
Development of New 1,3-Diazaphenoxazine Derivatives (ThioG-Grasp) to Covalently Capture 8-Thioguanosine

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Results and Discussion
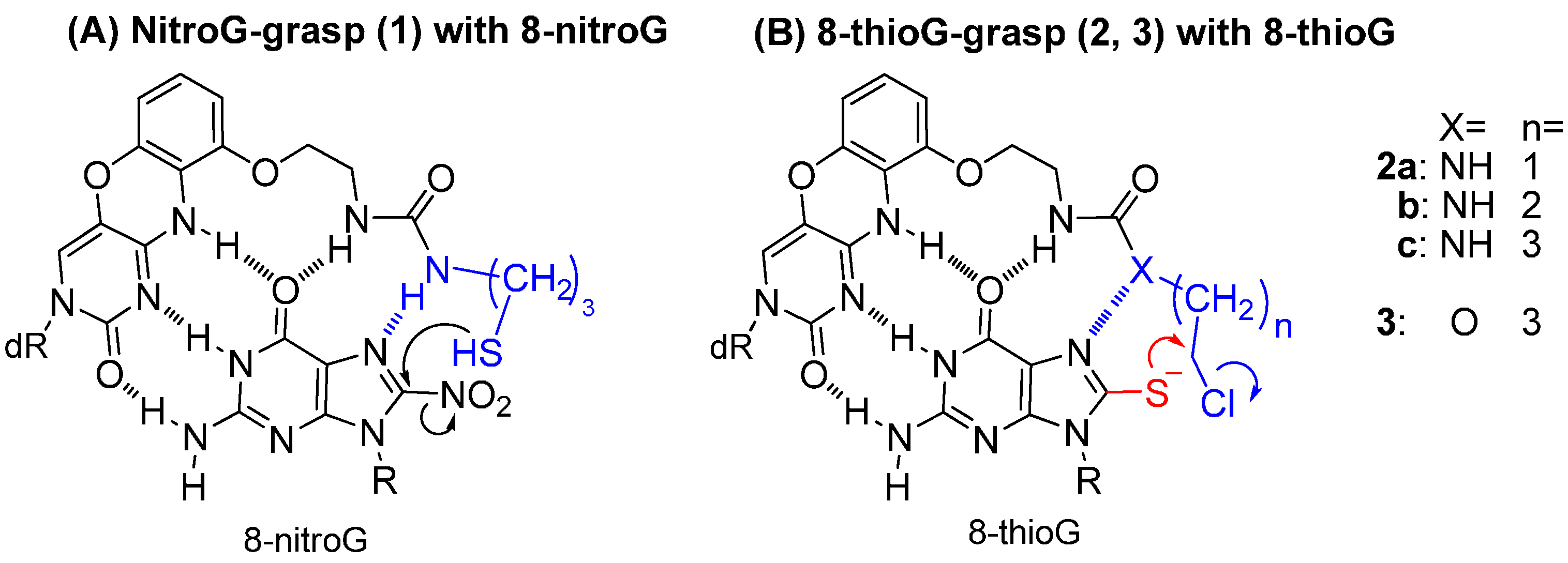
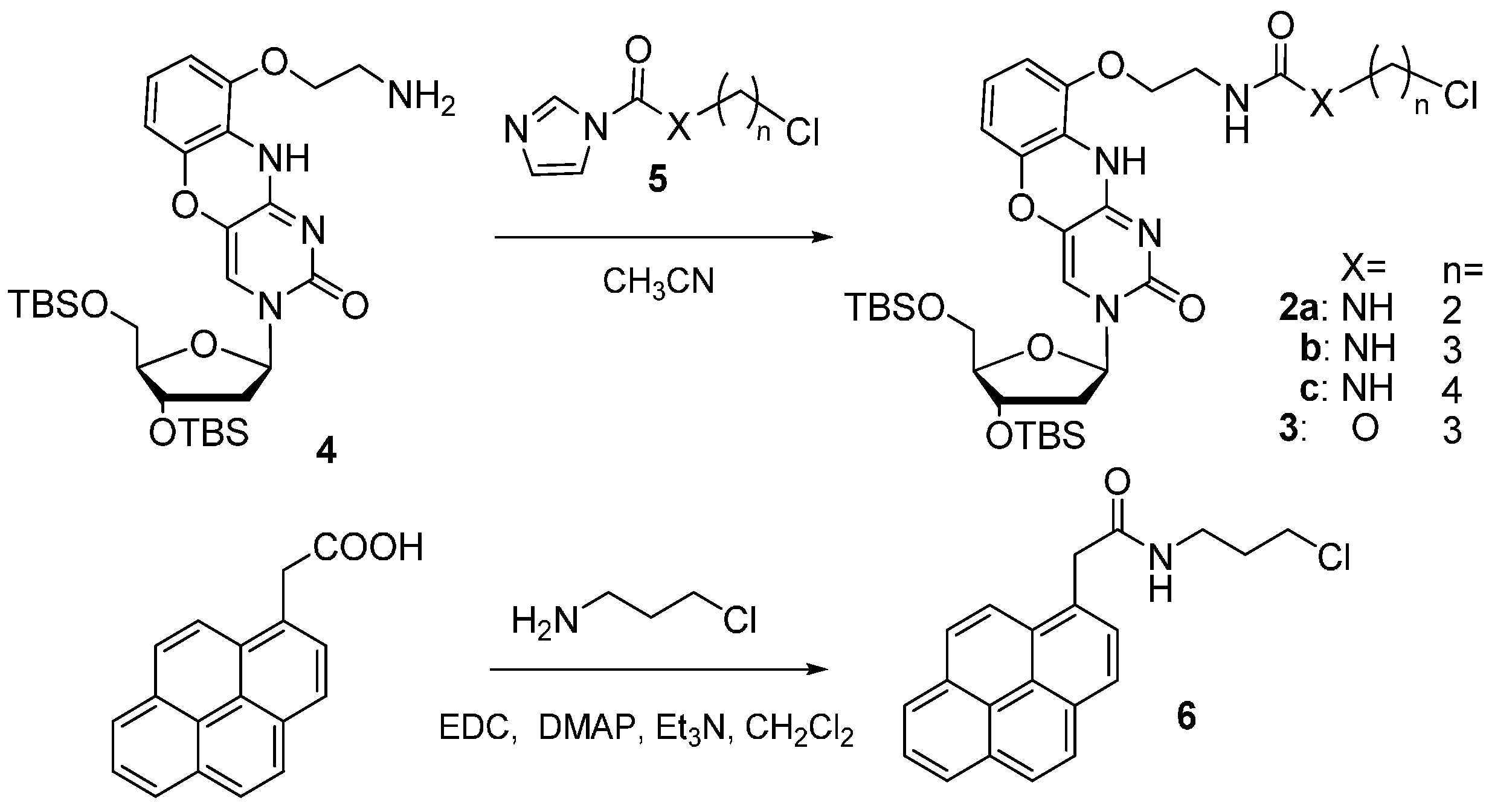
2.1. Chemistry

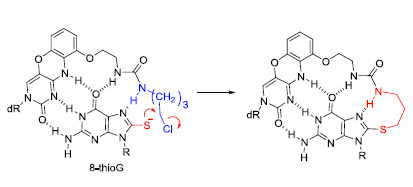
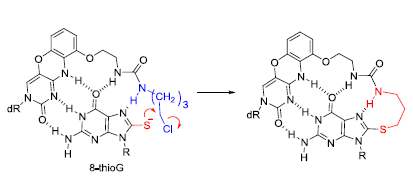
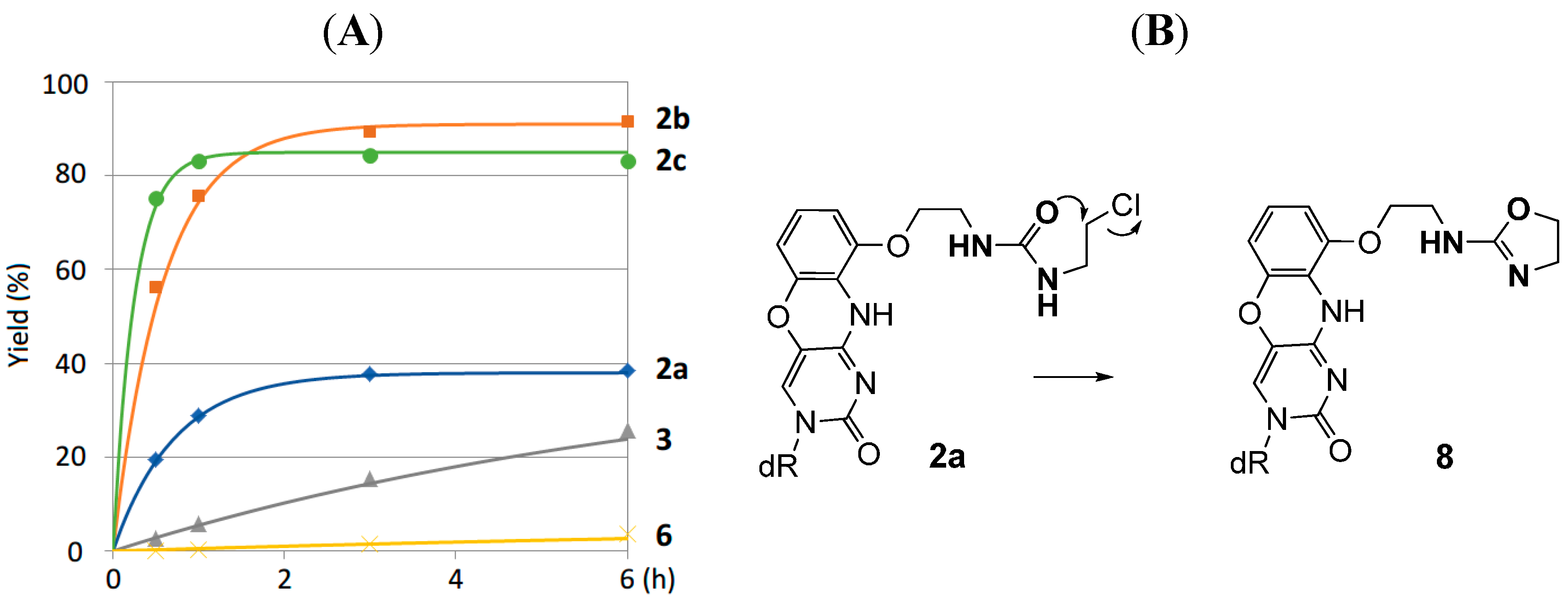
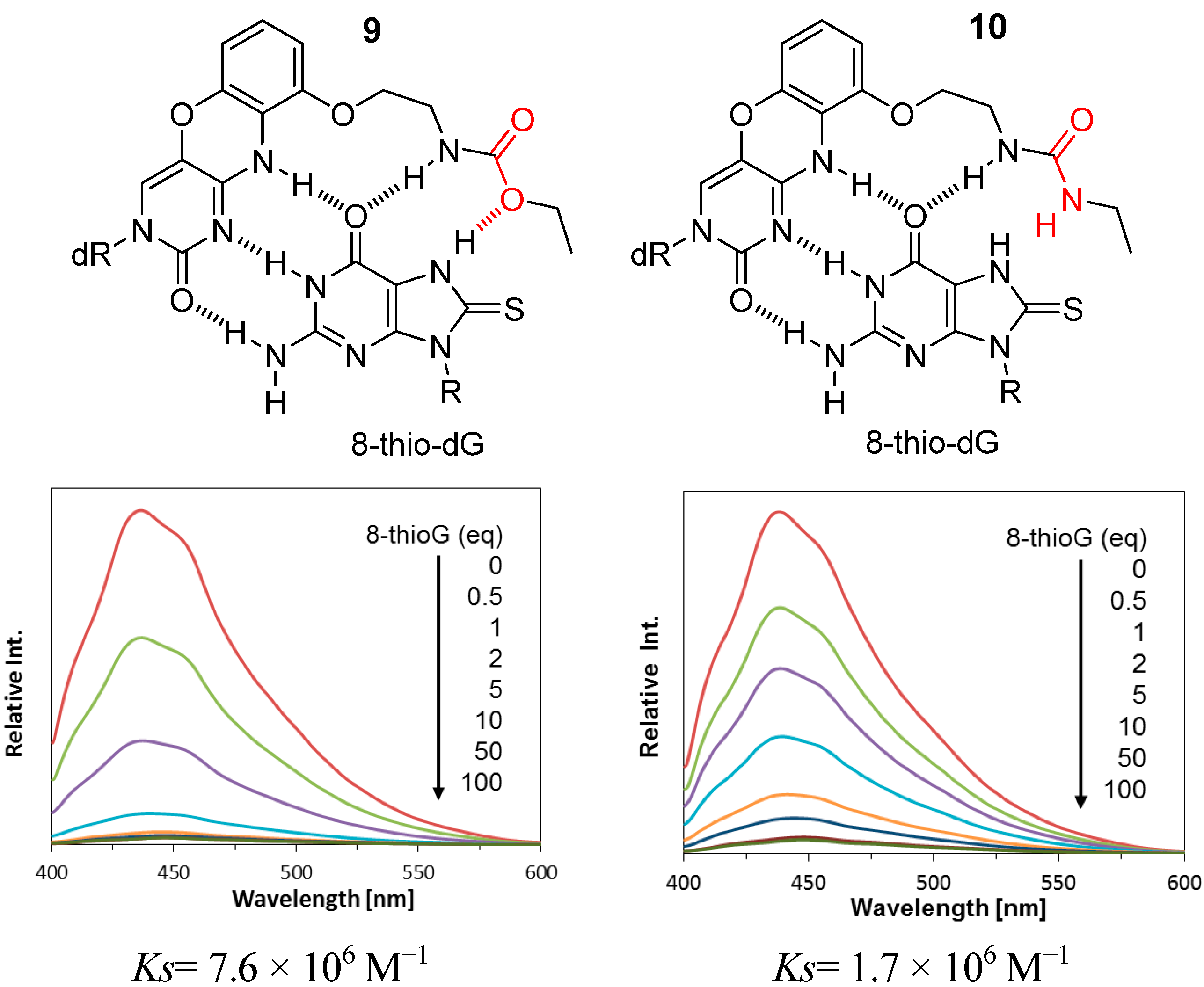
2.2. Reaction of 8-ThioG-Grasp with 8-ThioG
2.3. Comparison of the Reactivity between 8-ThioG-Grasp and the Control Compound



3. Experimental Section
3.1. General Information
3.2. Chemistry
3.2.1. General Synthesis of 8-ThioG-Grasp Derivatives
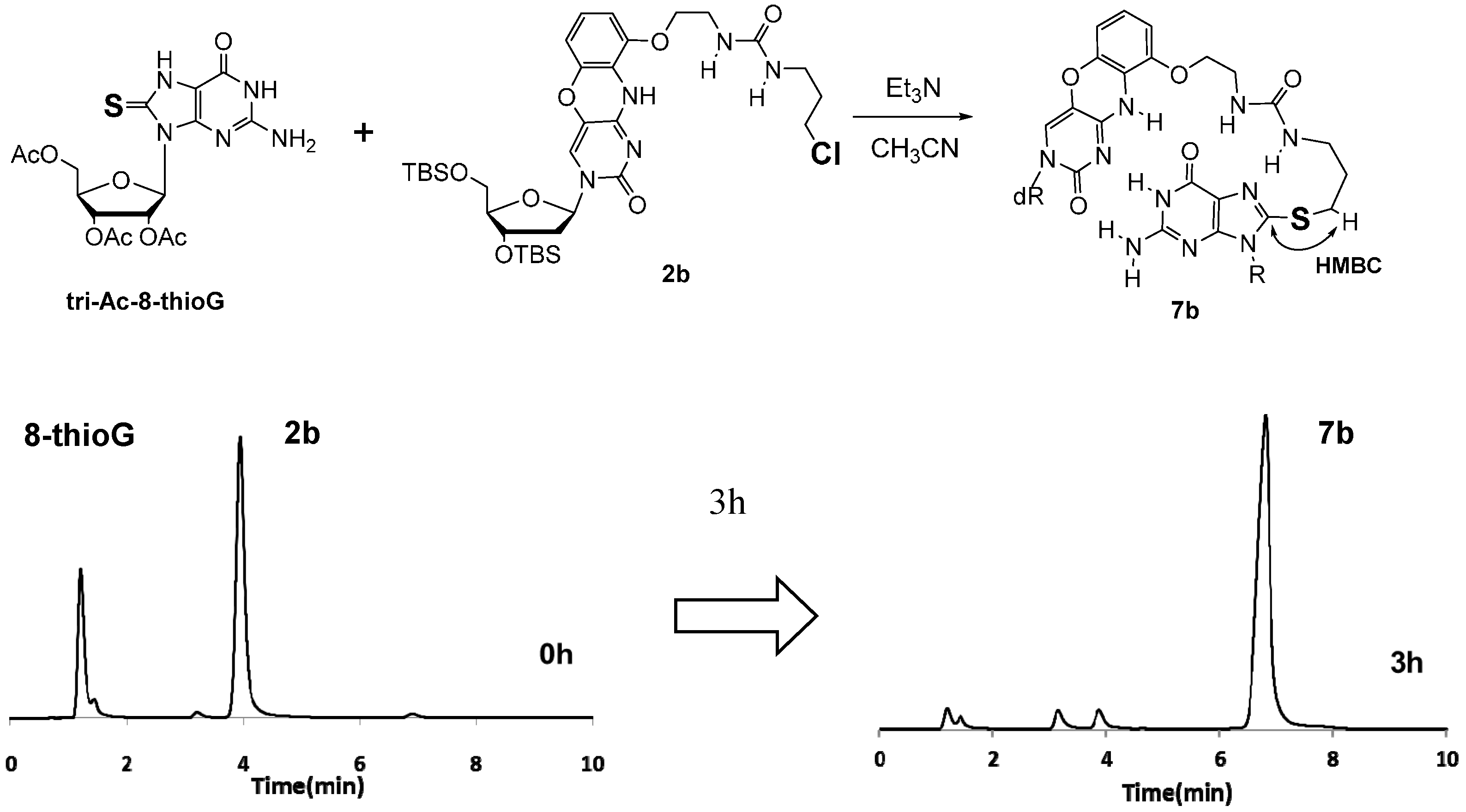
3.2.2. Synthesis of 8-ThioG Adduct 7b
3.2.3. N-(3-Chloropropyl)-2-(pyren-1-yl)acetamide (6)
3.2.4. Determination of the Structure of the Byproduct 8
3.2.5. General Procedure of Reaction Monitoring by HPLC
