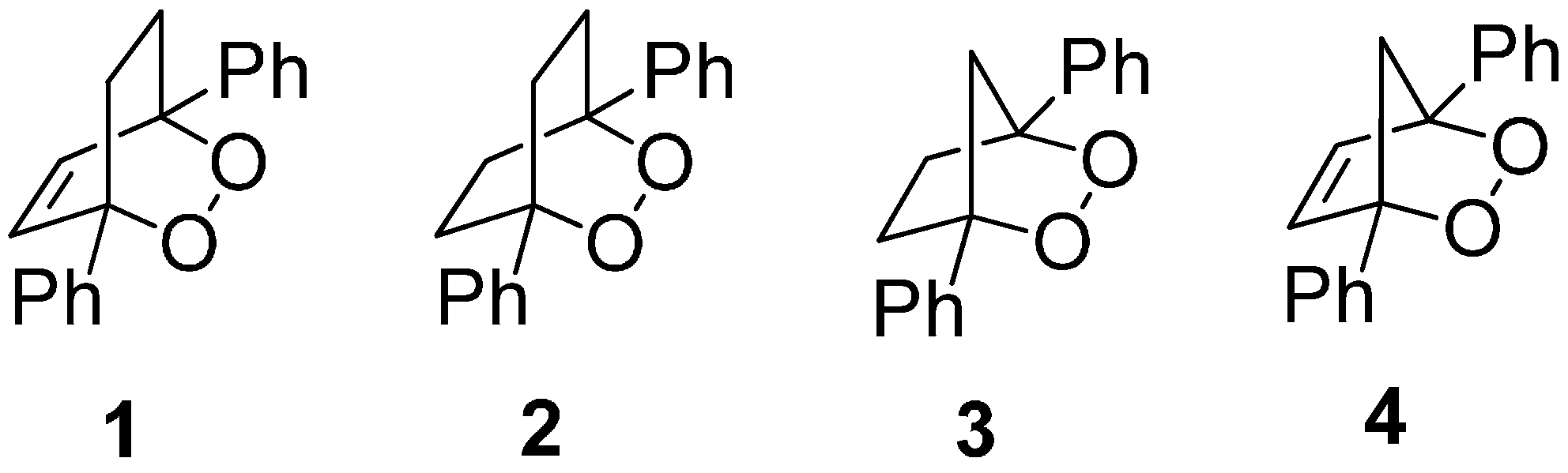
:
1. Introduction



2. Results and Discussion
2.1. Cyclic Voltammetry and Constant Potential Electrolyses
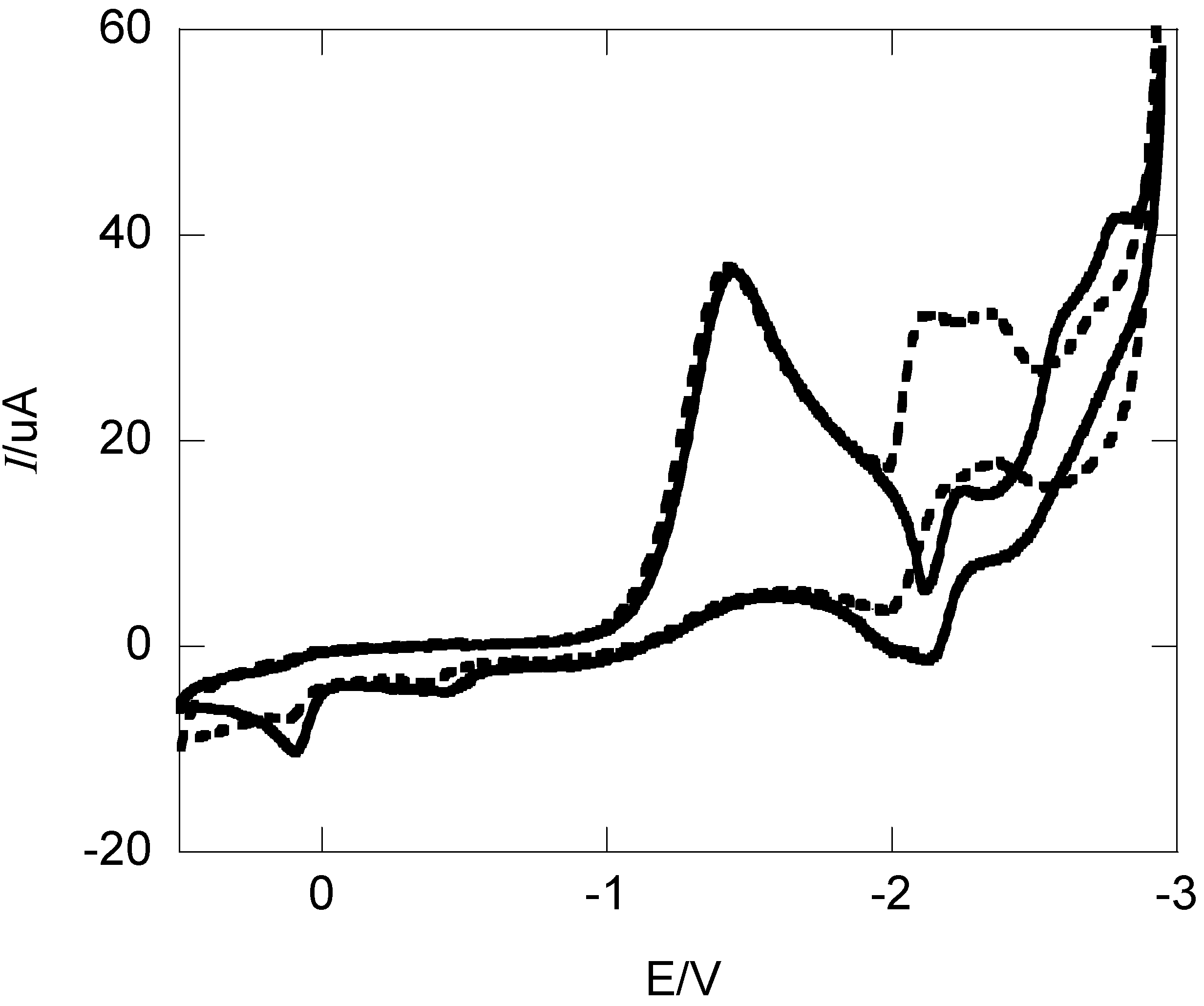

{kind=link}
{kind=link}
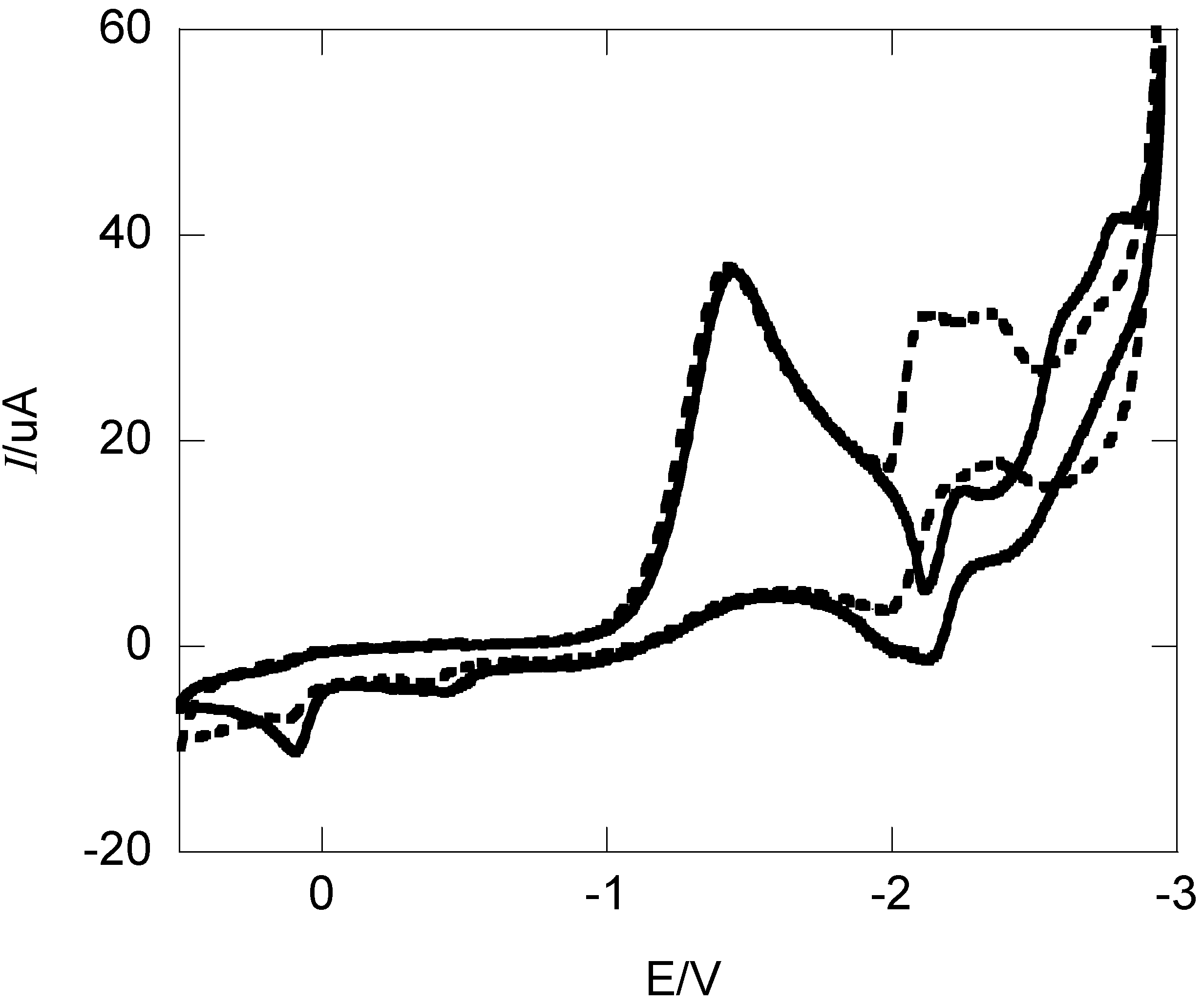{kind=link}
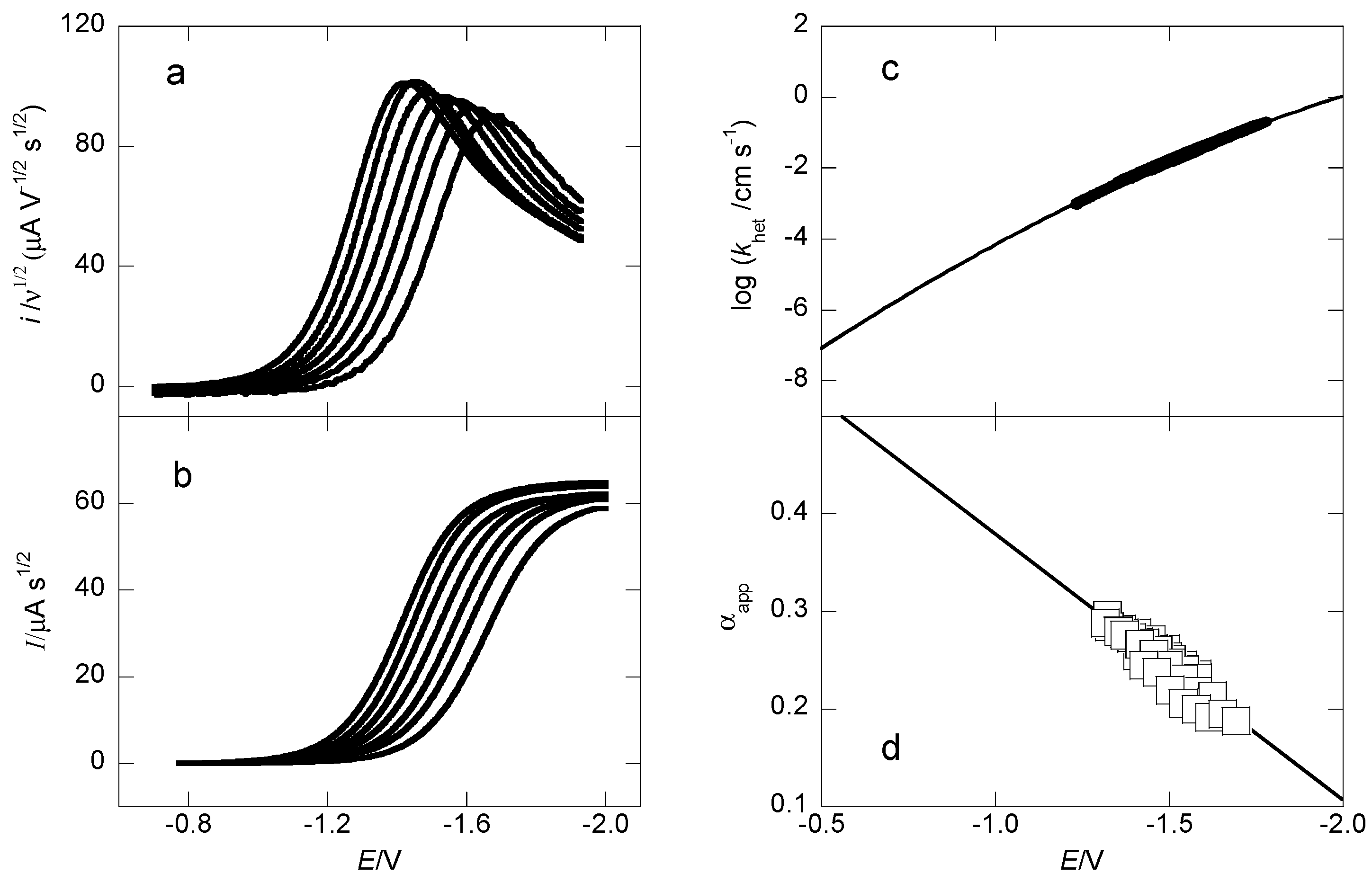{kind=link}
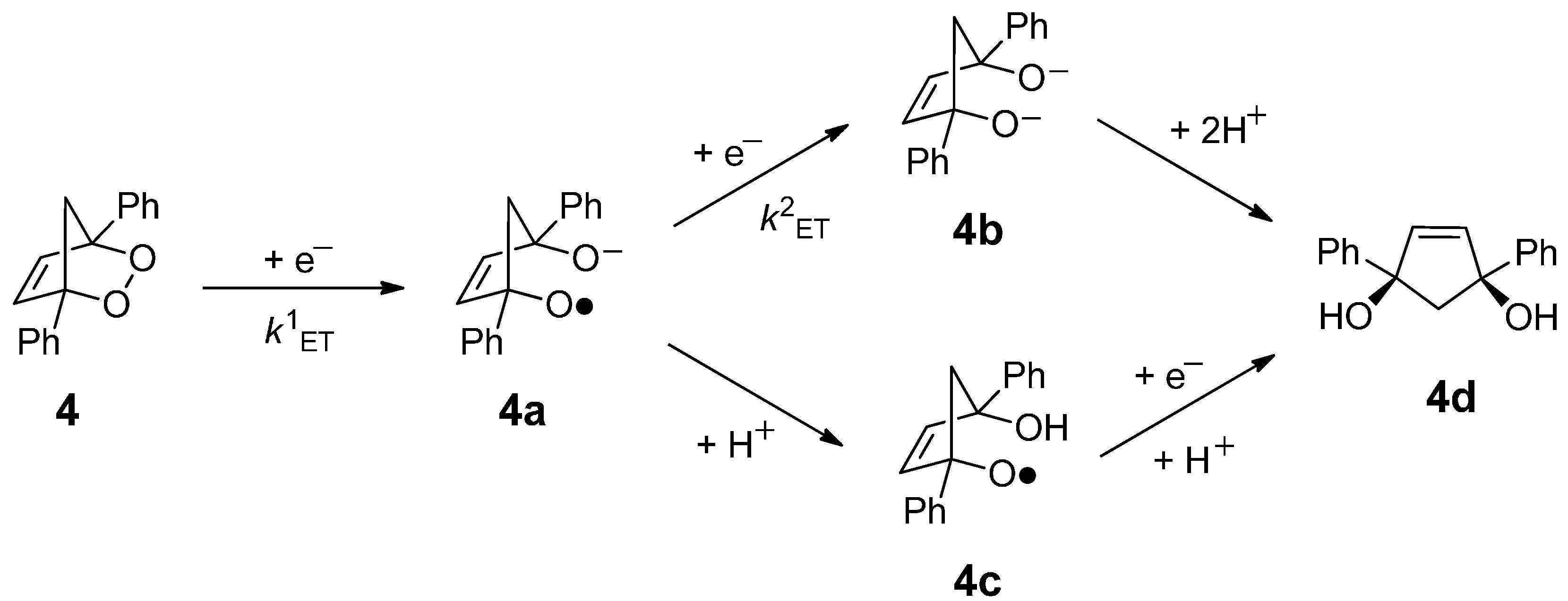{kind=link}
| Experimental CV Data | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Ep/V @ 0.1 V s−1 | −1.27 | −1.45 | −1.34 | −1.42 |
| ΔEp/2/mV @ 0.1 V s−1 | 136 | 142 | 116 | 178 |
| α @ 0.1 V s−1 | 0.351 | 0.336 | 0.411 | 0.268 |
| (dEp/dlog ν)/mV−1 a | −118 | −105 | −132 | −115 |
| α = 1.15RT/F(dEp/dlog ν) a | 0.25 | 0.28 | 0.23 | 0.26 |
| n @ Ep (no acid) b | 1.98 | 1.94 | 1.97 | 1.73 |
| n @ Ep (acid) b | 2.08 | 1.96 | 1.92 | 1.74 |
| n @ E (ca. 200 mV past dip) b | 1.96 | 1.3 | 0.8 | 0.8 |
2.2. Heterogeneous Kinetics and Thermochemical Parameters




| Thermodynamic Data | 1 | 2 | 3 g | 4 |
|---|---|---|---|---|
| D/cm2 s−1 a | 7.7 ° 10−6 | 7.4 ° 10−6 | 6.5 ° 10−6 | 6.0 ° 10−6 |
| E°diss/V b | −0.61 | −0.51 | −0.54 | −0.56 |
| log (k°het/cm s−1) c | −6.1 | −6.5 | −6.8 | −6.7 |
| ΔGo≠/kcal mol−1 d | 9.8 | 10.4 | 11.8 | 10.6 |
| λhet/kcal mol−1 e | 20 | 20 | 18 | 19 |
| BDE/kcal mol−1 f | 19 | 22 | 20 h | 20 i |

3. Experimental Section
3.1. General Information
3.2. Synthesis of 1,4-Diphenyl-2,3-dioxabicyclo[2.2.1]hept-5-ene (4)
3.3. Electrochemistry
