In the first approach, this mutation was designed to predict the relative interaction preferences of the trace residues on the conserved inhibition motifs (residues 639–652 of LDDALDQLSDSLGQ motif of subdomain A, and residues 678–691 of KLGERDDTIPPKYQ motif of subdomain B) at each position within subdomains A and B of CAST4 to the overall CAPN1 binding affinity. In the same pocket, the mutated subdomain B of CAST4 had few conformer changes (from the wild-type form) due to this single mutation effect do little consider the conformational properties of the polypeptide backbone than the interaction of both side chains in the bovine CAPN1/CAST4 complex. However, regarding the mutation energy function, the conformer change derived from alanine scanning was reflected to a side chain and backbone entropy term of the polypeptide dependent on temperature.
2.1.1. Predicting Mutation Effects on the KLGERDDTIPPKYQ Motif in Subdomain B of Bovine CAST4
Dramatic effects of these alanine replacements were observed; the Leu679 and Ile686 mutants of CAST4 (Leu679Ala and Ile686Ala of the subdomain B) permitted the peptide backbone to be more flexible and significantly reduced the hydrophobic interactions with the complementary binding sites (Cys115, Gly208, Gly271 and Ala273 of CAPN1). Each wild-type of the two key residues (Leu679, Ile686) was buried in two hydrophobic pockets (key hydrophobic contact residues; Gly271, Ala273 for Leu679 and Ala111, Leu112 for Ile686) to provide further stabilization via van der Waals contacts with the interface formed from domains DI and DII of the bovine CAPN1 enzyme (
Figure 1b). Regarding interaction preferences, these mutants do not even show two H-bonding interactions with the backbone of two glycine residues (Gly208 and Gly271) since the distance between them is too far to form any H-bonding (exceed the distance threshold 2.5 Å), in contrast to the wild-type complex. Consequently, the two mutants (Leu679Ala and Ile686Ala of CAST4) would be unfavorable for binding bovine CAPN1, destabilizer than the wild-type complex as 3.03 kcal/mol and 2.22 kcal/mol respectively. The prediction further performs a search for stabilizing multiple mutations on a set of any of 20 amino acids for saturation mutagenesis to determine the specificity of residues found at the positions (the positions of 679 and 686 for CAST4). In the cases where combinations of simultaneous mutations are generated, the mutations with the lowest mutation energy at the position from the wild-type protein are scanned. Then, a novel residue that satisfies the positions is identified, highlighting the importance of the sites for its inhibitor binding. This is reflected in the key factors in the active, site-directed CAST4 from the specific locations as shown in
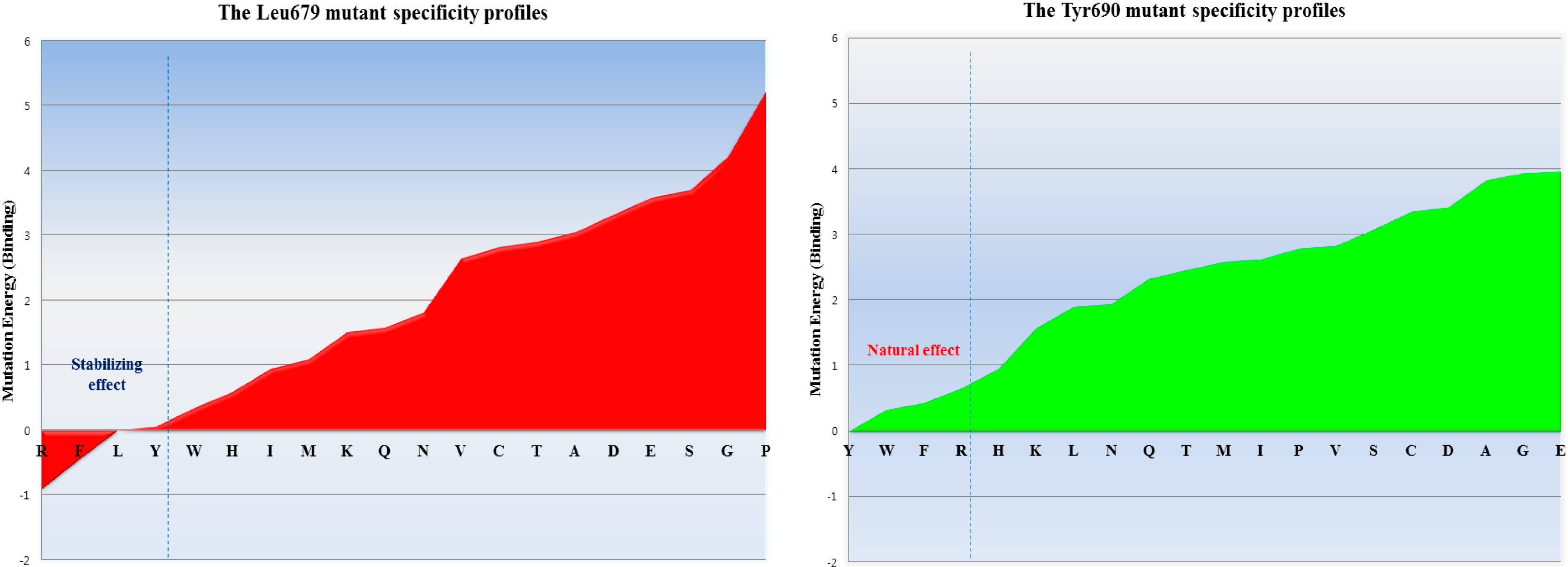
Figure 2. There seems to be a requirement for the formation of H-bonds between them as well as, contribution to essential hydrophobic interactions in the region of the active site for protease inhibition. Moreover, as the general preference, greater freedom of movement of the side chain for large side chain (in an inhibition potency in the order of arginine > phenylalanine > leucine > tyrosine) is required at the 679 position, and the bovine CAPN1 may tolerate tryptophan or, histidine within the hydrophobic binding site as shown in
Figure 2. The mutant specificity (
Figure 2) was defined by the highest selectivity of binding affinity from stabilizing to destabilizing effects at this position, but the priority of these residues seems unlikely to be very selective as they are also accommodated by the hydrophobic pocket of another CAPN member, CAPN2 (
Figure 1a). Interestingly, the residues (Leu679, Ile686) of CAST4 were positioned at the start and end points in a serial β-turn and kink construct, and their orientation in relation to each other was in the opposite direction for the wild-type. The predominant interactions are mediated by a complement to the distorted backbone conformation as a local kink of residues 679–686 directly compact interacting with the active core cleft of bovine CAPN1. For these specificity profiles of bovine CAST4, the frame of the local kink at the positions must be even more important if other three-dimensional structures (for example, an extended β-strand conformation consisting of proline peptide [
36], which arise from nucleophilic attack by the thiol group of Cys115, preferably located Leu679 and Ile686 residues closer to the Cys115 residue, where it can easily undergo local folding prior to cleavage in a substrate-like manner. The binding mode of residues 679–686 points away from the enzyme active site and thus avoids cleavage that is apparent from the inhibitor capabilities (as shown in
Figure 1).
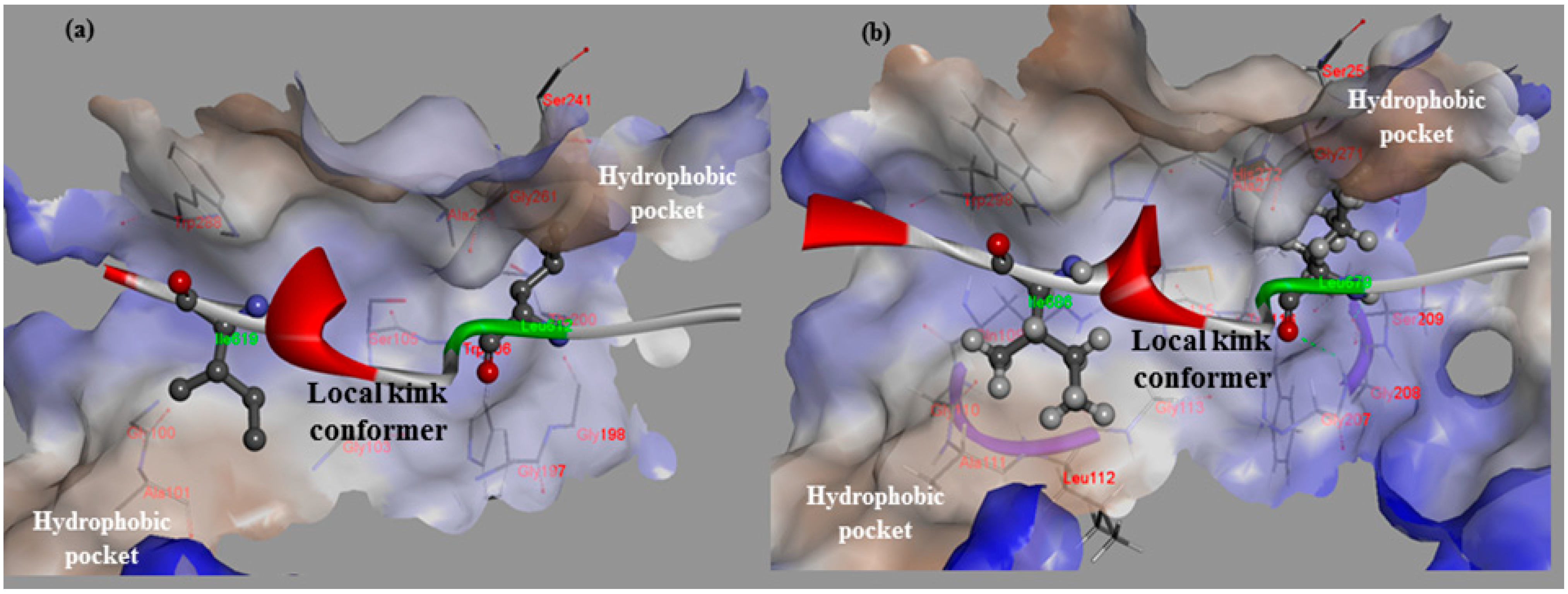
Figure 1.
The hydrophobic interaction surfaces of the binding-site residues near the location at which the local kink conformer (residues 611–624 of rat CAST4 and 678–691 of bovine CAST4) binds. (a) Close-up views of the residues (Leu612 and Ile619) of CAST4 binding at the key contacts of rat CAPN2; and (b) the corresponding residues (Leu679 and Ile686) bound to the active site of bovine CAPN1. Both the subdomains B of CAST4 assume a similar backbone conformation for the distorted local kink. These local kink conformers show the most similar patterns of hydrophobic contact distribution across the CAPN subgroups. Hydrogen bonds are represented by green dashed lines.
Figure 1.
The hydrophobic interaction surfaces of the binding-site residues near the location at which the local kink conformer (residues 611–624 of rat CAST4 and 678–691 of bovine CAST4) binds. (a) Close-up views of the residues (Leu612 and Ile619) of CAST4 binding at the key contacts of rat CAPN2; and (b) the corresponding residues (Leu679 and Ile686) bound to the active site of bovine CAPN1. Both the subdomains B of CAST4 assume a similar backbone conformation for the distorted local kink. These local kink conformers show the most similar patterns of hydrophobic contact distribution across the CAPN subgroups. Hydrogen bonds are represented by green dashed lines.
Figure 2.
