The Toxicity Mechanisms of Action of Aβ25–35 in Isolated Rat Cardiac Myocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
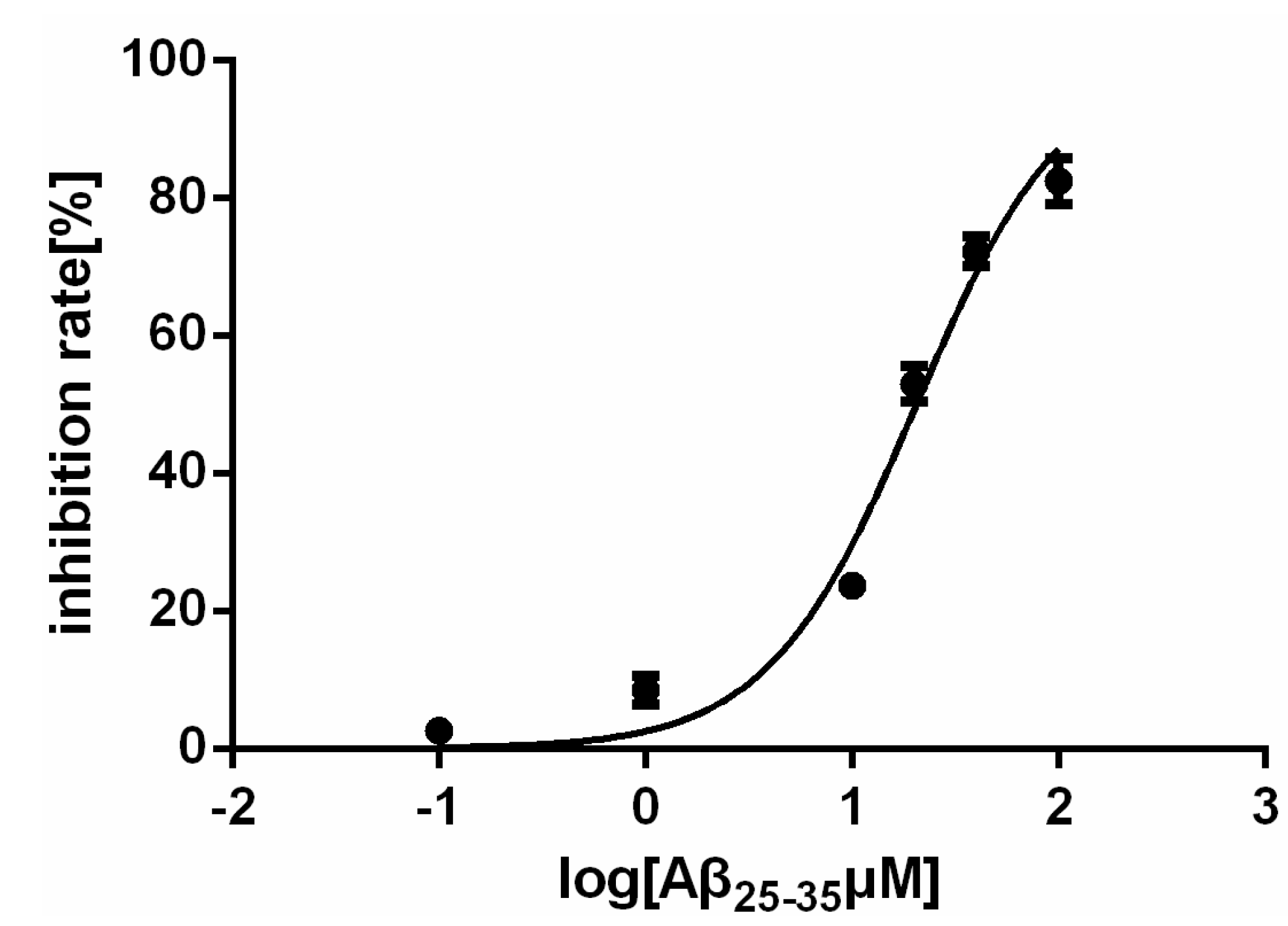
2.1. Effect of Aβ25–35 on the Viability of Rat Cardiac Myocyte in Vitro

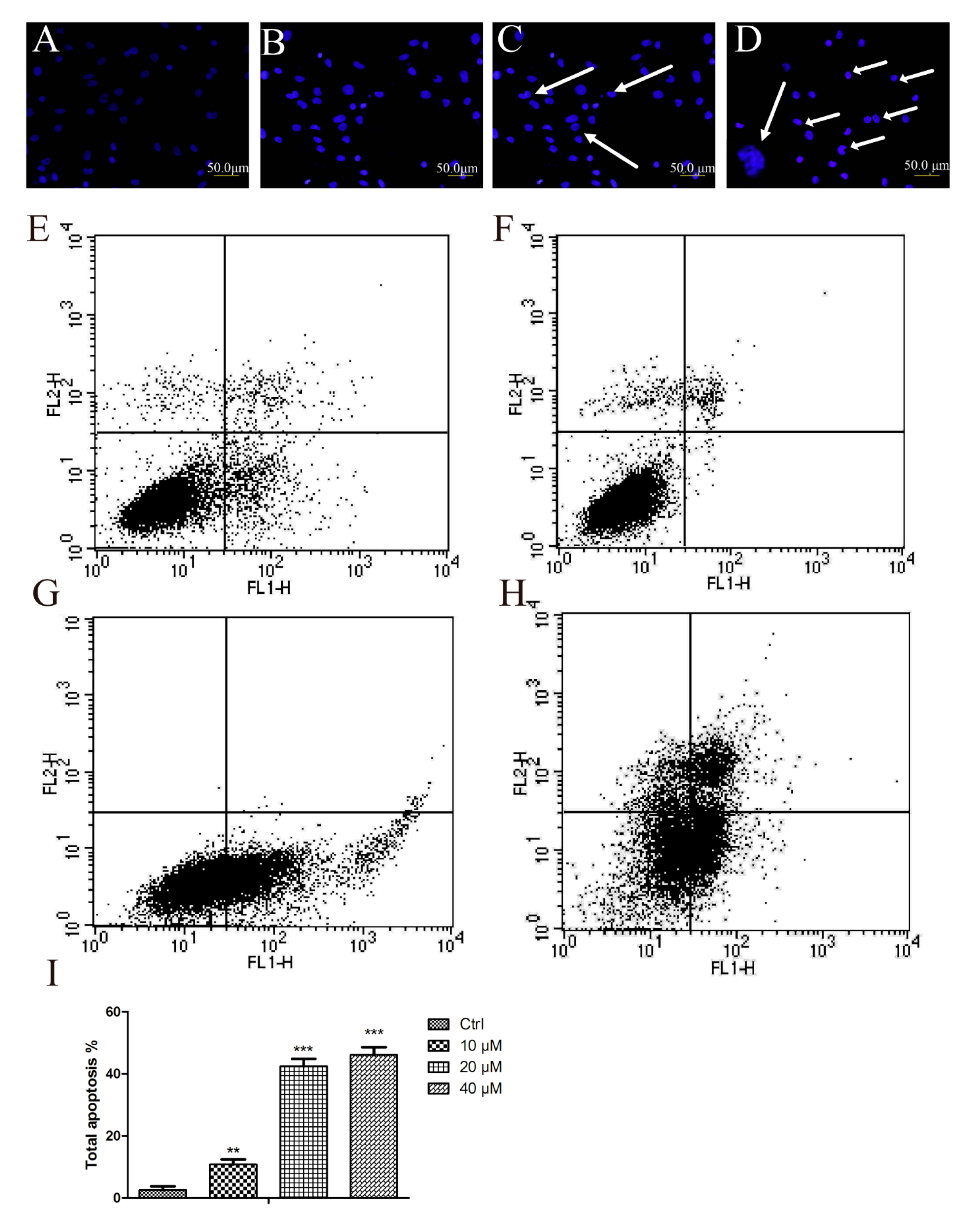
2.2. Aβ25–35 Induced Cardiac Myocytes Apoptosisfigure

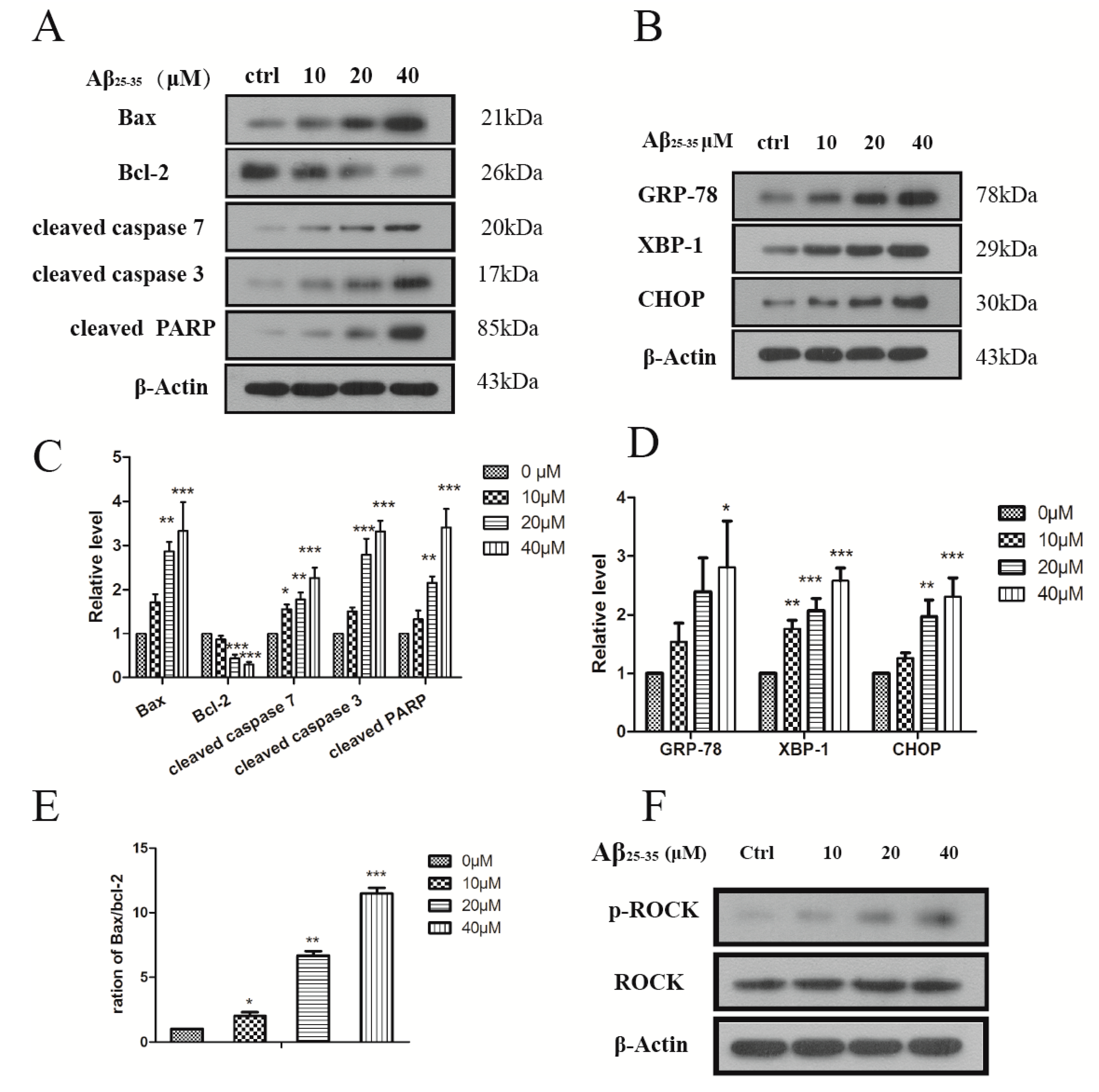
2.3. Aβ25–35 Induced ER Stress in Cardiac Myocytes Cell

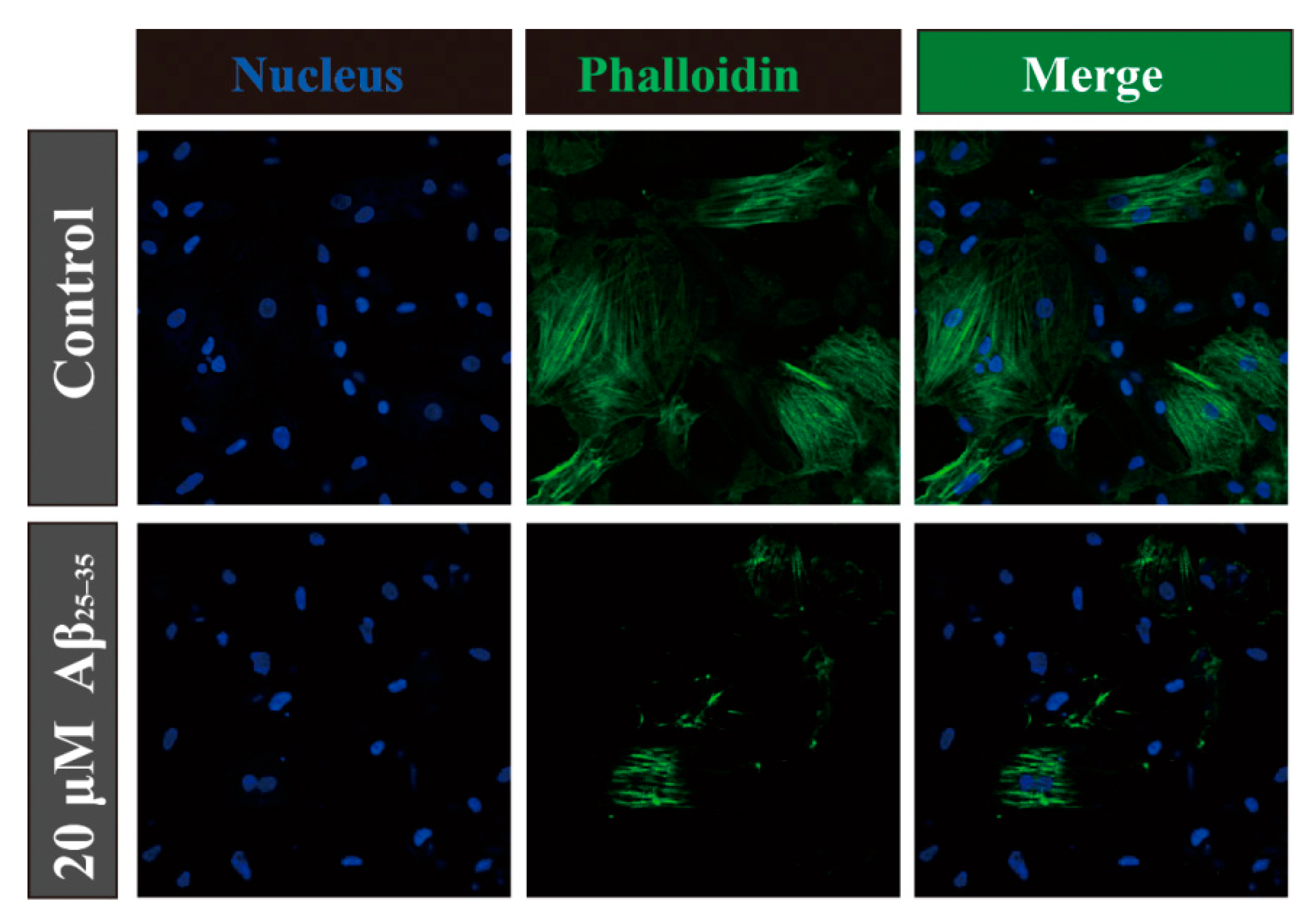
2.4. Aβ25–35 Affects the Cytoskeleton Assembly in Cardiac Myocyte Cells

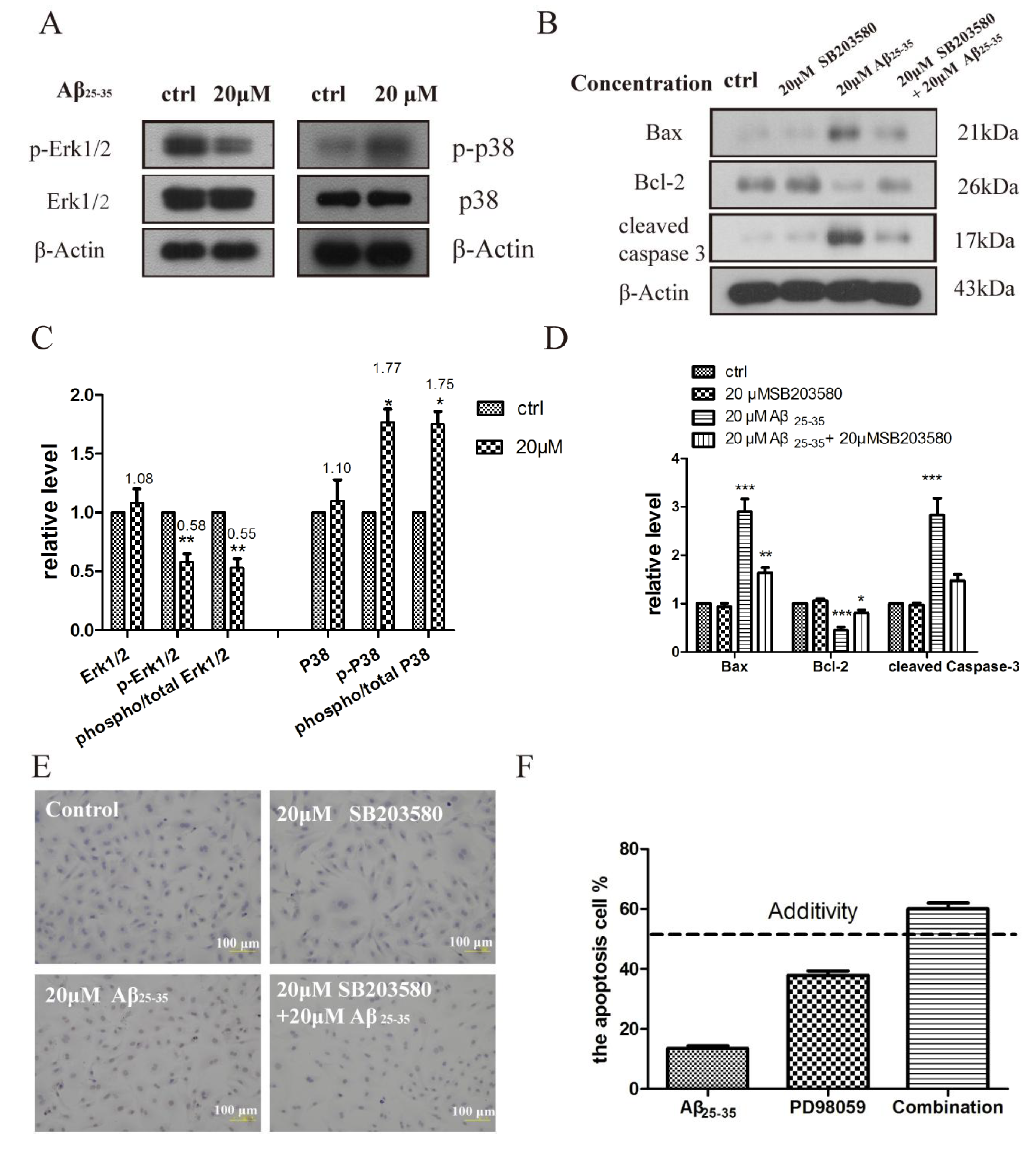
2.5. Activation of p38 and ERK1/2 by Aβ25–35 in Cardiac Myocytes Cell

3. Discussion
4. Experimental
4.1. Cells and Culture Conditions
4.2. Reagents
4.3. Viability Assay
4.4. Cell Morphology Assessment
4.5. TUNEL-Based Assay
4.6. Apoptosis Assay
4.7. Immunofluorescence Assays
4.8. Immunoblotting
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Zheng, H.; Koo, E.H. Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 2011, 6, 27. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Knauer, M.F.; Soreghan, B.; Burdick, D.; Kosmoski, J.; Glabe, C.G. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid a4/beta protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7437–7441. [Google Scholar] [CrossRef]
