
TDAE Strategy in the Benzoxazolone Series: Synthesis and Reactivity of a New Benzoxazolinonic Anion

Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Mono and Dibromide Substrates

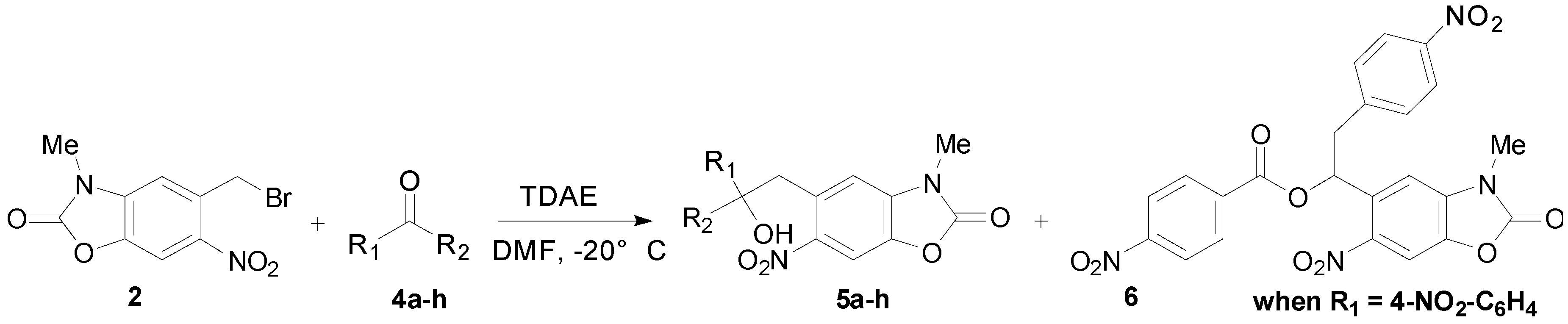
2.2. TDAE Reactivity of 5-(Bromomethyl)-3-methyl-6-nitrobenzoxazolone (2)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | Aromatic Carbonyl | R1 | R2 | Product Number | Yield (%) b |
|---|---|---|---|---|---|
| 1 | 4-Nitrobenzaldehyde | 4-NO2-C6H4 | H | 5a | 52 |
| 2 | 4-Bromobenzaldehyde | 4-Br-C6H4 | H | 5b | 49 |
| 3 | 4-Cyanobenzaldehyde | 4-CN-C6H4 | H | 5c | 31 |
| 4 | 2-Nitrobenzaldehyde | 2-NO2-C6H4 | H | 5d | 44 |
| 5 | 2-Bromobenzaldehyde | 2-Br-C6H4 | H | 5e | 49 |
| 6 | 3-Bromobenzaldehyde | 3-Br-C6H4 | H | 5f | 43 |
| 7 | Ethyl glyoxylate | CO2C2H5 | H | 5g | 72 |
| 8 | Diethyl ketomalonate | CO2C2H5 | CO2C2H5 | 5h | 62 |


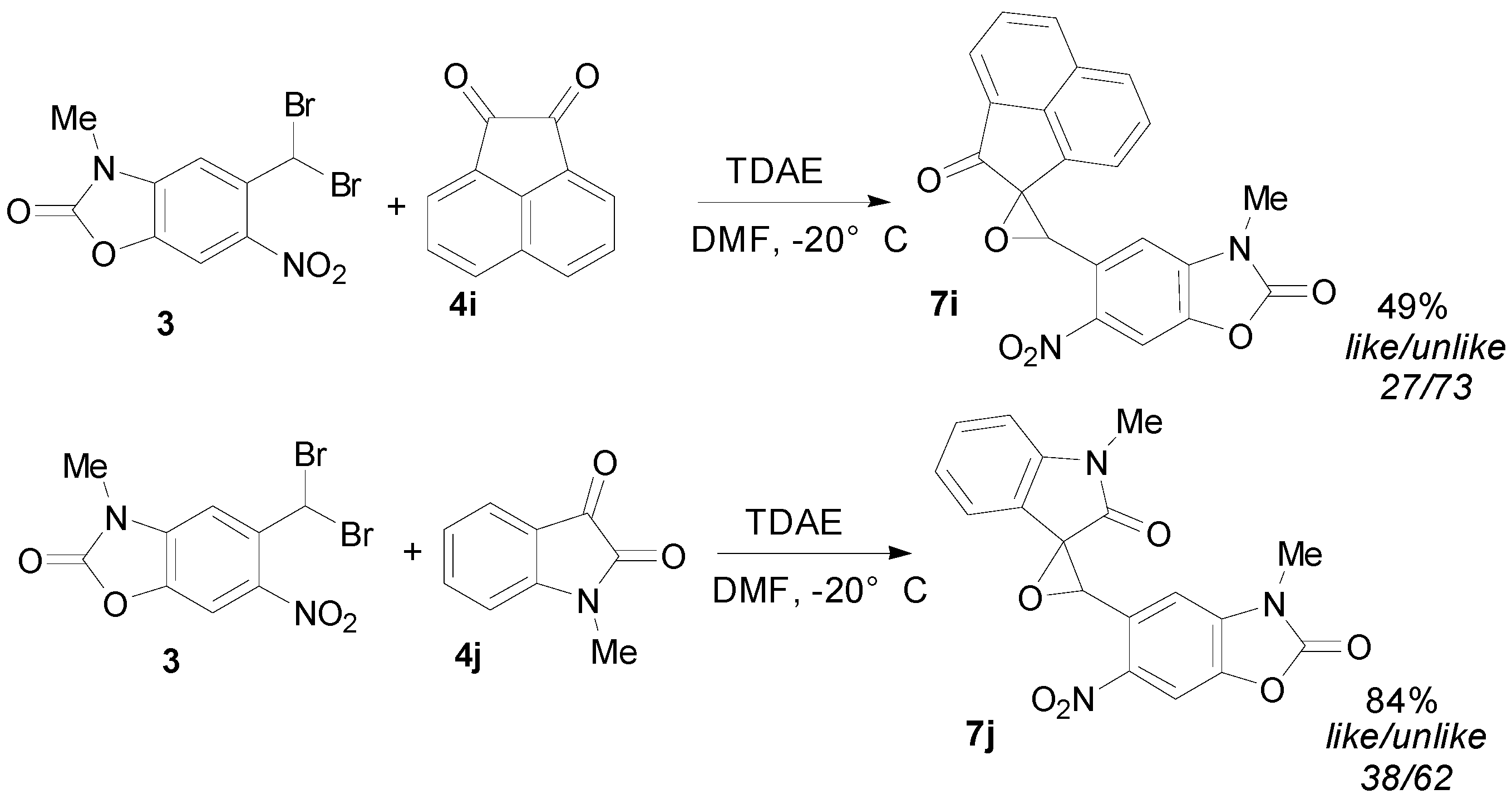
2.3. TDAE Reactivity of 5-(Dibromomethyl)-3-methyl-6-nitrobenzoxazolone (3)
| Entry a | Aromatic Carbonyl | R1 | R2 | Oxirane | Cis/Trans Isomers % b | Yield (%) c |
|---|---|---|---|---|---|---|
| 1 | 4-Nitrobenzaldehyde | 4-NO2-C6H4 | H | 7a | 15/85 | 63 |
| 2 | 4-Bromobenzaldehyde | 4-Br-C6H4 | H | 7b | 7/93 | 55 |
| 3 | 4-Cyanobenzaldehyde | 4-CN-C6H4 | H | 7c | 15/85 | 72 |
| 4 | 2-Nitrobenzaldehyde | 2-NO2-C6H4 | H | 7d | 32/68 | 46 |
| 5 | 2-Bromobenzaldehyde | 2-Br-C6H4 | H | 7e | 19/81 | 64 |
| 6 | 3-Bromobenzaldehyde | 3-Br-C6H4 | H | 7f | 7/93 | 48 |
| 7 | Ethyl glyoxylate | CO2C2H5 | H | 7g | 0/100 | 26 |
| 8 | Diethyl ketomalonate | CO2C2H5 | CO2C2H5 | 7h | 0/100 | 37 |


3. Experimental Section
3.1. General Information
3.2. Synthesis of Substrates 1–3
3.3. General Procedure for the Reaction of 2 and Aromatic Carbonyl Derivatives 4a–f, α-Carbonyl Ester 4g, Ketomalonate 4h, Acenaphtenedione 4i and Ketolactam 4j Using TDAE