The inhibitor specificity of the residue preferences observed for each position in subdomain B of bovine CAST4. The Leu679 mutant shows preferences for the residues Arg, Phe, Leu, and Tyr (had stabilizing effects), whereas the residues Tyr, Trp, Phe, and Arg of the Tyr690 mutant had natural effects of the almost apparent wild-type complex (from most to least stabilizing). Those mutants show distinct preferences for residues bearing hydrophobic side chains (rather than hydrophobic ones) indicating specificity for both positions. In the single-point mutation study, the mutant conformer contributes considerably to the difference in mutation binding energy from the wild-type complexes. The energy difference of each mutation on binding affinity is the difference between the binding free energy in mutated and wild-type proteins, which are predicted to cause destabilizing effects such that mutation energies greater than 0.5 kcal/mol are designated as destabilizing.
Figure 2.
The inhibitor specificity of the residue preferences observed for each position in subdomain B of bovine CAST4. The Leu679 mutant shows preferences for the residues Arg, Phe, Leu, and Tyr (had stabilizing effects), whereas the residues Tyr, Trp, Phe, and Arg of the Tyr690 mutant had natural effects of the almost apparent wild-type complex (from most to least stabilizing). Those mutants show distinct preferences for residues bearing hydrophobic side chains (rather than hydrophobic ones) indicating specificity for both positions. In the single-point mutation study, the mutant conformer contributes considerably to the difference in mutation binding energy from the wild-type complexes. The energy difference of each mutation on binding affinity is the difference between the binding free energy in mutated and wild-type proteins, which are predicted to cause destabilizing effects such that mutation energies greater than 0.5 kcal/mol are designated as destabilizing.
On the other side of the binding mode, the Thr685 residue does not show stronger interaction with specific residues Gly271 and Trp298 than the main chain interactions between Gly680 and Gly271 residues in the bovine CAPN1/CAST4 complex owing to a relatively disordered and steric orientation to the rear of the protruding local kink. This was well documented by the alanine mutation effect when the Thr685Ala mutant was introduced by hydrophobic substitution from the polar group in its side chain; the mutation newly occurred due to hydrophobic contact with both Val269 and Trp298, in contrast to the Gly680Ala mutant, which was designed to abolish the electrostatic interaction with Gly113 and Cys115 of CAPN1. The entrance into the opposed chemical property of the variants (the Thr685 and Gly680 mutants to alanine) appeared that the forehand variant replenished for the backbone’s entropic penalty into less destabilizing effect (0.80 kcal/mol) than latter variant was most significantly reducing a relative of backbone flexibility (3.96 kcal/mol).
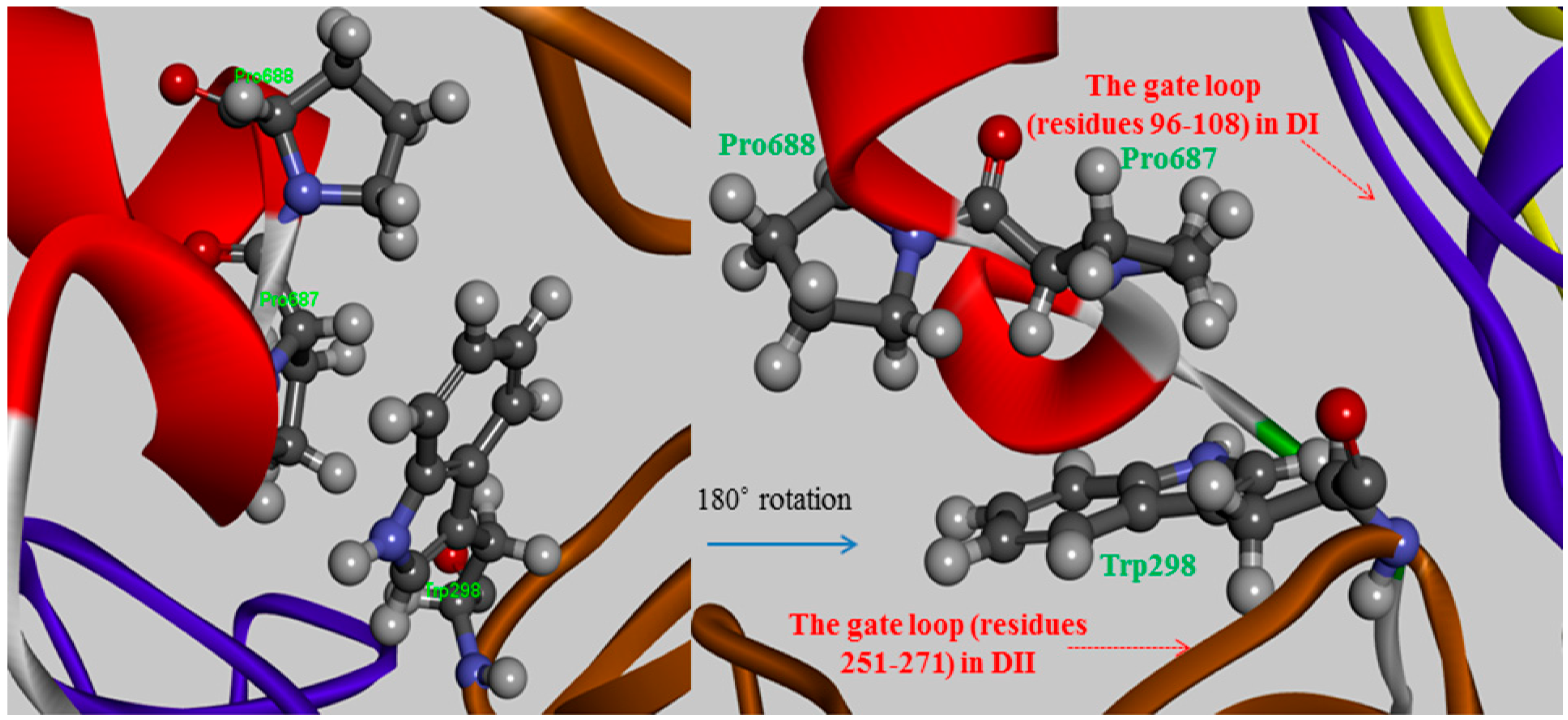
The Pro687 and Pro688 mutants demonstrated that the π-π stacking interactions with the Trp298 residue play major roles in stabilizing their substructure and maximizing its binding affinity, the interaction strength of which depends on their relative positions. The Pro687mutant not settle down than the neighboring Pro688 mutant as a difference of greater than 3-fold magnitude as 2.12 kcal/mol and 0.66 kcal/mol for their mutants respectively. In comparing the two proline variants, the degree of destabilization effects between them could possibly originate from whether a parallel or diagonal π-π stacking interacts with the indole ring of the Trp298 residue via stacking conformation and stabilization effect for their wild-type. The side chain of Pro687 stacks in a coplanar conformation as a parallel π-stacking with respect to the indole group of Trp298, and it has a generate effect than that of the Pro688 mutant (
Figure 3). Though these π-π stacking interactions are physically remote from the Ca
2+-binding-induced to the active conformer, the alanine scanning effects at residues 687–688 do not account for this coplanar stacking into destabilizing the dipole moment of the Trp298 residue, or any non-polar contacts observed in the absence of those π-π stacking interactions. Therefore, if either of the relevant proline to alanine substitutions made no direct contact with the Trp298 residue that prevents access of the inhibitor to the active center with direct blockage of the catalytic residues, the alternation effects might interpose to some extent an additive hindrance to the mutated complex. Two proline residues (687–688) have a restrained backbone conformation, which likely contributes to maintaining an unstructured conformer of the inhibitor protein. These residues also served specificity profile for the positions is not cleaved unlike the substrate of the enzyme. The observed overall mutation effect using virtual alanine scanning for the conserved TIPP peptide the mediating the inhibitory activity of CAST4 showed good agreement with the results of earlier studies by Betts
et.al. [
37]. One major structural attribute in the TIPP sequence is likely to bind inhibitor, that is, the backbone disorder of the peptide after all.
Figure 3.
The π-π stacking interactions between two proline residues (Pro687 and Pro688) of CAST4 and a key tryptophan residue, Trp298 of CAPN1 from bovine in which Trp298 moved to tuck into a hydrophobic patch formed by the Ca2+-binding induced rearrangement of the gating loops (residues 96–108 in DI and 251–271 in DII).
Figure 3.
The π-π stacking interactions between two proline residues (Pro687 and Pro688) of CAST4 and a key tryptophan residue, Trp298 of CAPN1 from bovine in which Trp298 moved to tuck into a hydrophobic patch formed by the Ca2+-binding induced rearrangement of the gating loops (residues 96–108 in DI and 251–271 in DII).
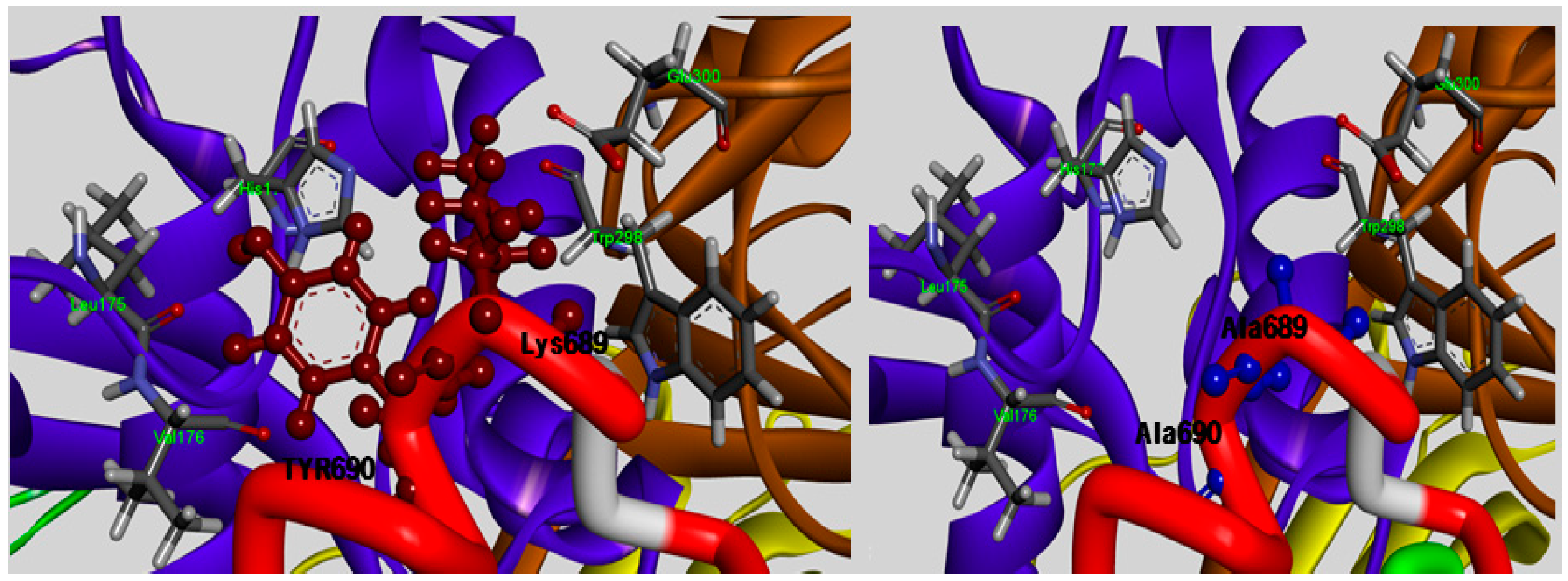
The TLPPKYK sequence in CAST1 and TIPPDYR sequence in CAST3 can be found in the N-terminal of the conserved TIPPXYR motif, but they are not detected in CAST2 corresponding to the TIPPKYQ inhibitory motif within the subdomain B of bovine CAST4. The TIPP peptide (residues 685–688 of the bovine CAST4) resulted in no obvious difference between its inhibitory profile and those of other mammal CAST but not the KYQ peptide (residues 689–691) located in the two turn helix conserved across the species. The position of the KYQ peptide-bond makes its lining out of the activated pocket cleft, whereby it may indirectly affect CAST inhibitor activity through interactions with the flexible regions on both sides of the entrance of the protease core of CAPN1. The comparison of the binding affinity of both Lys689 and Tyr690 mutations to the wild-type, showed very interesting results in this study. The substitution of Tyr690 with alanine showed more than a 1.7-fold decrease from 2.29 to 3.38 kcal/mol in the CAPN1 binding, as shown in
Figure 2, relative to the Lys689 modification (1.62 kcal/mol); these wild-type residues are bound to the enzyme surface in each position that corresponds to the localization of the exposed reactive site loop in the domains DII for Lys689 and DI for Tyr690 residue, and their mode of interaction with the protease binding loop is substantially different (
Figure 4).
