2.1. HPLC-DAD and UPLC-ESI(+)-HRMS/MS Profiling of Micromelum falcatum Extracts
In order to selectively detect coumarin derivatives, the profiling of different
M. falcatum extracts was performed using HPLC-DAD and UPLC-ESI(+)-HRMS/MS instruments. Two different extraction methods were followed and the obtained extracts were compared based on chromatographic (Rt), spectral (UV) and spectrometric (HR full scan and MS/MS) features. In particular, a part of the plant material was extracted using DCM, MeOH and H
2O successively as extraction solvents. In parallel, since
Micromelum is reported to contain alkaloids, another part of the plant material was extracted following a specific protocol for alkaloids. This protocol involves the extraction of the plant material with EtOAc and then alkalinization of the plant residue and successive extraction with EtOAc and MeOH. The derived extracts were partitioned using HCl solution and after a second alkalinization step organic (EtOAc_A, MeOH_A) and aqueous (EtOAc_B, MeOH_B) fractions were obtained, respectively (see
Section 3.3. for further details). Since, this study was focused on the identification of coumarin constituents the alkaloid content was not further investigated.
All the extracts were analyzed by HPLC-DAD (
Figure S1) and the comparison of their chromatograms revealed that the second extraction approach was qualitatively more efficient regarding the secondary metabolite content. As expected, the organic fractions (EtOAc_A, MeOH_A) were found more enriched in lipophilic compounds while the more polar constituents were concentrated in the aqueous fractions (EtOAc_B, MeOH_B) (
Figure S2). In addition, concerning the classical extraction approach, the DCM extract was found more rich compared to the MeOH and H
2O extracts which were found less abundant in secondary metabolites. It is worth noting that in all extracts a dominant metabolite was observed at 21 min (
Figure S1) with characteristic UV absorption maxima at 206 and 321 nm indicative of 7-oxygenated coumarins. Based on UV spectra, all extracts were screened and mainly in DCM, EtOAc, EtOAc_A, EtOAc_B, MeOH_A and MeOH_B extracts peaks providing similar absorption maxima were found.
Thereafter, the aforementioned extracts were analyzed by UPLC-ESI(+)-HRMS/MS in order to obtain characteristic spectrometric data for coumarin compounds and diagnostic fragment ions which could facilitate their detection in complex mixtures. To the best of our knowledge, no previous data have been reported concerning the analysis of 7-oxygenated coumarins by UPLC-ESI(+)-HRMS/MS. The LC-MS-based profiling of
M. falcatum extracts revealed several peaks in the base peak chromatograms (
Figure 1). In addition, the data dependent acquisition method using dynamic exclusion enabled the collection of numerous HRMS/MS spectra for each resolved peak. Taking advantage of the accurate mass measurements and the high resolution of the Orbitrap analyzer, a specific strategy was followed [
16]. Initially, descriptors such as the suggested elemental composition (EC), the RDB eq. values and the Δm (ppm) between the theoretical and measured values were calculated for each detected peak. For the prioritization of the suggested ECs, analysis of the reported 7-oxygenated coumarins from
Micromelum species led to the determination of certain thresholds. Assuming that 7-oxygenated coumarins present in the different extracts are substituted structures of the simple 7-oxygenated coumarin, specific limitations were set. In particular, a minimum number of 10 C atoms, seven H atoms and three O atoms were selected and a RDB eq. value greater than 7 was defined. The Δm was set ≤ 3 ppm since the analysis was performed using the Orbitrap analyzer routinely operating in 1–2 ppm accuracy window, at low masses [
17].
Figure 1.
Base peak chromatograms of each extract obtained from M. falcatum in positive ionization mode.
Figure 1.
Base peak chromatograms of each extract obtained from M. falcatum in positive ionization mode.
Armed with the information from the UV spectra as a starting point, the most abundant peak at 6.42 min (21 min in the HPLC-DAD chromatogram) which presented characteristic absorption maxima of a 7-oxygenated coumarin, was analyzed. The main ion in the full scan spectrum revealed the presence of an adduct ion with sodium [M+Na]
+ while the corresponding [M+H]
+ wasn’t detected. Based on this observation and the abovementioned criteria, all the peaks satisfying both conditions were defined and listed. Scrutinizing the literature, in combination with UPLC-ESI(+)-HRMS/MS data, 10 known coumarins were detected and eight possibly new coumarins were proposed in all selected extracts (
Table 1).