- Pike, C.J.; Walencewicz-Wasserman, A.J.; Kosmoski, J.; Cribbs, D.H.; Glabe, C.G.; Cotman, C.W. Structure-activity analyses of beta-amyloid peptides: Contributions of the beta 25–35 region to aggregation and neurotoxicity. J. Neurochem. 1995, 64, 253–265. [Google Scholar]
- Halverson, K.; Fraser, P.E.; Kirschner, D.A.; Lansbury, P.T., Jr. Molecular determinants of amyloid deposition in Alzheimer’s disease: Conformational studies of synthetic beta-protein fragments. Biochemistry 1990, 29, 2639–2644. [Google Scholar] [CrossRef]
- Shanmugam, G.; Polavarapu, P.L. Structure of Aβ(25–35) peptide in different environments. Biophys. J. 2004, 87, 622–630. [Google Scholar] [CrossRef]
- Turdi, S.; Guo, R.; Huff, A.F.; Wolf, E.M.; Culver, B.; Ren, J. Cardiomyocyte contractile dysfunction in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. PLoS One 2009, 4, e6033. [Google Scholar]
- Fidzianska, A.; Walczak, E.; Bekta, P.; Chojnowska, L. Are cardiomyocytes able to generate pre-amyloid peptides? Folia Neuropathol. 2011, 49, 64–70. [Google Scholar]
- Chen, W.; Dilsizian, V. Molecular imaging of amyloidosis: Will the heart be the next target after the brain? Curr. Cardiol. Rep. 2012, 14, 226–233. [Google Scholar] [CrossRef]