Figure 4.
Detailed view of the interaction sites of the Lys689 and Tyr690 residues of subdomain B, which are in contact with the entryway of the protease core DI and DII (shown in ribbon represented in blue and brown, respectively) and were mutated to alanine to reduce the size of its side-chain, rendering CAST4 less space-filling mode from the active site.
Figure 4.
Detailed view of the interaction sites of the Lys689 and Tyr690 residues of subdomain B, which are in contact with the entryway of the protease core DI and DII (shown in ribbon represented in blue and brown, respectively) and were mutated to alanine to reduce the size of its side-chain, rendering CAST4 less space-filling mode from the active site.
The Lys689 mutant causes its side chain to hold more rigidly in the hydrophilic contacts to considerably decrease electrostatic interactions with the polar loop region (Cys108, Gln109, His179, Trp298 and Glu300 residues) in DII, in particular to the primary amino group with a positively charged in Lys689 no longer enhances the electrostatic contact with side chain of Glu300 as a result of the replacement of the methyl group. More surprising was that the strongest stacking between Tyr690 and His179 in DI occurred in the presence of Trp298, an apparent consequence of the large surface area of their large side chain, and the dipole moments were devoid of change with alanine at the position. The absence of this staking in the Tyr690 mutant may alter a set of direct and indirect contacts that affect the positioning and orientation of the π-π stacking done by Trp298, which results in acting for the statically blocking access to the binding loop residue, and also electrostatic properties of both the His179 and Tyr690 residues. In contrast, the mutation of Tyr690, in which a methyl group was inserted between Leu175and Val176, the shielded the hydrophobic residues from the aqueous solvent but was insufficient alone. In particular, large amino acids, such as tryptophan or phenylalanine, at the position should contribute greatly to preference by CAPN1 interaction with the electron-delocalized π-system showed a relative preference in
Figure 2. The Tyr690 residue, which shows more than twice the inhibition of Pro687 residue, implies other preferences to an affinity of CAPN for the flexible binding loop in DI, especially on the C-terminal side. The Tyr690 mutant resulted in a dramatic loss of binding affinity at the position. This suggests that simultaneous π-π stacking interactions, in the presence of Trp298 residue with active site assembly seem particularly important for the inactivation of bovine CAPN1 by CAST4. The data in
Figure 2 suggests that the order of suitable fitting possesses a variety of key features, such as three-dimensional structure, and the physical and chemical properties within the binding pocket revealed some preferences at the 679 and 690 positions. The mutant of the same residue, arginine, which when compared with the mutant complexes from its wild-type has an unequal response to non-polar binding sites. Thus, it is important to characterize their similarities and differences in positions; there is a far greater stabilizing effect for the 679 (−0.9 kcal/mol in the Leu679Arg variant) than 690 (0.0 kcal/mol, without the mutation effect of the Tyr690Arg variant) position based on the free energy difference of the binding of two molecular partners due to single-point mutation of selected residues. The difference in the stabilizing effect for the arginine mutants (Leu679Arg, Tyr690Arg) according to their locations has been made from the backbone conformer. This appears to be more important in defining the preference profile within the subdomain B of bovine CAST4. Inhibition could occur at the positions more or less independently of the specific residues presently bound to the subsites as it fulfills the fundamental factor of being a distorted structure, whereas the scope of inhibition might be less affected by the primary residues. Though both structures are required to confirm the mode of action with the complex, this should provide interaction contacts to stabilize the bound form of CAPN1 as a crucial printing improving inhibitory activity. Notably, the tendency of binding preferences at those positions (the 679 and 690 positions) of CAPN1 appear to be more important in defining the most potent inhibitors of the enzyme share Arg-Trp moieties. This was also characterized by Cuerrier
et al. [
27]. When compared, even if two positions are not responsive to the P3 and P4 positions in the peptidomimetic inhibitors, if sufficient features similarly are observed there is some difference in the relative preferences toward CAPN1, possibly explained by the stabilizing contributions of the other domains (DIII, DIV of CAPN1, and subdomain A of CAST4) of the complex in our model system.
A further interesting point is that Gln691 is only bovine-species-specific and it was not preserved across the subdomain B of each of the four repeating domains of the protein CAST (lysine in CAST1, arginine in the CAST3 and glutamine in the CAST4 from bovine). However, the hydrophilic residue should be at the position at which it could possibly result in the adaptation of alanine, which would otherwise have a poor fit. The position of the Gln691 residue is not subject to direct contact with the matching domain DI of the CAPN1, but the aqueous environment where the mutation renders it apparent as a lesser extent of decreasing binding (0.02 kcal/mol) at the subsite compared with the wild-type protein including some impact on the specificity of its polar property of the side chain. The bovine CAST4 specificity of both the Lys689 and Gln691 residues towards bovine CAPN1 is a difference of more than ten-orders of a decreased affinity that remains between their mutants, which is highly dependent on these positions. On the other hand, the Tyr690 residue should only be made with its nonspecific-inhibitor caution by the conserved motif of the subdomain B on the C-terminus of the CAST4.
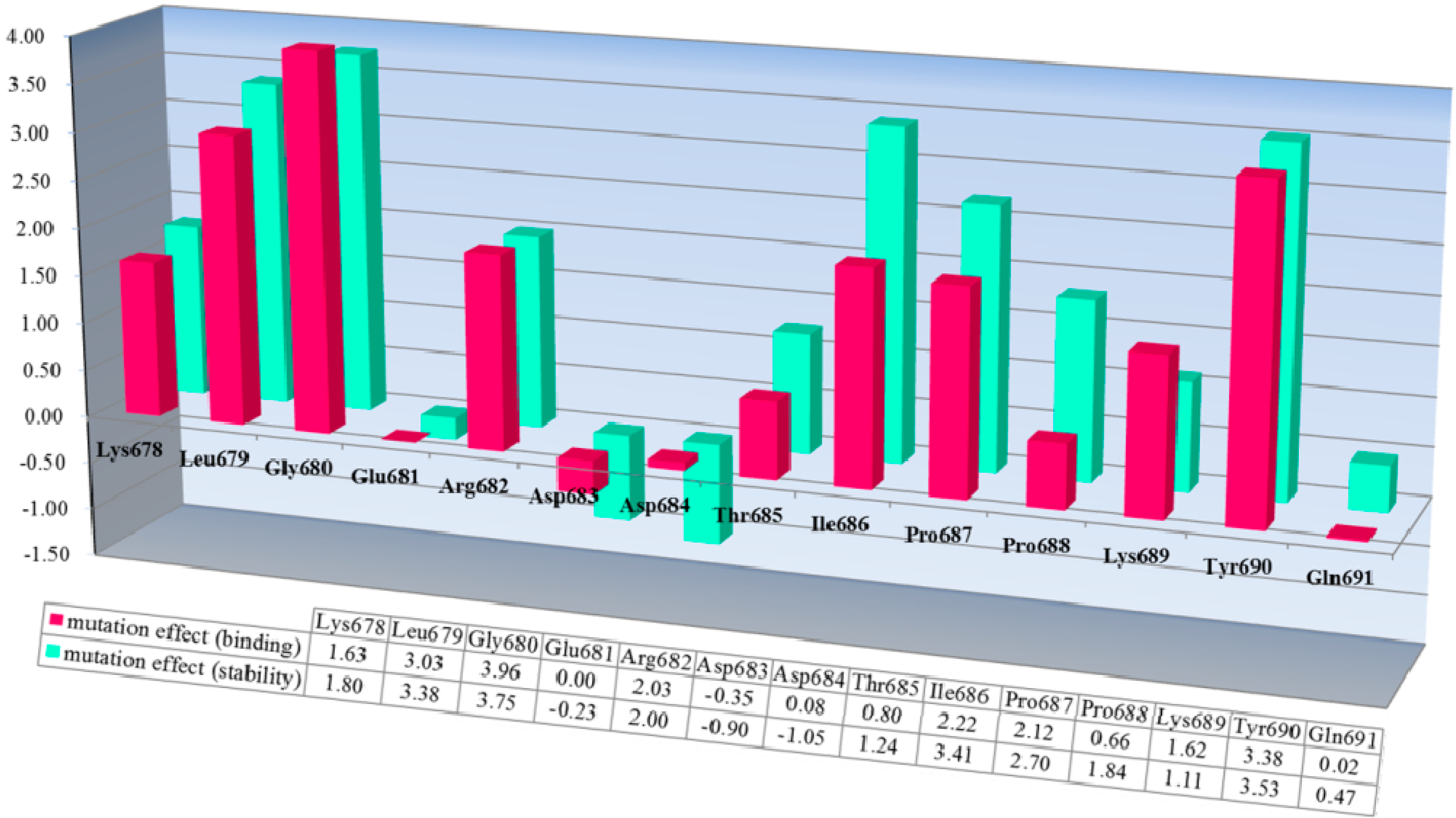
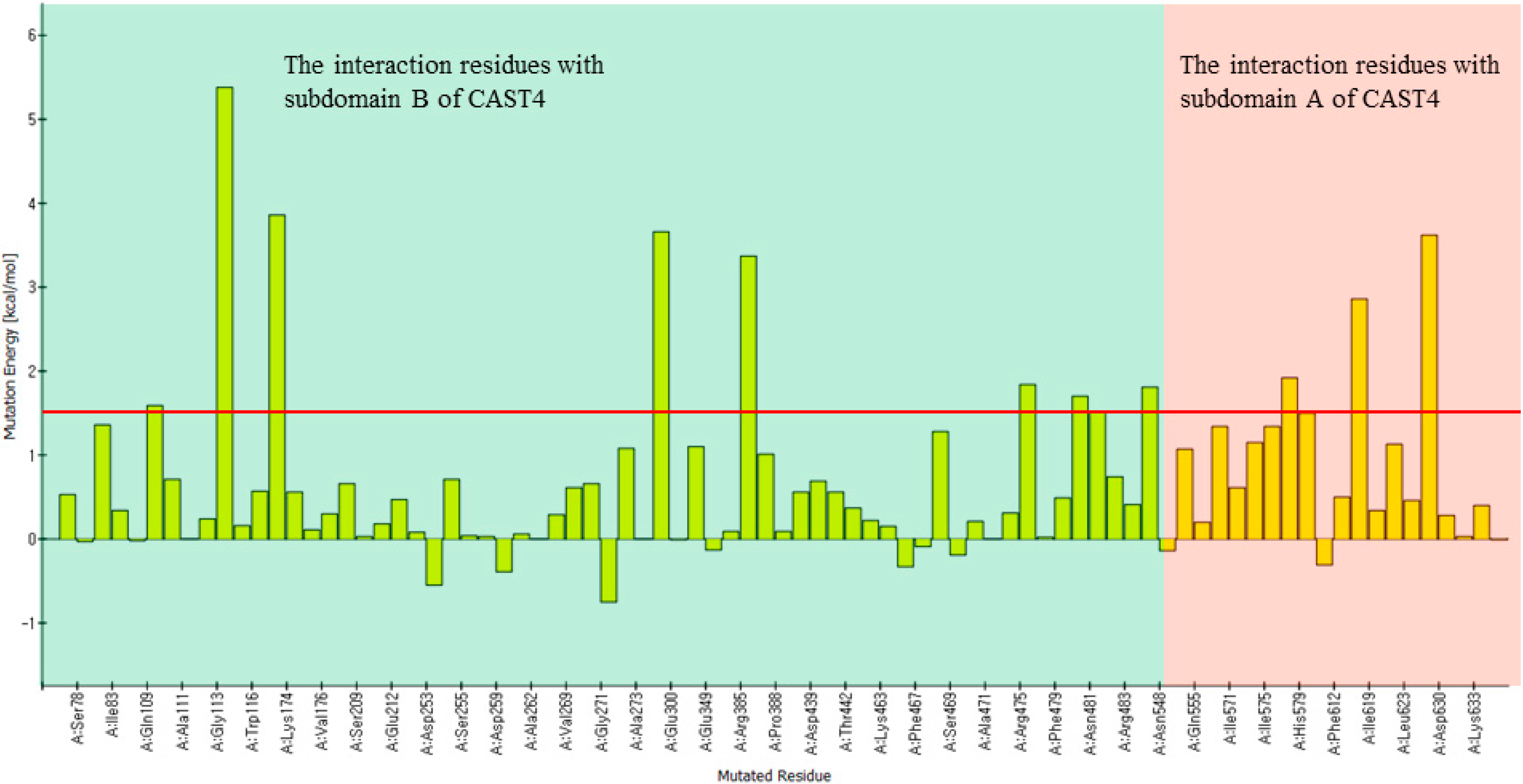
The corresponding mutation effects (calculated energy effects) in terms of the stability of the bovine CAPN1/CAST4 complex structure (a green bar chart) and binding affinity (a red bar chart) with the bovine CAST4 are shown in
Figure 5.