Table 1.
7-Oxygenated coumarin derivatives detected in M. falcatum extracts based on literature and UPLC-ESI(+)-HRMS/MS data.
Table 1.
7-Oxygenated coumarin derivatives detected in M. falcatum extracts based on literature and UPLC-ESI(+)-HRMS/MS data.
| No | Rt (min) | [M+Na]+ | Molecular Formula | RDB | Delta (ppm) | MS/MS (% Intensity) | Suggested Compounds (Annotation) | Extract |
|---|
| 1 | 4.10 | 315.0842 | C15H16O6Na | 7.5 | 1.018 | 297.0737 (24) | Unknown (Coumarin with aliphatic chain) | EtOAc, DCM, EtOAc_A |
| 285.0737 (5) |
| 227.0317 (100) |
| 2 | 4.37 | 315.0842 | C15H16O6Na | 7.5 | 0.922 | 297.0737 (17) | Unknown
(Microfalcrin,
1) | EtOAc, DCM, EtOAc_A, EtOAc_B |
| 285.0737 (3) |
| 227.0316 (100) |
| 3 | 5.20 | 357.0947 | C17H18O7Na | 8.5 | 0.633 | 297.0736 (100) | Micromeloside D | DCM, EtOAc_A |
| 4 | 5.68 | 297.0737 | C15H14O5Na | 8.5 | 1.196 | | Micromeloside A/Hopeyhopin
(Micromeloside A, 4) | EtOAc, DCM, EtOAc_A, EtOAc_B |
| 5 | 5.88 | 357.0948 | C17H18O7Na | 8.5 | 1.053 | 297.0737 (56) | Micromeloside D | DCM, EtOAc_A |
| 227.0317 (100) |
| 6 | 6.06 | 299.0893 | C15H16O5Na | 7.5 | 0.887 | 281.0788 (100) | Hydramicromelin A/Murrangatin | EtOAc |
| 269.0788 (81) |
| 7 | 6.12 | 311.0529 | C15H12O6Na | 9.5 | 1.095 | | Micromelin
(Microcoumaririn, 2) | DCM, EtOAc_A |
| 8 | 6.29 | 301.1050 | C15H18O5Na | 7.5 | 1.312 | 283.0945 (100) | Unknown | DCM, EtOAc_A |
| 9 | 6.30 | 315.0843 | C15H16O6Na | 7.5 | 1.113 | 297.0852 (100) | Unknown | EtOAc, EtOAc_B |
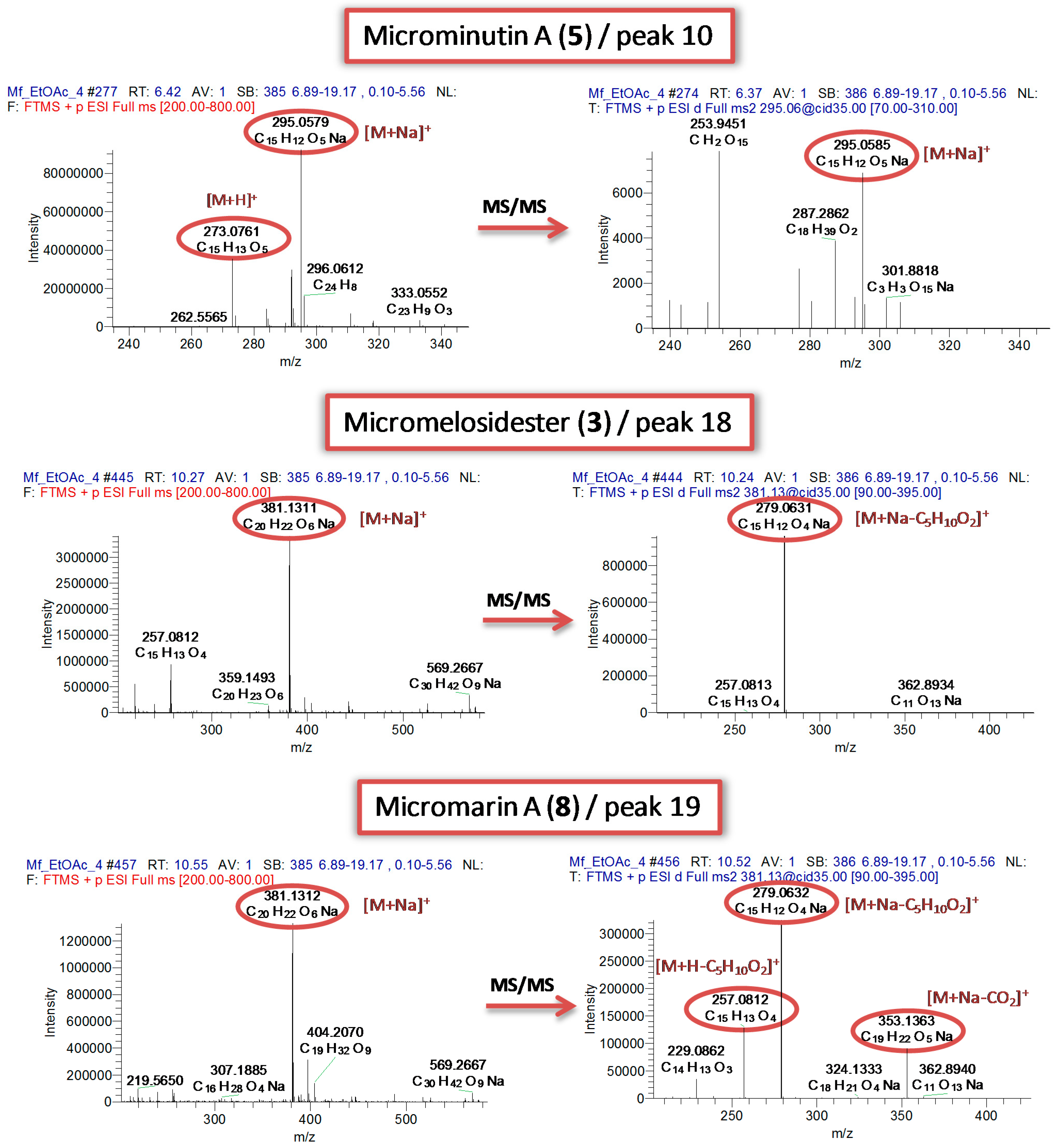