- Yan, J.; Young, M.E.; Cui, L.; Lopaschuk, G.D.; Liao, R.; Tian, R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 2009, 119, 2818–2828. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, C.; Sheng, X.; Zhang, X.; Wang, B.; Zhang, G. Peripheral expression of mapk pathways in Alzheimer’s and Parkinson’s diseases. J. Clin. Neurosci. 2014, 21, 810–814. [Google Scholar] [CrossRef]
- Davis, J.; Cribbs, D.H.; Cotman, C.W.; van Nostrand, W.E. Pathogenic amyloid beta-protein induces apoptosis in cultured human cerebrovascular smooth muscle cells. Amyloid 1999, 6, 157–164. [Google Scholar] [CrossRef]
- Tong, L.; Thornton, P.L.; Balazs, R.; Cotman, C.W. Beta-amyloid-(1-42) impairs activity-dependent camp-response element-binding protein signaling in neurons at concentrations in which cell survival is not compromised. J. Biol. Chem. 2001, 276, 17301–17306. [Google Scholar]
- Bashir, M.; Parray, A.A.; Baba, R.A.; Bhat, H.F.; Bhat, S.S.; Mushtaq, U.; Andrabi, K.I.; Khanday, F.A. Beta-amyloid-evoked apoptotic cell death is mediated through MKK6-p66SHC pathway. Neuromol. Med. 2014, 16, 137–149. [Google Scholar] [CrossRef]
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science 1989, 245, 417–420. [Google Scholar]
- Costa, R.O.; Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. Inhibition of mitochondrial cytochrome C oxidase potentiates abeta-induced er stress and cell death in cortical neurons. Mol. Cell. Neurosci. 