Figure 5.
Inhibition profiles of its native inhibitor CAST4. The conserved KLGERDDTIPPKYQ motif in the bovine CAST4 (residues 678–691) was screened using a computational, alanine scanning mutagenesis. The results are shown as the residue preference from a wild-type complex with the corresponding energy effect (kcal/mol) of alanine mutation at the single-point position for the stability (green) and binding affinity (red) of the bovine CAPN1/CAST4 complex. Each bar chart of mutation energy is created to allow for comparing the different effects of mutation, when the mutation effects are defined as “stabilization” (mutation energy less than −0.5 kcal/mol), “natural” (mutation energy between −0.5 and 0.5 kcal/mol), and “destabilizing” (mutation energy greater than 0.5 kcal/mol).
Figure 5.
Inhibition profiles of its native inhibitor CAST4. The conserved KLGERDDTIPPKYQ motif in the bovine CAST4 (residues 678–691) was screened using a computational, alanine scanning mutagenesis. The results are shown as the residue preference from a wild-type complex with the corresponding energy effect (kcal/mol) of alanine mutation at the single-point position for the stability (green) and binding affinity (red) of the bovine CAPN1/CAST4 complex. Each bar chart of mutation energy is created to allow for comparing the different effects of mutation, when the mutation effects are defined as “stabilization” (mutation energy less than −0.5 kcal/mol), “natural” (mutation energy between −0.5 and 0.5 kcal/mol), and “destabilizing” (mutation energy greater than 0.5 kcal/mol).
Both mutation effects (stability and binding affinity) displayed analogical inhibitory preference results that generally show almost identical relative CAST4 mutation effects having a correlative tendency (the correlation coefficient is observed by 0.92) toward bovine CAPN1, consistent with their binding site. Interestingly, the differences between the effects of the Asp 683 and 684 mutations to alanine at the positions for stability and binding affinity suggest that the other domains in full-length have a greater impact on the complex stability than binding affinity related to the inhibition specificity. These positions are far enough from the hydrophobic sites that they must be influenced by the local kink construct. Conversely, the general preference for bulk hydrophobic residues, Ile, Leu, and Tyr (at positions 679, 686, and 690) is consistent with fitting into the equivalent large hydrophobic pockets within the protease core of bovine CAPN1, where their inhibitory efficiency is similar to that of the other CAPN2 subgroup. Indeed, the Gly680Ala mutant suggests that this β-turn backbone portion of the inhibitor is highly flexible when complex with the protease core to avoid its steric packing (
Figure 1 and
Figure 5). However, additional inhibitory preferences were observed with the KYQ peptide (residues 689–691) of bovine-specific residues. The unfixed sequence (KYQ in the conserved TIPPXYR motif) influences the selectivity of bovine CAST4 at these positions, but it does not need to be similar to optimized inhibitor candidates, because the leading structure may orient to an extent sufficient to direct the enzyme inhibition independent of whether the primary sequence is adequate for the active site. Consistent with the central roles of the residues at the binding pocket, active site-directed inhibitors [
26,
27,
28,
29,
37] block their susceptibility to CAPN1 similar to the CAST4 preference in the corresponding positions (
Figure 6) throughout the same non-covalent interactions found in the rat, humans and our bovine model. The generating specificity of both active site-directed inhibitors is illustrated in
Figure 6.
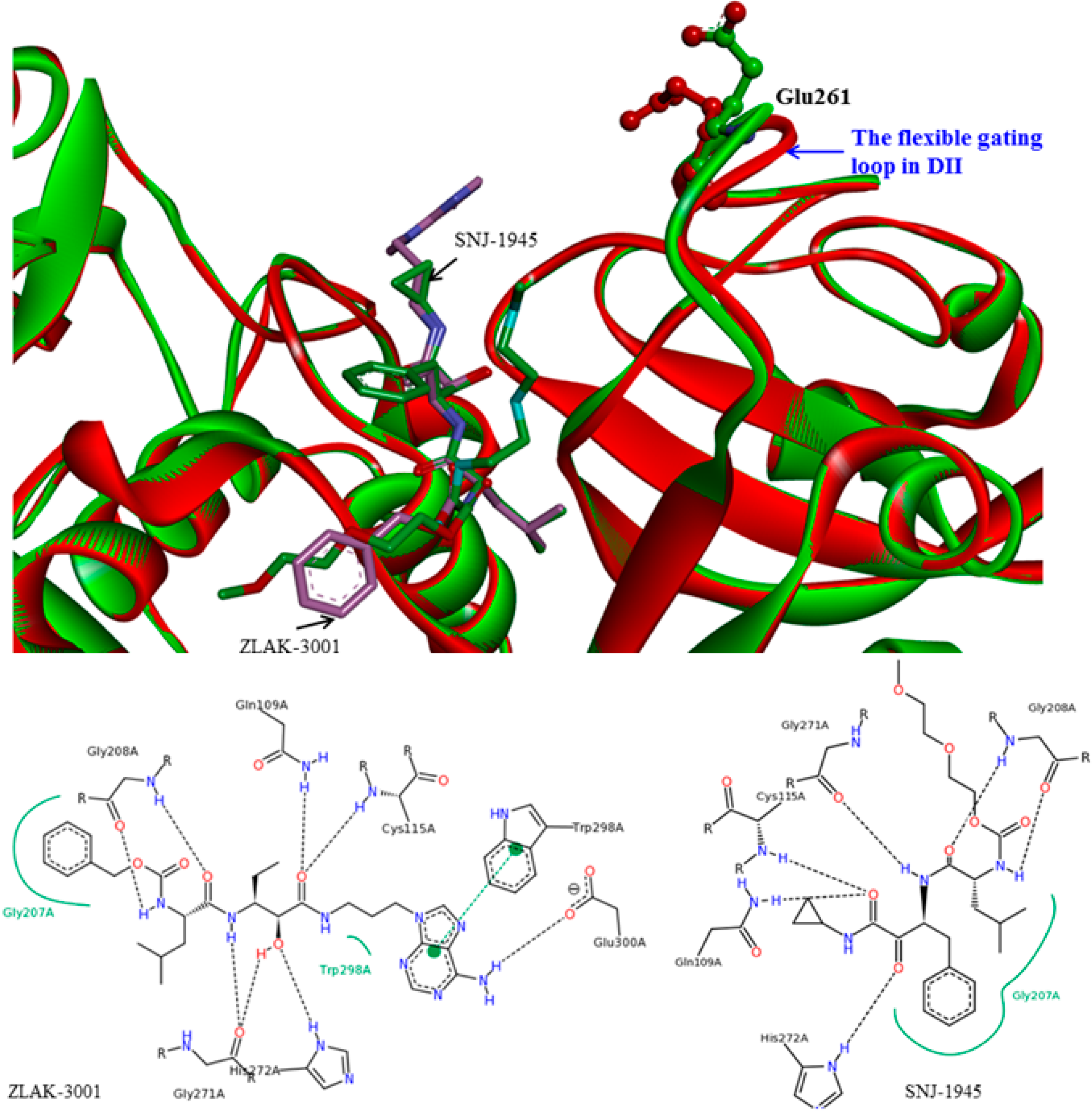
Figure 6.