3.4. Heterogeneous Electrolysis Products
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Todres, Z.V. Organic Ion Radicals: Chemistry and Applications; Marcel Dekker: New York, NY, USA, 2003. [Google Scholar]
- Chatgilialoglu, C.; Studer, A. Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Inc.: Chichester, UK, 2012. [Google Scholar]
- Zhang, N.; Shampa, R.; Rosen, B.M.; Percec, V. Single electron transfer in radical ion and radical-mediated organic, materials and polymer synthesis. Chem. Rev. 2014, 114, 5848–5958. [Google Scholar] [CrossRef]
- Stevenson, J.P.; Jackson, W.F.; Tanko, J.M. Cyclopropylcarbinyl-type ring openings. Reconciling the chemistry of neutral radicals and radical anions. J. Am. Chem. Soc. 2002, 124, 4271–4281. [Google Scholar]
- Tanko, J.M.; Phillips, J.P. Rearrangements of radical ions: What it means to be both a radical and an ion. J. Am. Chem. Soc. 1999, 121, 6078–6079. [Google Scholar] [CrossRef]
- Chahma, M.; Li, X.; Phillips, P.; Schwartz, P.; Brammer, L.E.; Wang, Y.; Tanko, J.M. Activation/driving force relationships for cyclopropylcarbinyl-homoallyl-type rearrangements of radical anions. J. Phys. Chem. A 2005, 109, 3372–3382. [Google Scholar]
- Tanko, J.M.; Li, X.; Chahma, M.; Jackson, W.F.; Spencer, J.N. Cyclopropyl conjugation and ketyl anions: When do things begin to fall apart? J. Am. Chem. Soc. 2007, 129, 4181–4192. [Google Scholar]