2013, 52, 1–8. [Google Scholar]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. Xbp-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Ward, M.W.; Concannon, C.G.; Whyte, J.; Walsh, C.M.; Corley, B.; Prehn, J.H. The amyloid precursor protein intracellular domain(aicd) disrupts actin dynamics and mitochondrial bioenergetics. J. Neurochem. 2010, 113, 275–284. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of mapk signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar]
- Li, D.Y.; Tao, L.; Liu, H.; Christopher, T.A.; Lopez, B.L.; Ma, X.L. Role of ERK1/2 in the anti-apoptotic and cardioprotective effects of nitric oxide after myocardial ischemia and reperfusion. Apoptosis 2006, 11, 923–930. [Google Scholar] [CrossRef]
- Jones, E.V.; Dickman, M.J.; Whitmarsh, A.J. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem. J. 2007, 405, 617–623. [Google Scholar] [CrossRef]
- Goldoni, M.; Johansson, C. A mathematical approach to study combined effects of toxicants in vitro: Evaluation of the bliss independence criterion and the loewe additivity model. Toxicol. In Vitro 2007, 21, 759–769. [Google Scholar] [CrossRef]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar]
- Rees, K.; Dyakova, M.; Wilson, N.; Ward, K.; Thorogood, M.; Brunner, E. Dietary advice for reducing cardiovascular risk. Cochrane Database Syst. Rev. 2013, 12, CD002128. [Google Scholar]