3.4. General Procedure for the Reaction of 3 and Aromatic Carbonyl Derivatives 4a–f, α-Carbonyl Ester 4g, Ketomalonate 4h, Acenaphtenedione 4i and Keto-lactam 4j Using TDAE
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lesieur, D.; Carato, P.; Bonte, J.-P.; Depreux, P.; Caignard, D.-H.; Millan, M.; Newman-Tancredi, A.; Renard, P.; Rettori, M.-C. Preparation of Piperazinylmethylbenzothiazolinones, -Benzoxazolinones, -Benzoxazinones, and Related Compounds as Central Nervous System Agents. EP 841330, 13 May 1998. [Google Scholar]
- Fukaya, T.; Ishiyama, T.; Baba, S.; Masumoto, S. Identification of a novel benzoxazolone derivative as a selective, orally active 18 kDa translocator protein (TSPO) ligand. J. Med. Chem. 2013, 56, 8191–8195. [Google Scholar] [CrossRef] [PubMed]
- Aichaoui, H.; Poupaert, J.H.; Lesieur, D.; Henichart, J.-P. Regioselectivity in the C-acylation of 2(3H)-benzoxazolones. Tetrahedron 1991, 47, 6649–6654. [Google Scholar] [CrossRef]
- Moussavi, Z.; Depreux, P.; Lesieur, D.; Cotelle, N.; Sauzieres, J.; Plancke, M.O.; Fruchart, J.C. Pharmacomodulation of 7-(2-methylenebutyryl)-2,3-dihydrobenzoxazin-[1,4]-3-one structure and normolipemic activity. Farmaco 1991, 46, 339–355. [Google Scholar] [PubMed]
- Ucar, H.; van derpoorten, K.; Cacciaguerra, S.; Spampinato, S.; Stables, J.P.; Depovere, P.; Isa, M.; Masereel, B.; Delarge, J.; Poupaert, J.H. Synthesis and anticonvulsant activity of 2(3H)-benzoxazolone and 2(3H)-benzothiazolone derivatives. J. Med. Chem. 1998, 41, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Courtois, M.; Mincheva, Z.; Andreu, F.; Rideau, M.; Viaud-Massuard, M.C. Synthesis and biological evaluation with plant cells of new fosmidomycin analogues containing a benzoxazolone or oxazolopyridinone ring. J. Enzym. Inhib. Med. Chem. 2004, 19, 559–565. [Google Scholar] [CrossRef]