- Gryn’ova, G.; Marshall, D.L.; Blanksby, S.J.; Coote, M.L. Switching radical stability by pH-induced orbital conversion. Nat. Chem. 2013, 5, 474–481. [Google Scholar] [CrossRef]
- Gryn’ova, G.; Coote, M.L. Origin and scope of long-range stabilizing interactions and associated SOMO-HOMO conversion in distonic radical anions. J. Am. Chem. Soc. 2014, 135, 15392–15403. [Google Scholar]
- Magri, D.C.; Workentin, M.S. Model dialkyl peroxides of the Fenton mechanistic probe 2-methyl-1-phenyl-2-propyl hydroperoxide (MPPH): Kinetic probes for dissociative electron transfer. Org. Biomol. Chem. 2003, 1, 3418–3429. [Google Scholar] [CrossRef]
- Donkers, R.L.; Maran, F.; Wayner, D.D.M.; Workentin, M.S. Kinetics of the reduction of dialkyl peroxides. New insights into the dynamics of dissociative electron transfer. J. Am. Chem. Soc. 1999, 121, 7239–7248. [Google Scholar] [CrossRef]
- Antonello, S.; Musumeci, M.; Wayner, D.D.M.; Maran, F. Electroreduction of dialkyl peroxides. Activation-driving force relationships and bond dissociation free energies. J. Am. Chem. Soc. 1997, 119, 9541–9549. [Google Scholar] [CrossRef]
- Workentin, M.S.; Maran, F.; Wayner, D.D.M. Reduction of di-tert-butyl peroxide—Evidence for nonadiabatic dissociative electron transfer. J. Am. Chem. Soc. 1995, 117, 2120–2121. [Google Scholar] [CrossRef]
- Antonello, S.; Formaggio, F.; Moretto, A.; Toniolo, C.; Maran, F. Intramolecular, intermolecular, and heterogeneous nonadiabatic dissociative electron transfer to peresters. J. Am. Chem. Soc. 2001, 123, 9577–9584. [Google Scholar]