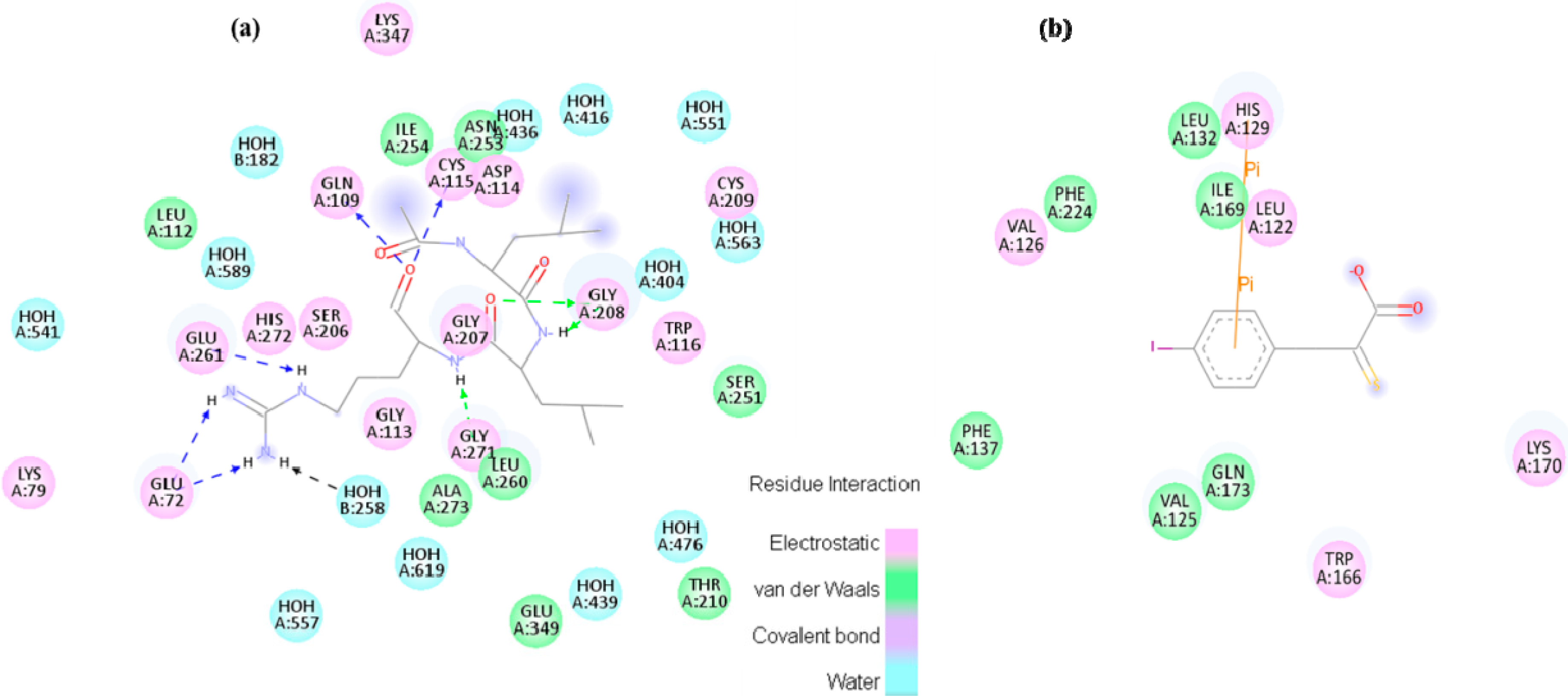
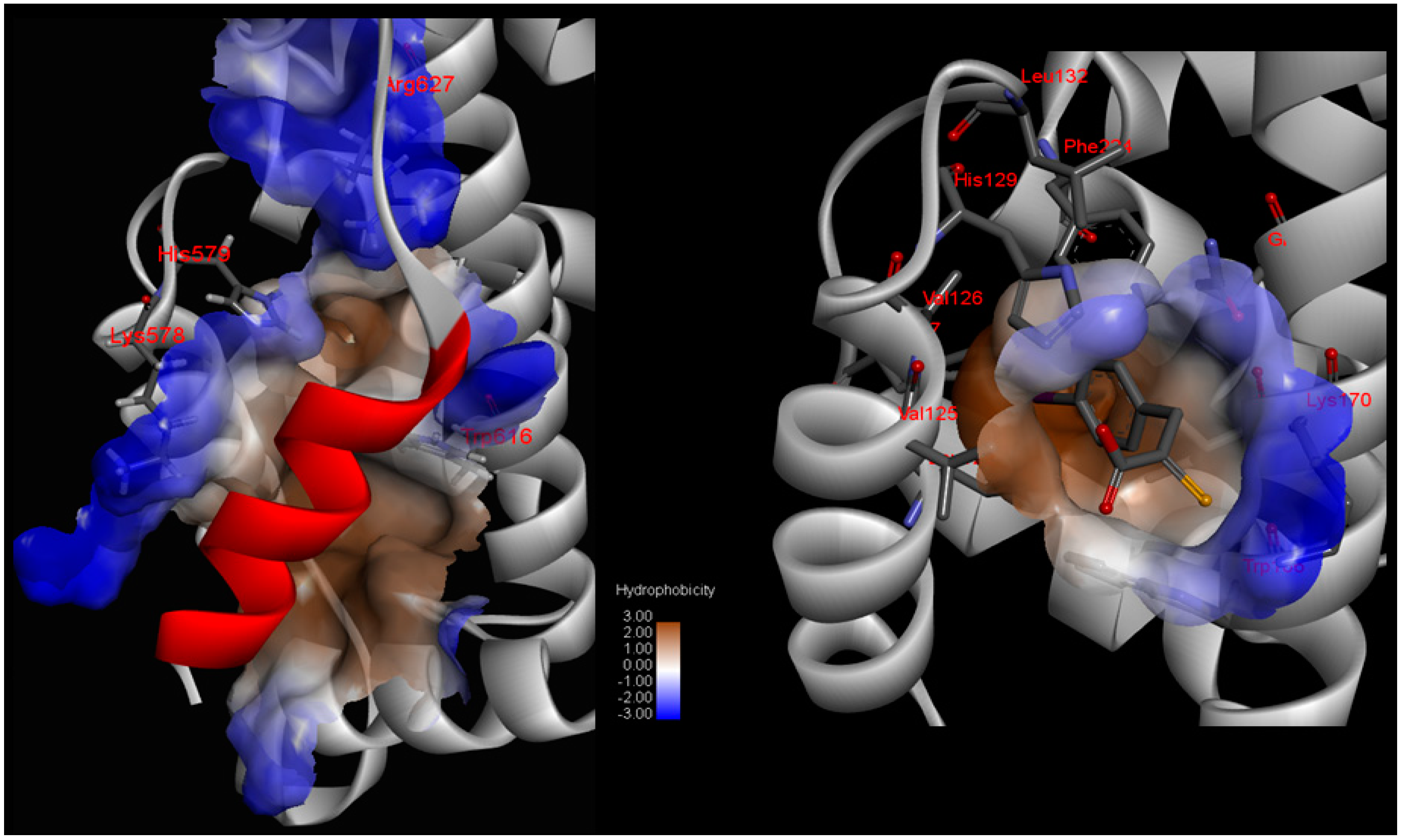
Small molecule inhibitor interaction diagrams for the binding pocket of the mammalian CAPN subunits (PDB code: 1TL9, 1NX3). For both inhibitor-bound structures, inhibitors are shown as sticks accompanied by key residue representation of 2D-CAPN interacting interface. The complex structures reveal the defining their physicochemical properties for CAPN selectivity and specificity: (
a) In the complex structure between rat CAPN1 and leupeptin (1TL9) at the protease core, extensive interactions help stabilize leupeptin at the active site by eight hydrogen bonds with the side-chains of Glu72, Glu261, Gln109, Cys115 residues (a blue dashed arrow) and backbone of Gly208, Gly271 residues (a green dashed arrow) and hydrophobic interactions with Leu260, Ser251, Ala273, Asn253, Ile264 residues. The domain-leupeptin complex overlaps with those of the CAPN/CAST complex structure (PDB code 3DF0) as main chain atoms r.m.s.d of 1.4 Å [
38]; (
b) The interaction site for the crystal complex structure (1NX3) of CAPN domain DVI (of pig) and its inhibitor PD150606 bond in a hydrophobic pocket (Val125, Leu132, Phe137, Ile169, Gln173, Phe224) of DVI where it makes favorable π-π interaction with the side-chain of His129.
Figure 6.
Small molecule inhibitor interaction diagrams for the binding pocket of the mammalian CAPN subunits (PDB code: 1TL9, 1NX3). For both inhibitor-bound structures, inhibitors are shown as sticks accompanied by key residue representation of 2D-CAPN interacting interface. The complex structures reveal the defining their physicochemical properties for CAPN selectivity and specificity: (
a) In the complex structure between rat CAPN1 and leupeptin (1TL9) at the protease core, extensive interactions help stabilize leupeptin at the active site by eight hydrogen bonds with the side-chains of Glu72, Glu261, Gln109, Cys115 residues (a blue dashed arrow) and backbone of Gly208, Gly271 residues (a green dashed arrow) and hydrophobic interactions with Leu260, Ser251, Ala273, Asn253, Ile264 residues. The domain-leupeptin complex overlaps with those of the CAPN/CAST complex structure (PDB code 3DF0) as main chain atoms r.m.s.d of 1.4 Å [
38]; (
b) The interaction site for the crystal complex structure (1NX3) of CAPN domain DVI (of pig) and its inhibitor PD150606 bond in a hydrophobic pocket (Val125, Leu132, Phe137, Ile169, Gln173, Phe224) of DVI where it makes favorable π-π interaction with the side-chain of His129.
![Molecules 19 14316 g006]()
The major interactions, during which H-bonding and hydrophobic interactions between active site residues and the inhibitors serve to stabilize the complex during Ca2+-binding and to orient the inhibitor as a common feature, and other marked deviations from the structural characteristic are also observable in both complex structures. In the immediate vicinity of peptidomimetic inhibitors, the secondary structure, on the other hand, behaves exclusively as a major obstacle where CAPN attack occurs since these portions of the inhibitors are highly flexible when competitively bound in the active site to the enzyme substrates. The β-strand conformation (PDB code 1TL9 and 1TL0) is apparent from these specificity profiles for protease recognition as one of the major factors that is not considered when deciding whether to develop reversible or irreversible inhibitors. The conformation accelerates additional H-bonding and an antiparallel β-sheet of both backbones with the key residues (the Gly271, Gly208, Cys115 residues), and then the ability of the peptide to inhibit CAPN is altered. This further makes bovine CAPN1 a target for the computationally aided molecular design of novel active site-directed inhibitors. A potent inhibitor specific for bovine CAPN1 could be designed to scrutinize allosteric or other binding sites such as domain DIV, which when ligated by CAST4 imitation, renders the enzymes unable to achieve their proteolysis function. It must be carefully considered whether the targeting interaction sites would be subject to either autolysis or subunit dissociation or aggregation.