- Raju, B.G.; Ciabatti, R.; Maffioli, S.I.; Singh, U.; Romano, G.; Micheluucci, E.; Tiseni, P.S.; Candiani, G.; Kim, B.; O’Dowd, H. Ramoplanin Derivatives Possessing Antibacterial Activity. US 0211603, 21 September 2006. [Google Scholar]
- Jadhav, J.S.; Chatpalliwar, V.A.; Khadse, S.C.; Patil, R.R. Synthesis and screening of some new 2-(3H)-benzoxazolone derivatives for analgesic, antiinflammatory, and skeletal muscle relaxant activity. Indian J. Heterocycl. Chem. 2008, 17, 343–346. [Google Scholar]
- Köksal, M.; Kelekci, N.G.; Mercanoglu, G.O.; Erdoğan, H. Synthesis and evaluation of analgesic, anti-inflammatory and antioxidant activities of new 6-acyl-3-alkyl-5-methyl-2(3H)-benzoxazolones. Arzneim. Forsch. 2008, 58, 398–404. [Google Scholar]
- Mésangeau, C.; Narayanan, S.; Green, A.M.; Shaikh, J.; Kaushal, N.; Viard, E.; Xu, Y.; Fishback, J.A.; Poupaert, J.H.; Matsumoto, R.R.; et al. Conversion of a highly selective sigma-1 receptor-ligand to sigma-2 receptor preferring ligands with anticocaine activity. J. Med. Chem. 2008, 51, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Poupaert, J.H.; Carato, P.; Colacino, E. 2(3H)-benzoxazolone and bioisosters as “privileged scaffold” in the design of pharmacological probes. Curr. Med. Chem. 2005, 12, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Diouf, O.; Carato, P.; Depreux, P.; Bonte, J.P.; Caignard, D.H.; Guardiola-Lemaître, B.; Rettori, M.C.; Belzung, C.; Lesieur, D. 5-HT1A and 5-HT2A ligands with anxiolytic and antipanic-like properties. Bioorg. Med. Chem. Lett. 1997, 7, 2579–2584. [Google Scholar] [CrossRef]
- Diouf, O.; Carato, P.; Lesieur, I.; Rettori, M.C.; Caignard, D.H. Synthesis and pharmacological evaluation of novel 4-(4-fluorobenzoyl)piperidine derivatives as mixed 5-HT1A/5-HT2A/D2 receptor ligands. Eur. J. Med. Chem. 1999, 34, 69–73. [Google Scholar] [CrossRef]