- Antonello, S.; Maran, F. The role and relevance of the transfer coefficient alpha in the study of dissociative electron transfers: Concepts and examples from the electroreduction of perbenzoates. J. Am. Chem. Soc. 1999, 121, 9668–9676. [Google Scholar] [CrossRef]
- Antonello, S.; Maran, F. Evidence for the transition between concerted and stepwise heterogeneous electron transfer bond fragmentation mechanisms. J. Am. Chem. Soc. 1997, 119, 12595–12600. [Google Scholar] [CrossRef]
- Antonello, S.; Crisma, M.; Formaggio, F.; Moretto, A.; Taddei, F.; Toniolo, C.; Maran, F. Insights into the free-energy dependence of intramolecular dissociative electron transfers. J. Am. Chem. Soc. 2002, 124, 11503–11513. [Google Scholar] [CrossRef]
- Magri, D.C.; Workentin, M.S. A Radical-anion chain mechanism initiated by dissociative electron transfer to a bicyclic endoperoxide: Insight into the fragmentation chemistry of neutral biradicals and distonic radical anions. Chem. Eur. J. 2008, 14, 1698–1709. [Google Scholar]
- Magri, D.C.; Workentin, M.S. A radical-anion chain mechanism following dissociative electron transfer reduction of the model prostaglandin endoperoxide, 1,4-diphenyl-2,3-dioxabicyclo[2.2.1]heptane. Org. Biomol. Chem. 2008, 18, 3354–3361. [Google Scholar] [CrossRef]