- Liu, G.; Yao, L.; Liu, J.; Jiang, Y.; Ma, G.; Chen, Z.; Zhao, B.; Li, K. Cardiovascular disease contributes to Alzheimer’s disease: Evidence from large-scale genome-wide association studies. Neurobiol. Aging 2014, 35, 786–792. [Google Scholar]
- Martins, I.J.; Hone, E.; Foster, J.K.; Sunram-Lea, S.I.; Gnjec, A.; Fuller, S.J.; Nolan, D.; Gandy, S.E.; Martins, R.N. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol. Psychiatry 2006, 11, 721–736. [Google Scholar] [CrossRef]
- Van Kan, A.G.; Rolland, Y.; Nourhashemi, F.; Coley, N.; Andrieu, S.; Vellas, B. Cardiovascular disease risk factors and progression of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2009, 27, 240–246. [Google Scholar] [CrossRef]
- Rosendorff, C.; Beeri, M.S.; Silverman, J.M. Cardiovascular risk factors for Alzheimer’s disease. Am. J. Geriatr. Cardiol. 2007, 16, 143–149. [Google Scholar] [CrossRef]
- Epstein, N.U.; Xie, H.; Ruland, S.D.; Pandey, D.K. Vascular risk factors and cardiovascular outcomes in the Alzheimer’s disease neuroimaging initiative. Am. J. Alzheimer’s Dis. Other Dement. 2012, 27, 275–279. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; Veerhuis, R.; van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Resende, R.; Ferreiro, E.; Pereira, C.; Oliveira, C.R. ER stress is involved in abeta-induced Gsk-3beta activation and TAU phosphorylation. J. Neurosci. Res. 2008, 86, 2091–2099. [Google Scholar] [CrossRef]