| 10 | 6.42 | 295.0579 | C15H12O5Na | 9.5 | 0.662 | | Microminutin(Microminutin, 5) | EtOAc, DCM, EtOAc_A, EtOAc_B, MeOH_A, MeOH_B |
| 11 | 6.55 | 311.0527 | C15H12O6Na | 9.5 | 1.053 | | Micromelin
(Micromelin, 6) | EtOAc, DCM, EtOAc_B |
| 12 | 7.54 | 399.1416 | C20H24O7Na | 8.5 | 0.365 | 297.0736 (100) | Micromarin B/C | EtOAc, DCM, EtOAc_A, EtOAc_B, MeOH_A, MeOH_B |
| 13 | 7.73 | 399.1416 | C20H24O7Na | 8.5 | 0.516 | 297.0736 (100) | Micromarin B/C | EtOAc, EtOAc_B |
| 14 | 8.17 | 399.1416 | C20H24O7Na | 8.5 | 0.516 | 381.1312 (5) | Micromarin B/C
(Micromarin B, 7) | EtOAc, DCM, EtOAc_A, EtOAc_B, MeOH_A, MeOH_B |
| 297.0737 (75) |
| 227.0317 (100) |
| 15 | 8.44 | 343.1156 | C17H20O6Na | 7.5 | 1.138 | 315.0843 (100) | Micromeloside B | DCM, EtOAc_A, EtOAc_B |
| 297.0737 (60) |
| 16 | 8.81 | 413.1573 | C21H26O7Na | 8.5 | 0.522 | 311.0894 (100) | Micromeloside C | EtOAc_A, MeOH_A |
| 17 | 9.52 | 409.1624 | C22H26O6Na | 9.5 | 0.465 | 289.1050 (100) | Unknown
(Coumarin with isovaleryl chain) | EtOAc, DCM, EtOAc_A, EtOAc_B, MeOH_A, MeOH_B |
| 18 | 10.27 | 381.1311 | C20H22O6Na | 9.5 | 0.710 | 279.0630 (100) | Micromarin A
(Micromelosidester, 3) | EtOAc, DCM, EtOAc_B, MeOH_A |
| 19 | 10.55 | 381.1312 | C20H22O6Na | 9.5 | 0.893 | 353.1363 (28) | Micromarin A
(Micromarin A, 8) | EtOAc, EtOAc_B, MeOH_A |
| 279.0630 (100) |
| 257.0812 (41) |
Based on these data targeted isolation of representative known and unknown coumarins were followed. Furthermore, the purified coumarins were used as reference compounds using the direct infusion method in order to investigate their spectrometric behavior and characteristics.