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yermilov, V.; Rubio, J.; Ohshima, H. Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett. 1995, 376, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat. Res. 1997, 387, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. The link between infection and cancer: Tumor vasculature, free radicals, and drug delivery to tumors via the EPR effect. Cancer Sci. 2013, 104, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Zaki, M.H.; Okamoto, T.; Akuta, T.; Tokutomi, Y.; Kim-Mitsuyama, S.; Ihara, H.; Kobayashi, A.; Yamamoto, M.; Fujii, S.; et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclic monophosphate. Nat. Chem. Biol. 2007, 3, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, M.; Sawa, T.; Kitajima, N.; Ono, K.; Inoue, H.; Ihara, H.; Motohashi, H.; Yamamoto, M.; Suematsu, M.; Kurose, H.; et al. Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nat. Chem. Biol. 2012, 8, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Matteucci, M.D. A Cytosine analogue capable of clamp-like binding to a guanine in helical nucleic acids. J. Am. Chem. Soc. 1998, 120, 8531–8532. [Google Scholar] [CrossRef]
- Flanagan, W.M.; Wolf, J.J.; Olson, P.; Grant, D.; Lin, K.Y.; Wangner, R.W.; Matteucci, M.D. A cytosine analog that confers enhanced potency to antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1999, 96, 3513–3518. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.A.; Leeds, J.M.; Balow, G.; Springer, R.H.; Bharadwaj, R.; Manoharan, M. Nuclease resistance of oligonucleotides containing the tricyclic cytosine analogues phenoxazine and 9-(2-aminoethoxy)-phenoxazine (“G-clamp”) and origins of their nuclease resistance properties. Biochemistry 2002, 41, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Wilds, C.J.; Maier, M.A.; Tereshko, V.; Manoharan, M.; Egli, M. Direct