- Dalton, L.E.; Clarke, H.J.; Knight, J.; Lawson, M.H.; Wason, J.; Lomas, D.A.; Howat, W.J.; Rintoul, R.C.; Rassl, D.M.; Marciniak, S.J. The endoplasmic reticulum stress marker CHOP predicts survival in malignant mesothelioma. Br. J. Cancer 2013, 108, 1340–1347. [Google Scholar] [CrossRef]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef]
- Scull, C.M.; Tabas, I. Mechanisms of er stress-induced apoptosis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2792–2797. [Google Scholar] [CrossRef]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting er stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef]
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; del Monte, F.; Ward, J.E.; Connors, L.H.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Padilla, J.; Jenkins, N.T. Induction of endoplasmic reticulum stress impairs insulin-stimulated vasomotor relaxation in rat aortic rings: Role of Endothelin-1. J. Physiol. Pharmacol. 2013, 64, 557–564. [Google Scholar]
- Haase, N.; Herse, F.; Spallek, B.; Haase, H.; Morano, I.; Qadri, F.; Szijarto, I.A.; Rohm, I.; Yilmaz, A.; Warrington, J.P.; et al. Amyloid-beta peptides activate alpha1-adrenergic cardiovascular receptors. Hypertension 2013, 62, 966–972. [Google Scholar] [CrossRef]