- Stringle, D.L.B.; Magri, D.C.; Workentin, M.S. Efficient homogeneous radical-anion chain reactions initiated by dissociative electron transfer to 3,3,6,6-tetraaryl-1,2-dioxanes. Chem. Eur. J. 2010, 16, 178–188. [Google Scholar] [CrossRef]
- Donkers, R.L.; Workentin, M.S. Elucidation of the electron transfer reduction mechanism of anthracene endoperoxides. J. Am. Chem. Soc. 2004, 126, 1688–1698. [Google Scholar] [CrossRef]
- Donkers, R.L.; Workentin, M.S. Kinetics of dissociative electron transfer to ascaridole and dihydroascaridole—Model bicyclic endoperoxides of biological relevance. Chem. Eur. J. 2001, 7, 4012–4020. [Google Scholar] [CrossRef]
- Donkers, R.L.; Workentin, M.S. First determination of the standard potential for the dissociative reduction of the antimalarial agent artemisinin. J. Phys. Chem. B 1998, 102, 4061–4063. [Google Scholar]
- Najjar, F.; André-Barrès, C.; Lacaze-Dufaure, C.; Magri, D.C.; Workentin, M.S.; Tzèdakis, T. Electrochemical reduction of G3-factor endoperoxide and its methyl ether: Evidence for a competition between concerted and stepwise dissociative electron transfer. Chem. Eur. J. 2007, 13, 1174–1179. [Google Scholar] [CrossRef]
- Costentin, C.; Hajj, V.; Robert, M.; Savéant, J.-M.; Tard, C. Concerted heavy-atom bond cleavage and proton and electron transfers illustrated by proton-assisted reductive cleavage of an O-O bond. Proc. Natl. Acad. Sci. USA 2011, 108, 8559–8564. [Google Scholar] [CrossRef]