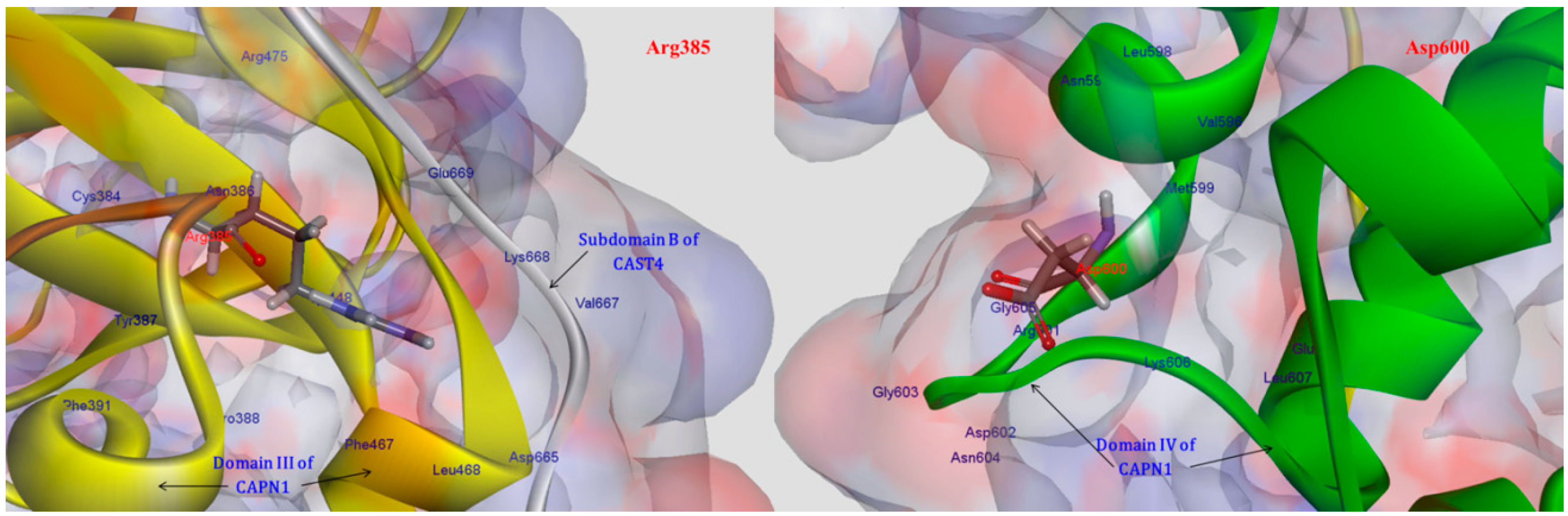
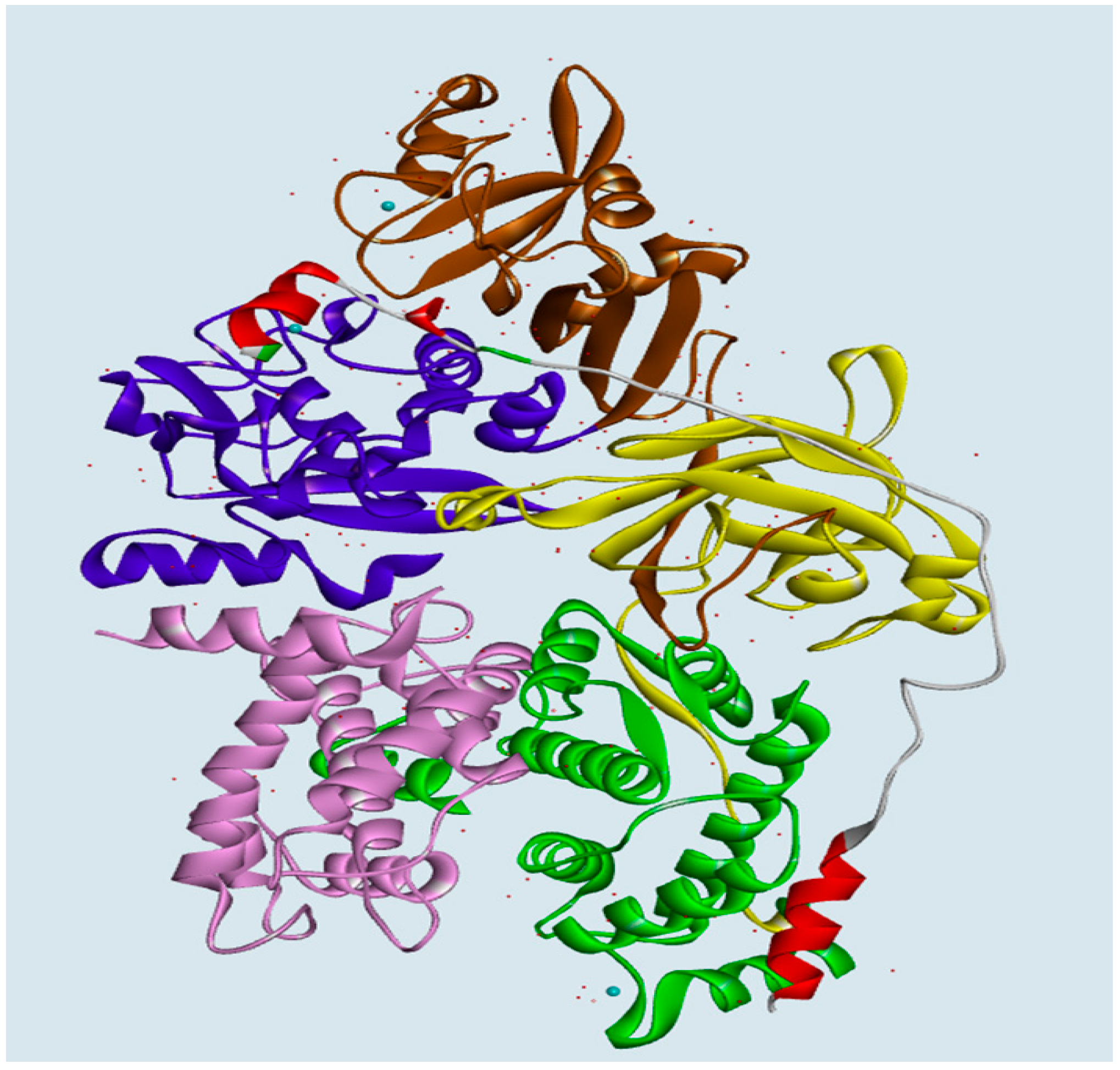
2.1.2. Predicting Mutation Effects on the Two LGMD2A-Accociated Mutations (R385H and D600G in the Corresponding Positions in the Bovine CAPN1)
More importantly, additional tendencies of preferences were observed with the subsite of an exposed bent loop (residues 663–669) on the N-terminus of subdomain B in CAST4, which is adjacent to the interface of the Arg385 residue related to the LGMD-2A mutation’s position (R448H in CAPN3). These tendencies were not predicted resulting in a distinct difference of specificity from previous studies on the inhibitor selectivity of CAPN1 [
34]. We were interested in two LGMD2A-associated mutants, R448H and D705G, which retain the proteolysis activity of either of these CAPN isoforms (CAPN3, CAPN2); however, they significantly affect the stability of the protein itself and are also diseases causing [
8]. In the current study, of known pathogenic mutations in CAPN3, it was possible to predict the effects of two mutants for pathogenic missense mutations (R448H and D705G) in LGMD2A in terms of the bovine CAPN1 inactivation and its complex stability through the CAPN1/CAST4 complex model. We made an assumption based on a sequence homology (more than 45%) that the structure should be similar to CAPN1 and should share biochemical properties, such as Ca
2+-dependent activation (in the nanomolar range of Ca
2+ ions concentration) and maximal activity at natural pH, despite the presence of three exclusive sequence inserts (NS at the N-terminus, IS1 in the domain DII, and IS2 between domain DII and DIII) without small subunits [
7]. Both mutants R448H and D705G in CAPN3 should be projected onto R385H and D600G in the corresponding positions in the bovine CAPN1. The Arg385 residue is situated on a loop of domain DII that makes intramolecular domain contacts (DII/DIII) and intermolecular interfaces (CAPN1/CAST4) on either side of the center of the loop, and it may leaven the assembly and activation of the enzyme. On the contrary, the Asp600 residue is located elsewhere in the EF-hand2 motif (residues 587–620) of domain DIV; it is exposed to solvent in both the absence and presence of Ca
2+ ions and has no interactions with reactive residues of the CAST4 (
Figure 7). Surprisingly, the Asp600Gly mutant adjoins to the dimerized interfaces (domains DIV/DVI) between CAPN1 molecules through the fifth-EF-hand motif such that its stability must be dominated by the heterodimer formed.
One possibility, we considered was that the Arga385His mutant could affect stabilizing, electrostatic interactions (salt-bridge) with the enzyme itself and some binding partner, CAST4, in such a way that it altered the coupling strengths of partners (the residues Asp665, Val667, Lys668, Glu669 on N-terminus of subdomain B) interacting with Arg385 residue in both interaction regions (DII/DIII) and that it could play an important transfer part in Ca
2+-induced activation signaling. When we analyzed the complex model, the residues Asp665 and Glu669 located in the bent loop of CAST4 were considered potential partners for salt-bridge formation according to the strength governed by their positions on the flexible loop. As another factor, both residues of subdomain B are also localized at the external surface of the CAST4 across the domain DIII of CAPN1 as seen in
Figure 7. The mutation of arginine to histidine at the 385 position could decrease electrostatic interactions with the adjacent hydrophilic residues within both the CAPN1 and N-terminus subdomain B of CAST4 originating from its basic property and charge variation. In particular, the internal salt-bridge would finally result in a much greater reduction in the structural stability of the complex (2.70 kcal/mol) than of the enzyme itself (0.20 kcal/mol) in relation to both their wild-types (
Table 1).
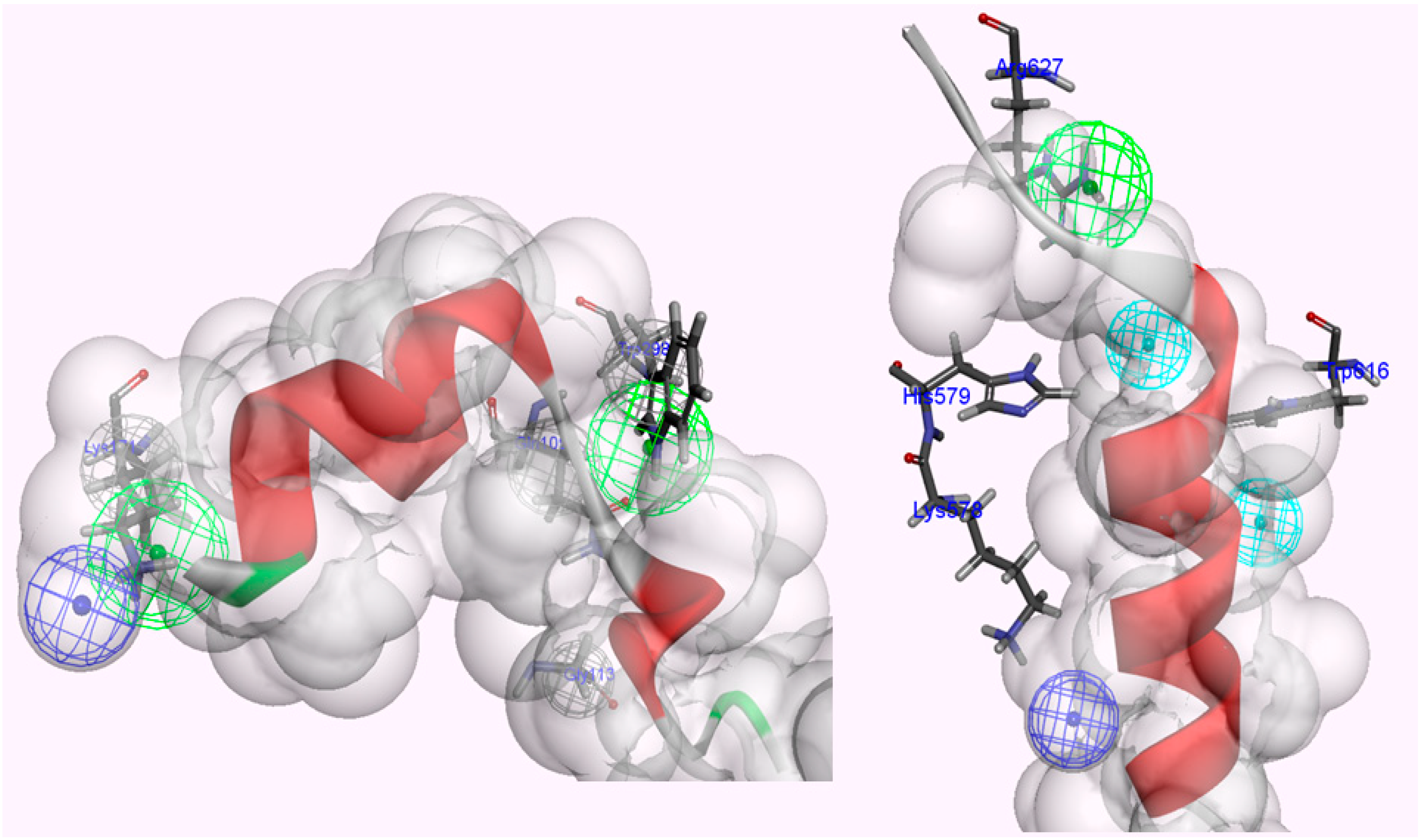
Figure 7.