2.2. Isolation and Structure Elucidation of 7-Oxygenated Coumarin Derivatives
Based on the profiling of
M. falcatum extracts, specific peaks were selected for targeted isolation of representative coumarins. The most promising extracts (DCM/EtOAc/EtOAc_A) were subjected to HPLC-DAD semi-preparative isolation. Specifically, eight peaks were targeted and finally five known coumarins of the genus together with three new coumains were purified (
Figure 2). For structure determination and unambiguous identification 1 and 2 D NMR spectra were acquired verifying in all cases the initial predictions.
Figure 2.
Structures of compounds 1–8.
Figure 2.
Structures of compounds 1–8.
Compound
1 (peak 2, Rt = 4.37 min) was isolated as a white amorphous powder. The UV spectrum in methanol revealed the characteristic absorption maxima for 7-oxygenated coumarins at 206 and 319 nm [
5]. Its molecular formula was deduced as C
15H
16O
6, implying eight degrees of unsaturation, based on the adduct ion [M+Na]
+ at
m/z 315.0835 as determined from the ESI-(+)-HRMS data. The presence of an OH at position C-1' gave rise to the fragment ion at
m/z 227.0315 attributed to the C
11H
8O
4Na
+ as observed in the HRMS/MS spectrum. In addition, the C
15H
14O
5Na
+ fragment ion was observed in the MS/MS spectrum of compound
1, indicating the presence of two oxygen atoms in positions C-1' and C-2'.
The NMR spectra of compound
1 (
Table 2) revealed the presence of a 7-methoxylated coumarin and in particular the occurrence of a 3-methylene-1,2,4-
n-butanotriol chain in position 8 of the core coumarin structure. Specifically, in the
1H-NMR spectrum of
1, two spin systems in the aromatic region were observed which were present in all isolated 8-substituted 7-methoxylated coumarins (compounds
1–
5,
7 and
8). The spin system, corresponding to the aromatic protons H-5, H-6 consists of two doublets observed at δ 7.55 and 7.05, respectively, with an 8.8 Hz coupling constant. The respective aromatic carbons C-5 and C-6 resonate at δ 130.7 and 109.9, respectively (HMQC spectrum). The second spin system was comprised of two doublets at δ 6.25 (H-3) and 7.87 (H-4) with a coupling constant of 9.4 Hz typical for olefinic protons of an α-pyrone ring. Correlations of these protons with carbons at δ 113.6 and 146.5 were observed in the HSQC spectrum, leading to their characterization as C-3 and C-4, respectively. The methoxy group was deduced from the presence of a singlet integrating for three protons and resonating at δ 3.96. The carbon at δ 57.0 was attributed to the oxygenated carbon of the methoxy group by the cross peak correlation of this carbon with the methoxy protons detected in the HSQC spectrum. In addition, the quaternary carbons of the 7-methoxylated coumarin structure were revealed by the cross peaks detected in the HMBC spectrum.
Table 2.
NMR Spectroscopic Data (600 MHz, CD3OH) for compounds 1–3.
Table 2.