- Cuenda, A.; Rouse, J.; Doza, Y.N.; Meier, R.; Cohen, P.; Gallagher, T.F.; Young, P.R.; Lee, J.C. Sb 203580 is a specific inhibitor of a map kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995, 364, 229–233. [Google Scholar] [CrossRef]
- Goldstein, D.M.; Gabriel, T. Pathway to the clinic: Inhibition of p38 MAP kinase. A review of ten chemotypes selected for development. Curr. Top. Med. Chem. 2005, 5, 1017–1029. [Google Scholar]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Verma, A.; Prasad, K.N.; Singh, A.K.; Nyati, K.K.; Gupta, R.K.; Paliwal, V.K. Evaluation of the MTT lymphocyte proliferation assay for the diagnosis of neurocysticercosis. J. Microbiol. Methods 2010, 81, 175–178. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, B.; Bian, X.; He, P.; Fu, X.; Higuchi, K.; Yang, X.; Li, D. The Toxicity Mechanisms of Action of Aβ25–35 in Isolated Rat Cardiac Myocytes. Molecules 2014, 19, 12242-12257. https://doi.org/10.3390/molecules190812242
Zhang B, Bian X, He P, Fu X, Higuchi K, Yang X, Li D. The Toxicity Mechanisms of Action of Aβ25–35 in Isolated Rat Cardiac Myocytes. Molecules. 2014; 19(8):12242-12257. https://doi.org/10.3390/molecules190812242
Chicago/Turabian StyleZhang, Beiru, Xiaohui Bian, Ping He, Xiaoying Fu, Keiichi Higuchi, Xu Yang, and Detian Li. 2014. "The Toxicity Mechanisms of Action of Aβ25–35 in Isolated Rat Cardiac Myocytes" Molecules 19, no. 8: 12242-12257. https://doi.org/10.3390/molecules190812242