Interaction residues associated with two LGMD2A-related mutants (Arg385His, Asp600Gly).
Figure 7.
Interaction residues associated with two LGMD2A-related mutants (Arg385His, Asp600Gly).
The Arg385 residue is distant enough from the protease core that it should retain the proteolysis function of the enzyme, but the mutant considerably affects fissure of the CAPN1 structural integrity in the presence of the inhibitor CAST4. The structural stability was much lower in the unbound state than in the bound state, that is, almost 14-times lower. Moreover, the substitution did not directly change the enzyme folding but significantly disturbed the tighter association with subdomain B since, at the scanned position, which is not positioned and oriented at an optimal distance for the salt-bridge-interaction of acidic partners (the residues Asp665 and Glu669 of CAST4), so the wild-type salt-bridge is no longer available as a result of the Arg385His mutation. Correspondingly, the basic loop flexibility allows a small increase in the entropy of the complex, as if the conformer change of the local site caused by the mutation is not sufficient to influence for the Ca2+ion-binding of either the inactive or active enzyme form to any significant degree (mutation energy effect of −0.23 kcal/mol as neutral effect). However, in the case of the CAPN1/CAST4 complex, the reduction of electrostatic interactions (salt-bridge) with the inhibitor was more unstable in the bound state than in the unbound state (with increased binding free energy of 1.85 kcal/mol from that of the wild-type complex as a destabilizing effect) and could let down the binding affinity of CAST4. Thus the variation at the 385 position has a larger impact on the inhibition of CAST4 via the structural stability of the complex than the efficiency of association between them.
In contrast to the Arg385His mutant, it is feasible that the Asp600Gly variation would have a smaller impact on structural stabilization and would behave similarly in the absence or presence of protease inhibitor dominated by its ability to form a heterodimer. The stability of the variant at the 600 position was shown to be greater in its inhibitor complex (−0.99 kcal/mol) than in the enzyme itself (−0.55 kcal/mol) against either wild-type.
Table 1.
Evaluating effect of single-point mutations on stability and binding affinity.
Table 1.
Evaluating effect of single-point mutations on stability and binding affinity.
| | CAPN1 Arg385His | CAPN1 Asp600Gly | CAST4 Leu643Ala | CAST4 Leu646Ala | CAST4 Leu650Ala |
|---|
| † Mutation Energy (the enzyme itself stability) | 0.20 kcal/mol | −0.55 kcal/mol | - | - | - |
| * Stability effect | Neutral | Stabilizing | | | |
| † Mutation Energy (the complex stability) | 2.70 kcal/mol | −0.99 kcal/mol | 1.70 kcal/mol | 1.96 kcal/mol | 2.09 kcal/mol |
| * Stability effect | Destabilizing | Stabilizing | Destabilizing | Destabilizing | Destabilizing |
| ‡ Mutation Energy (Ca2+-binding) | −0.23 kcal/mol | −0.99 kcal/mol | - | - | - |
| * Ca2+-binding effect | Neutral | Stabilizing | - | - | - |
| ‡ Mutation Energy (CAST4-binding) | 1.85 kcal/mol | −0.43 kcal/mol | 2.39 kcal/mol | 2.88 kcal/mol | 2.50 kcal/mol |
| * CAST4-binding effect | Destabilizing | Neutral | Destabilizing | Destabilizing | Destabilizing |
The stability gain of the Asp600Gly mutant that was observed on heterodimerization with engaging the major hydrophobic interface that holds together through the pairing of their EF-hands motif of between CAPN1 molecules and not only that bound in a direction opposite to an amphipathic α-helices of the subdomain A of its inhibitor CAST4. The structural stability would be maintained by the CAPN1-induced helical anchors of subdomain A. After mutation, the structural stability became too high for effective protection through the hydrophobic residue’s packing form in an aqueous environment. Thus, the mutation energy for CAST4 binding was lower (−0.43 kcal/mol) than that for Ca
2+-binding (0.44 kcal/mol) for more increasing binding affinity with the former Arg385His. However, this would have a relatively small impact on the mutation energies, which were in the neutral range from −0.5 to 0.5 kcal/mol, which mean that the mutation had a very small effect on enzyme function. The variation of any acidic residue at the 600 position is required for the Ca
2+-chelating residues. As summarized in
Table 1, two variants (Areg385His, Asp600Gly) showed differences between the folding free energy of mutated structures and binding free energy with molecular partners (Ca
2+ ions and CAST4) in their mutated complex structures. The variants allowed CAPN1 to hold its active conformation within the Ca
2+-induced structure, but the preference for inhibition of CAST4 was different for each mutant; the Arg385His variation significantly decreased its binding affinity to CAST4 due to its complex’s instability.
On the other hand, the Asp600Gly variant was less efficient in forming the complex than a heterodimer with the other CAPN; it had a stabilizing effect on the complex but did not affect cohesion with the inhibitor. In addition, the Arg385His mutant contributed to revealing the important interaction sites (the bent loop of residues 663–669) on the N-terminus of subdomain B of CAST4 for enzyme regulation. Furthermore, the importance of the bent loop for the CAPN1-CAST4 binding was previously unknown.
2.1.3. Predicting Mutation Effects on the LDDALDQLSDSLGQ Motif in Subdomain A of Bovine CAST4
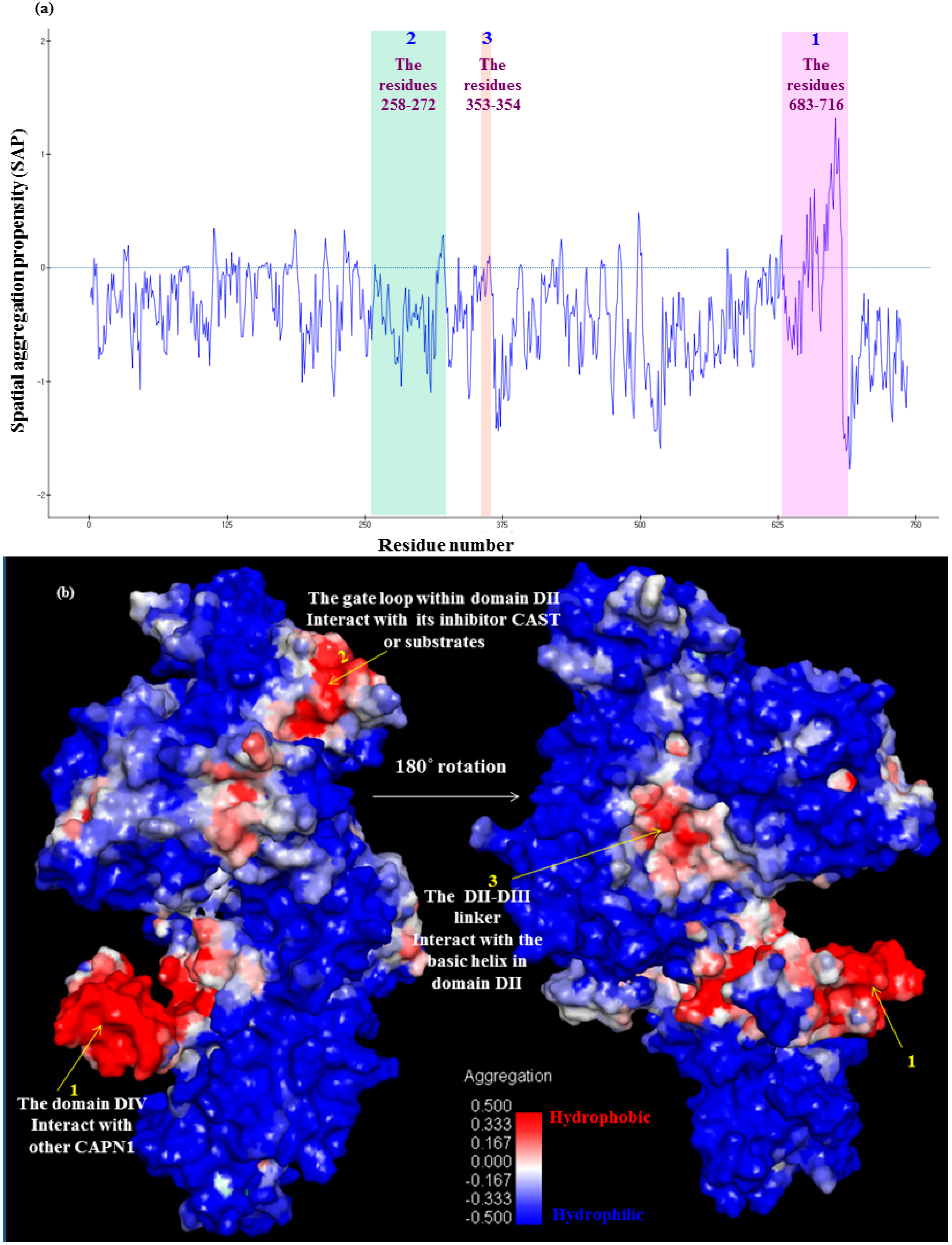
Subdomain A of CAST4 bound to a hydrophobic pocket formed by EF-hands one (the residues 543–578) and two (the residues 587–620) in the domain DIV filled by the inhibitor. Peculiarly, the conserved hydrophobic Leu residues 643, 646, and 650 inside an amphipathic helix (the residues 638–650) of subdomain A were buried deep in the allowed binding region, which helped to form strong hydrophobic interactions with the domain DIV. Several bulky residues (Leu552, Leu556, Phe612, Trp616, and Leu623 of DIV) in the hydrophobic pocket closely embrace the Leu residues to form favorable contacts with between side chains. The breadth of the hydrophobic groove seems like an opened baseball glove, within which the inhibitor subdomain A is somewhat flexible. The conservative interaction residues, both of them in the bovine complex, were further condensed as shown in
Figure 8 and
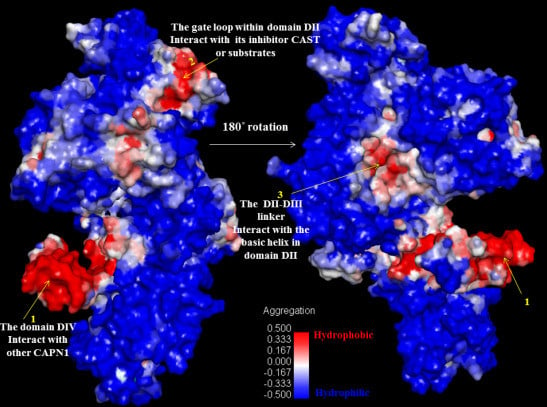
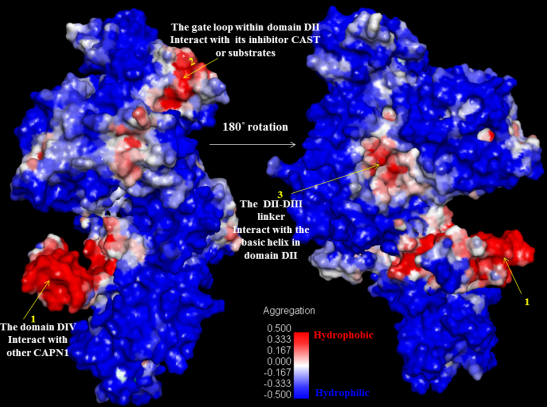
Figure 9. Even a single site mutation in either region could have an effect on the affinity of either region alone. This can be seen by estimating the SAP (Spatial Aggregation Propensity) whereby inhibition can occur as a complement to the subdomain A of CAST4, directly interacting with the key trace residues (colored blue and red) on the exposed hydrophobic region as shown in
Figure 9. While the Leu residues (Leu643, Leu646, and Leu650 in the residues 639–652 of LDDALDQLSDSLGQ motif of subdomain A) have always been thought to be embedded in the inhibition region, virtual alanine scanning (mutated to be less hydrophobic) was performed of either a single or multiple displacement to elicit an induced effect (for the complex’s stability and binding affinity) caused by the positional changes along with the hydrophobic interaction. The effect of each mutation of the three Leu residues (Leu643, Leu646, and Leu650) individually on alanine had varying results (
Table 1). The stability of the complex was greatly reduced, even further than the binding capacity to the enzyme in each variation, illustrating the sensitivity of this region to changes in the hydrophobic interactions. These mutants were expected to display a reducing flexibility of the side chain and van der Waals contacts with the companion residues (Leu552, Leu556, Phe612, Trp616, and Leu623 of DIV) in the wider bounding pocket, thereby showing weaker preferences for the positions. The effects of these mutations on the hydrophobic pocket of the enzyme could be directly predicted by multiple mutations of a set of the Leu residues for the complex’s stability, when double mutations are generated from the retained single mutations and then the triple mutations are generated from both the former mutations (the retained single mutations and double mutations). Their multiple displacements to alanine show a sharp stability drop in a mutually dependent manner to approximately an order of magnitude as a high order of repeated variations (single Leu646Ala to double Leu650Alal and Leu646Ala to triple Leu643Ala, Leu646Ala and Leu650Ala mutants leads to an increase in the folding free energy from the wild-type complex with values of 2.88, 5.21, and 7.11 kcal/mol respectively. The data is not shown in
Table 1) with a negative effect on the inhibition. The increase in the structural instability of these mutant complexes contributes to subdomain A binding to the hydrophobic pocket of the CAPN1 in the stabilization of the wild-type complex structure, resulting in stronger inhibition than that seen with the mutant complexes.