NMR Spectroscopic Data (600 MHz, CD3OH) for compounds 1–3.
| Position | 1 | 2 | 3 |
|---|
| δH (J in Hz) | δC | HMBC | δH (J in Hz) | δC | HMBC | δH (J in Hz) | δC | HMBC |
|---|
| 2 | | 163.1 | | | 163.0 | | | 162.6 | |
| 3 | 6.25, d (9.4) | 113.6 | 2, 9 | 6.29, d (9.4) | 114.0 | 2, 9 | 6.26,d (9.4) | 113.8 | 2, 9 |
| 4 | 7.87, d (9.4) | 146.5 | 2, 3, 5, 10 | 7.95, d (9.4) | 146.4 | 2, 5, 9, 10 | 7.90,d (9.4) | 146.5 | 2, 5, 9, 10 |
| 5 | 7.55, d (8.8) | 130.7 | 4, 7, 10 | 7.73, d (8.8) | 132.2 | 4, 7, 10 | 7.63,d (8.8) | 131.4 | 4, 7, 9, 10 |
| 6 | 7.05, d (8.8) | 109.9 | 8, 9 | 7.17, d (8.8) | 109.8 | 7, 8, 9 | 7.09,d (8.8) | 109.9 | 7, 9, 10 |
| 7 | | 162.4 | | | 162.3 | | | 162.9 | |
| 8 | | 117.9 | | | 107.5 | | | 109.2 | |
| 9 | | 114.8 | | | 115.0 | | | 114.8 | |
| 10 | | 154.5 | | | 154.4 | | | 154.9 | |
| 1' | 5.37, d (8.8) | 70.6 | 7, 8, 10, 2' | | | | 5.52, d (5.9) | 78.3 | 6, 7, 10, 2' |
| 2' | 4.94, d (8.8) | 77.2 | 1', 3', 4', 5' | | 172.6 | | 6.11, d (5.9/1.8) | 78.2 | 3', 5' |
| 3' | | 150.4 | | | 123.4 | | | 148.3 | |
| 4'a | 4.08, dt (15.3/1.8) | 62.8 | 3', 5' | | 165.1 | | 4.74, dt (13.0/2.0) | 72.5 | 8, 1', 3' |
| 4'b | 4.00, dt (15.3/1.8) | 5' | | 4.62, m | 8, 1', 3' |
| 5'a | 4.92, d (1.8) | 112.0 | 2', 4' | 6.18, brs | 101.4 | | 5.28, m | 109.6 | 2', 3', 4' |
| 5'b | 4.91,d (1.8) | 2', 4' | |
| 1'' | | | | | | | | 174.6 | |
| 2'' | | | | | | | 2.21, m | 44.5 | 1'', 3'', 4'', 5'' |
| 3'' | | | | | | | 2.01, m | 27.3 | 1'', 4'', 5'' |
| 4'' | | | | | | | 0.88, d (6.7) | 22.7 | 2'', 3'', 5'' |
| 5'' | | | | | | | 0.89, d (6.7) | 22.7 | 2'', 3'', 4'' |
| CH3O | 3.96, s | 57.0 | 7 | 3.93, s | 57.2 | 7 | 3.93, s | 57.1 | 7 |
| CH3 | | | | 1.96, s | 13.3 | 8, 2', 3', 4', 5' | | | |
Specifically, the aromatic proton H-5 displayed correlations with the downfield shifted oxygenated quaternary carbons C-7 at δ 162.4 and C-10 at δ 154.5 as well as with the olefinic carbon C-4. Carbon C-7 was also correlated with the protons of the methoxy group. Moreover, the quaternary carbon at δ 114.8 was determined as C-9 based on its correlations with the aromatic proton H-6 and the olefinic proton H-3 while the carbon at δ 117.9 was assigned as C-8 due to its correlations with H-6. Finally, the carbonyl carbon at δ 163.1 was revealed from its cross peak correlation with the olefinic H-3.
The aliphatic chain at position 8 on the core structure was deduced as 3'-methylene-1',2',4' n-butanetriol based on certain resonances. Specifically, H-1' and H-2' were observed as doublets at δ 5.37 and 4.94, respectively, with an 8.8 Hz coupling constant, while the absence of a NOE cross peak between them suggested the erythro configuration of the hydroxyl groups. The C-1' and C-2' carbons were detected at δ 70.6 and 77.2 and correlated with the respective protons in the HSQC spectrum. The quaternary carbon C-3' resonated at 150.4 ppm as deduced from the HMBC spectrum. Methylene protons H-4'a and H-4'b were observed at δ 4.08 and 4.00 as two doublets of triplets with coupling constants of 15.3 and 1.8 Hz, while C-4' was resonated at δ 62.8. Finally, the protons at δ 4.92 and 4.91 were attributed to H-5' which correlated with the C-5' at δ 112.0 as observed in the HSQC spectrum. Finally, the position of the side chain was determined based on the correlation of H-1' (δ 5.37) with C-8 observed in the HMBC spectrum. Thus, compound 1 was identified as 8-(5-hydroxy-4-methyl-2-oxo-5H-fur-3-yl)-7-methoxy-2-chromenone to which the trivial name microfalcrin was given.