observation of a cytosine analogue that forms five hydrogen bonds to guanosine: guanidino G-clamp. Angew. Chem. Int. Ed. 2002, 41, 115–117. [Google Scholar] [CrossRef]
- Holmes, S.C.; Arzumanov, A.A.; Gait, M.J. Steric inhibition of human immunodeficiency virus type-1 Tat-dependent trans-activation in vitro and in cells by oligonucleotides containing 2′-O-methyl G-clamp ribonucleoside analogues. Nucleic Acids Res. 2003, 31, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, O.; Ono, S.; Li, Z.; Tsujimoto, A.; Sasaki, S. Specific fluorescent probe for 8-Oxoguanosine. Angew. Chem. Int. Ed. Engl. 2007, 119, 4584–4587. [Google Scholar] [CrossRef]
- Li, Z.; Nakagawa, O.; Koga, Y.; Taniguchi, Y.; Sasaki, S. Synthesis of new derivatives of 8-oxoG-clamp for better understanding the recognition mode and improvement of selective affinity. Bioorg. Med. Chem. 2010, 18, 3992–3998. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Fuchi, Y.; Nakagawa, O.; Sasaki, S. Optimization of fluorescence property of the 8-oxodGclamp derivative for better selectivity for 8-oxo-2′-deoxyguanosine. Tetrahedron 2011, 67, 6746–6752. [Google Scholar] [CrossRef]
- Fuchi, Y.; Sasaki, S. Efficient covalent capture of 8-nitroguanosine via a multiple hydrogen-bonded complex. Org. Lett. 2014, 16, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Robins, K. Purine nucleosides. VII. Direct bromination of adenosine, deoxyadenosine, guanosine, and related purine nucleosides. J. Am. Chem. Soc. 1964, 86, 1242–1245. [Google Scholar] [CrossRef]
- Strassmaier, T.; Karpen, J.W. Novel N7- and N1-substituted cGMP derivatives are potent activators of cyclic nucleotide-gated channels. J. Med. Chem. 2007, 50, 4186–4194. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Walker, S.G.; Parker, K.A.; Sampson, N.S. Antibacterial studies of cationic polymers with alternating, random, and uniform backbones. ACS Chem. Biol. 2011, 6, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are available from authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchi, Y.; Obayashi, H.; Sasaki, S. Development of New 1,3-Diazaphenoxazine Derivatives (ThioG-Grasp) to Covalently Capture 8-Thioguanosine. Molecules 2015, 20, 1078-1087. https://doi.org/10.3390/molecules20011078
Fuchi Y, Obayashi H, Sasaki S. Development of New 1,3-Diazaphenoxazine Derivatives (ThioG-Grasp) to Covalently Capture 8-Thioguanosine. Molecules. 2015; 20(1):1078-1087. https://doi.org/10.3390/molecules20011078
Chicago/Turabian StyleFuchi, Yasufumi, Hideto Obayashi, and Shigeki Sasaki. 2015. "Development of New 1,3-Diazaphenoxazine Derivatives (ThioG-Grasp) to Covalently Capture 8-Thioguanosine" Molecules 20, no. 1: 1078-1087. https://doi.org/10.3390/molecules20011078