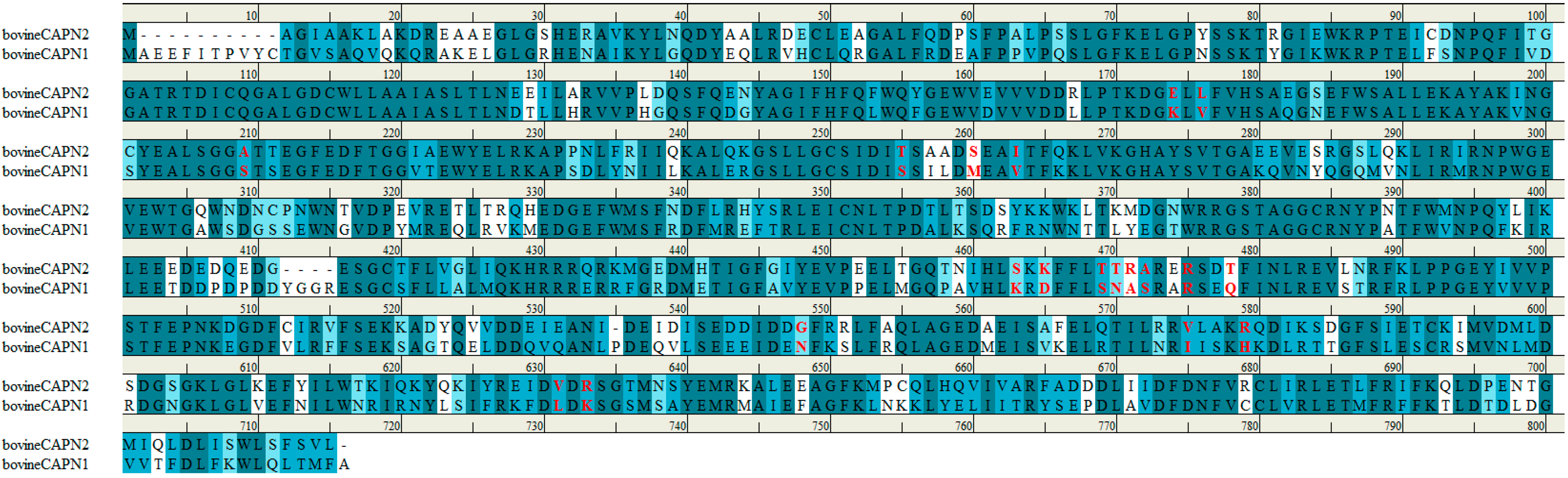
Figure 8.
Sequence alignment of bovine CAPN1 and CAPN2 large catalytic subunit. The accession numbers for these sequences are NP_776684.1 (NCBI reference sequence) and AAI34527.1 (GenBank), respectively. Bovine CAPN1 has 81.5% sequence homology to the subunit of CAPN2. The trace residues that made up the class-specificity of CAPN in the interaction sites with subdomains A and B of the CAST4 group are indicated in red. All positions are described in terms of the bovine CAPN1 amino acid sequence. For comparison, two bovine CAPN isoforms had similar specificity toward the enzyme CAST4.
Figure 8.
Sequence alignment of bovine CAPN1 and CAPN2 large catalytic subunit. The accession numbers for these sequences are NP_776684.1 (NCBI reference sequence) and AAI34527.1 (GenBank), respectively. Bovine CAPN1 has 81.5% sequence homology to the subunit of CAPN2. The trace residues that made up the class-specificity of CAPN in the interaction sites with subdomains A and B of the CAST4 group are indicated in red. All positions are described in terms of the bovine CAPN1 amino acid sequence. For comparison, two bovine CAPN isoforms had similar specificity toward the enzyme CAST4.
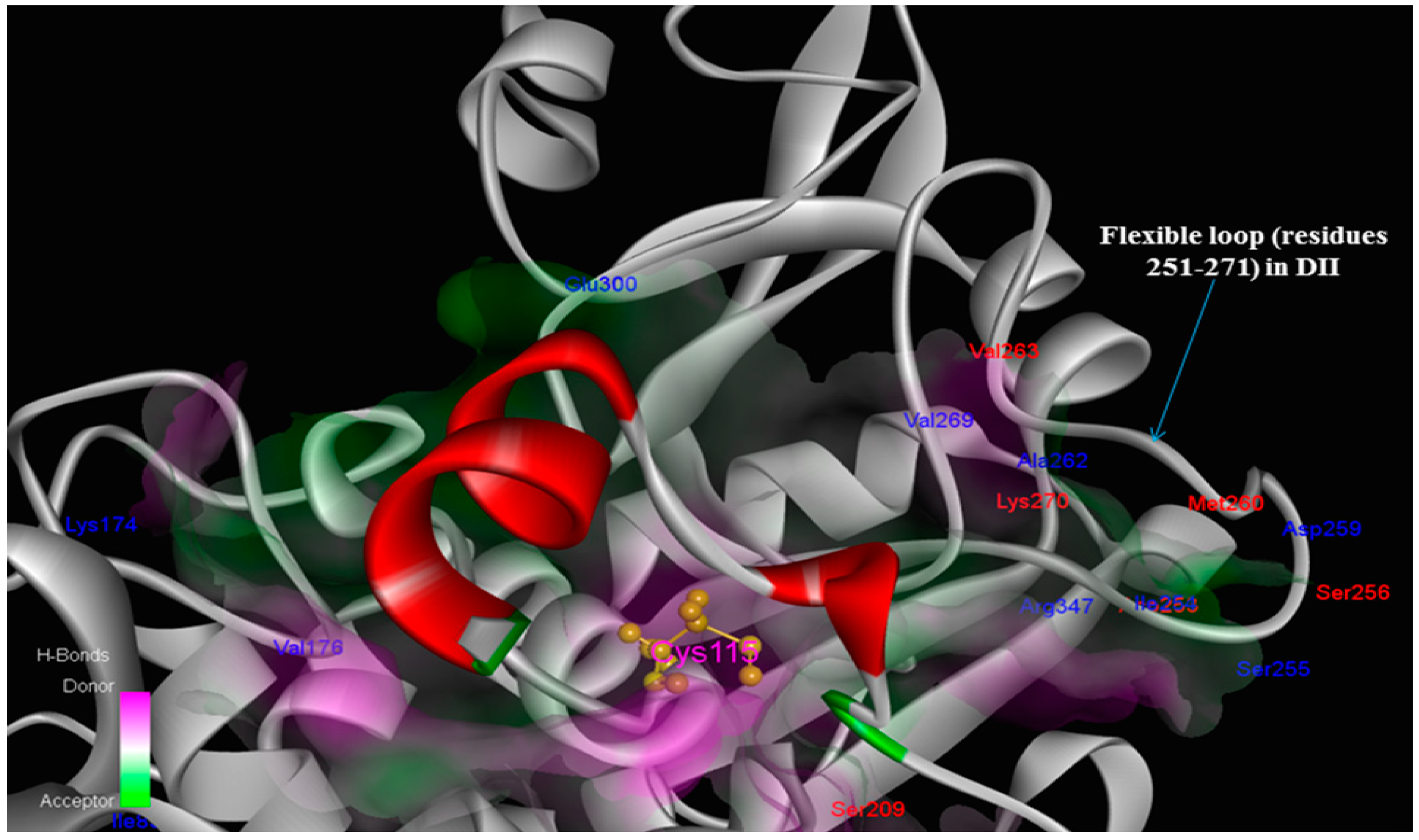
Figure 9.
Interaction sites within the protease core by residue character: class-specific (blue; Ile83, Lys174, Val176, Ile254, Ser255, Asp259, Ala262, Val269, Glu300, Arg347) and bovine only species-specific (red; Ser209, Asp253, Ser256, Met260, Val263, Lys270) trace residues among key interaction contacts made the CAPN1/CAST4 complex. Notably, not adjacent regions of the catalytic Cys115 residue, the positions of trace residues from bovine CAPN1 are location-focused within the open conformation of the flexible loop of domain DII (residues 251–271) which gates active site, consistent with their binding site of conserved KLGERDDTIPPXYX motif of CAST4 subdomain B having similar inhibitory preferences in the active site cleft, but will likely not be identical against two CAPN isoforms (CAPN1 and CAPN2) from the bovine.
Figure 9.
Interaction sites within the protease core by residue character: class-specific (blue; Ile83, Lys174, Val176, Ile254, Ser255, Asp259, Ala262, Val269, Glu300, Arg347) and bovine only species-specific (red; Ser209, Asp253, Ser256, Met260, Val263, Lys270) trace residues among key interaction contacts made the CAPN1/CAST4 complex. Notably, not adjacent regions of the catalytic Cys115 residue, the positions of trace residues from bovine CAPN1 are location-focused within the open conformation of the flexible loop of domain DII (residues 251–271) which gates active site, consistent with their binding site of conserved KLGERDDTIPPXYX motif of CAST4 subdomain B having similar inhibitory preferences in the active site cleft, but will likely not be identical against two CAPN isoforms (CAPN1 and CAPN2) from the bovine.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