- Magri, D.C.; Workentin, M.S. Kinetics of the photoinduced dissociative reduction of the Model alkyl peroxides di-tert-butyl peroxide and ascaridole. Mediterr. J. Chem. 2012, 6, 303–315. [Google Scholar] [CrossRef]
- Magri, D.C.; Donkers, R.L.; Workentin, M.S. Kinetics of the photoinduced electron transfer dissociative reduction of the antimalarial endoperoxide, artemisinin. J. Photochem. Photobio. A Chem. 2001, 138, 29–34. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M.; Tard, C. Breaking bonds with electrons and protons. Models and examples. Acc. Chem. Res. 2014, 47, 271–280. [Google Scholar] [CrossRef]
- Houmam, A. Electron transfer initiated reactions: Bond formation and bond dissociation. Chem. Rev. 2008, 108, 2180–2237. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Electron transfer and bond breaking: Recent advances. Chem. Phys. 2006, 324, 40–56. [Google Scholar]
- Savéant, J.-M. Electron transfer, bond breaking and bond formation. In Advances in Physical Organic Chemistry; Tidwell, T.T., Ed.; Academic Press: New York, NY, USA, 2000; Volume 35, pp. 117–192. [Google Scholar]
- Savéant, J.-M. Electron transfer, bond breaking and bond formation. Acc. Chem. Res. 1993, 26, 455–461. [Google Scholar] [CrossRef]
- Maran, F.; Wayner, D.D.M.; Workentin, M.S. Kinetics and mechanism of the dissociative reduction of C-X and X-X Bonds (X = O, S). In Advances in Physical Organic Chemistry; Tidwell, T.T., Ed.; Academic Press: New York, NY, USA, 2001; Volume 36, p. 85. [Google Scholar]