Compound 2 (peak 7, Rt 6.12 min) was isolated as a white amorphous powder presenting a characteristic UV spectrum (MeOH) with absorption maxima at 207 and 319 nm indicative of 7-oxygenated coumarin derivative. Its molecular formula was calculated as C15H12O6 from the adduct ion [M+Na]+ at m/z 311.0524 observed in the ESI-(+)-HRMS spectrum, implying eight degrees of unsaturation. The absence of characteristic fragment ion in MS/MS level is an indication of furan or lactone ring on the coumarin structure.
Like
1, in the NMR spectra of
2 the spin systems corresponding to a typical 8-substituted 7-methoxylated coumarin were obvious. In addition to these characteristic resonances certain signals indicative to a butenolide (2-furanone) ring were observed (
Table 2). Particularly, H-5' was detected as a broad singlet at δ 6.18 indicating the presence of a hydroxyl group on this carbon, and C-5' was observed at δ 101.4 in the HSQC spectrum. Furthermore, the protons of the methyl group in C-4' position resonated at δ 1.96 as a singlet and correlated with the upfield shifted carbon at δ 13.3 as observed in the HSQC spectrum. In order to determine the rest of the quaternary carbons of this ring and its position on the core structure, the HMBC spectrum was analyzed. Interestingly, the methyl protons displayed correlations with all the carbons of the butenolide ring (C-2', C-3', C-4', C-5') revealing the quaternary carbons C-2', C-3' and C-4' which resonated at δ 172.6, 123.4, and 165.1, respectively. In addition, another very useful correlation was observed between the same methyl protons and C-8 showing the position of the ring on the core structure. Hence, the 8-(5-hydroxy-4-methyl-2-oxo-5H-fur-3-yl)-7-methoxy-2-chromenone structure was deduced for compound
2 and the name microcoumaririn was given to it (
Figure 2).
Compound 3 (peak 18, Rt 10.27 min) was isolated as a white amorphous powder. The UV spectrum in methanol presented absorption maxima at 206 and 320 indicating the presence of a 7-oxygenated coumarin. Its molecular formula was deduced as C20H22O6, suggesting 10 degrees of unsaturation, based on the [M+Na]+ adduct ion observed at m/z 381.1306 in the ESI-(+)-HRMS spectrum. Compound 3 presented a characteristic fragment ion at m/z 279.0631 corresponding to the neutral loss of an isovaleryl chain.
The NMR spectra of
3 were similar to the aforementioned compounds concerning the coumarin skeleton while additional signals revealed the presence of a 1',2',3',4'-tetrahydro-3'-methylene-1'-furanyl-2'-isovalerylester substitute (
Table 2). In the
1H-NMR spectrum the H-1' of the tetrahydrofuran ring resonated at δ 5.52 as a doublet (
J = 5.9 Hz) and H-2' was observed at δ 6.11 as doublet of doublets (
J = 5.9 and 1.8 Hz). Based on the coupling constant value between H-1' and H-2', a
trans configuration is assumed [
8,
18]. In the HSQC spectrum the respective carbons at δ 78.3 and 78.2 were determined and from their ppm values it could be deduced that both were oxygenated. The position of the ring at C-8 of the core structure was determined from the cross peak correlations of the H-1' with C-6 (
4J), C-7 (
3J), and C-10 (
3J), observed in the HMBC spectrum. H-4'a was observed at δ 4.74 as a doublet of triplets (
J = 13.0 and 2.0 Hz) and H-4'b at δ 4.62 as a multiplet; both H-4'a and H-4'b were correlated with a carbon at δ 72.5 as detected in the HSQC spectrum. H-5' resonated at δ 5.28 as a multiplet and C-5' was observed at δ 109.6 in the HSQC spectrum. Finally, the presence of the quaternary C-3 at δ 148.3 was revealed from its cross peak correlation with H-4'a and H-4'b and H-5' in the HMBC spectrum. The isovaleryl moiety was deduced from the presence of a carbon resonated at δ 174.6 attributed to the carbonyl C-1'' as observed from the cross peaks of H-2'' and H-3'' in the HMBC spectrum. H-2'' and H-3'' were detected as multiplets at δ 2.21 and 2.01, respectively, with their corresponding carbons resonating at δ 44.5 and 27.3. Finally, H-4'' and H-5'' were observed at δ 0.88 and 0.89 as two doublets integrating for six protons and both C-4'' and C-5'' resonated at δ 22.7 as observed in the HSQC spectrum. Thus, compound
3 was identified as 2-(7-methoxy-8-coumarinyl)-4-methylene-3,5-dihydro-2
H-fur-3-yl 3-methylbutyrate and named micromelosidester (
Figure 2).
2.3. Mass Spectrometry of 7-Methoxylated Coumarin Derivatives
The UPLC-HRMS & HRMS/MS profiling of the
M. falcatum extracts as well as the analysis of the isolated 7-methoxylated coumarins by the direct infusion method revealed patterns that could be utilized for dereplication purposes (
Figure 3). Specifically, even if both ionization modes were assessed only in positive mode stable precursor ions were observed in the full scan spectra. Moreover, all isolated compounds formed sodium adduct ions [M+Na]
+ whereas the respective molecular or pseudomolecular ions were present in very low intensity or were even absent. Additionally, at the MS/MS level, diagnostic fragment ions were detected and attributed to specific structural features (
Figures S12–S32).
For instance, the presence of a free OH group at the C-2' position of 8-substituted 7-methoxylated coumarins seems to favor the cleavage of the C-1'/C-2' bond and the fission of the side chain generating stable C
11H
8O
4Na
+ ions observed at
m/z 227.0317. This diagnostic ion was observed only in the HRMS/MS spectra of isolated compounds
1 and
7 as well as in the HRMS/MS spectra of peaks 1, 2, 5, 14 (
Table 1). Another characteristic fragment ion was observed at
m/z 297.0736 in the MS/MS spectra and attributed to the C
15H
14O
5Na
+ ions. It seems that coumarins with oxygenated C-1' and C-2' generate this fragment ion through the loss of H
2O in a certain stage of their fragmentation. Only compounds
1 and
7 from isolated ones and peaks 1–3, 5, 9, 12–15 revealed this fragment ion as confirmed by the MS/MS spectra. It seems that the presence of 3'-methylen-1',2',4'-
n-butanotriol side chain is evident only when both fragment ions are present (
1 and
7). Moreover, compounds possessing an isovaleryl side chain (
3,
7,
8) presented a characteristic loss of 102 amu corresponding to the cleavage of the ester bond and the elimination of this chain. Independently of the substitute structure (chain or ring) the ester bond seems to be fragile even under the mild CID conditions. This loss was also observed in the HRMS/MS spectra of peaks 12–14 and 16–19 suggesting that these compounds incorporate an isovaleryl side chain in their structures (
Table 1).
Figure 3.
Characteristic HRMS and HRMS/MS spectra of representative 7-methoxylated coumarins.
Figure 3.
Characteristic HRMS and HRMS/MS spectra of representative 7-methoxylated coumarins.
Interestingly, the presence of an additional tetrahydrofuran or 2-furanone ring at position C-6 or C-8 lead to the formation of stable molecular adduct ions [M+Na]+ which did not give any fragment ion in MS/MS level in any CID conditions and this was observed in compounds 2, 4–6 and peaks 4, 7, 10, 11. Thus, the absence of fragment ions could be indicative for the presence of any abovementioned ring on the basic 7-methoxylated 8-substituted coumarin structure.
Summarizing these observations, the developed method enabled the detection of 8-substituted 7-methoxylated coumarins, the discrimination of coumarins with open side chains
vs. additional rings at C-8 as well as the identification of esterified derivatives. At this point, it is important to note that the proposed structures were verified after isolation and structure elucidation by NMR validating our approach. Finally, it is worth noting that LTQ-Orbitrap platform displayed excellent robustness and high reproducibility between LC-MS
n and direct infusion spectra confirming this statement from previous studies [
19].

 ,
, 

{kind=link}
{kind=link}
{kind=link}