- Imbeaux, J.C.; Savéant, J.-M. Convolutive Potential Sweep Voltammetry: Ι. Introduction. J. Electroanal. Chem. 1973, 44, 169–187. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods, Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Sample Availability: Samples of the compounds 1–4are available from the authors on request.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Magri, D.C.; Workentin, M.S. Dissociative Electron Transfer to Diphenyl-Substituted Bicyclic Endoperoxides: The Effect of Molecular Structure on the Reactivity of Distonic Radical Anions and Determination of Thermochemical Parameters. Molecules 2014, 19, 11999-12010. https://doi.org/10.3390/molecules190811999
Magri DC, Workentin MS. Dissociative Electron Transfer to Diphenyl-Substituted Bicyclic Endoperoxides: The Effect of Molecular Structure on the Reactivity of Distonic Radical Anions and Determination of Thermochemical Parameters. Molecules. 2014; 19(8):11999-12010. https://doi.org/10.3390/molecules190811999
Chicago/Turabian StyleMagri, David C., and Mark S. Workentin. 2014. "Dissociative Electron Transfer to Diphenyl-Substituted Bicyclic Endoperoxides: The Effect of Molecular Structure on the Reactivity of Distonic Radical Anions and Determination of Thermochemical Parameters" Molecules 19, no. 8: 11999-12010. https://doi.org/10.3390/molecules190811999