- Carato, P.; Depreux, D.; Lesieur, D.; Millan, M.; Newman-Tancredi, A.; Rettori, M.C.; Caignard, D.H. Synthesis and binding studies on a new series of arylpiperazino benzazol-2-one and benzoxazin-3-one derivatives as selective D4 ligands. Drug Des. Discov. 2000, 17, 173–181. [Google Scholar] [PubMed]
- Lesieur, D.; Delmas, E.; Yous, S.; Depreux, P.; Guillaumet, G.; Dacquet, C.; Levens, N.; Boutin, J.; Bennejean, C.; Renard, P. Preparation of Novel Heterocyclic Derivatives and Pharmaceutical Composition Containing Them as Hypoglycemic Agents. FR 2804431, 3 August 2001. [Google Scholar]
- Lesieur, D.; Blanc-Delmas, E.; Bennejean, C.; Chavatte, P.; Guillaumet, G.; Dacquet, C.; Levens, N.; Boutin, J.; Renard, P. Preparation of Azolylalkylphenylalkylindolinones as Hypoglycemic Andhypolipidemic Agents. FR 2830012, 28 March 2003. [Google Scholar]
- Takechi, N.; Ait-Mohand, S.; Medebielle, M.; Dolbier, W.R., Jr. Nucleophilic trifluoromethylation of acyl chlorides using the trifluoromethyl iodide/TDAE reagent. Tetrahedron Lett. 2002, 43, 4317–4319. [Google Scholar] [CrossRef]
- Pooput, C.; Médebielle, M.; Dolbier, W.F., Jr. A New and efficient method for the synthesis of trifluoromethylthio- and selenoethers. Org. Lett. 2004, 6, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Pooput, C.; Dolbier, W.F., Jr.; Médebielle, M. Nucleophilic perfluoroalkylation of aldehydes, ketones, imines, disulfides, and diselenides. J. Org. Chem. 2006, 71, 3564–3568. [Google Scholar] [CrossRef] [PubMed]
- Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Original reaction of p-nitrobenzyl chloride with aldehydes using tetrakis(dimethylamino)ethylene (TDAE). Tetrahedron Lett. 2003, 44, 6433–6435. [Google Scholar] [CrossRef]
- Giuglio-Tonolo, G.; Terme, T.; Médebielle, M.; Vanelle, P. Nitrobenzylation of α-carbonyl ester derivatives using TDAE approach. Tetrahedron Lett. 2004, 45, 5121–5124. [Google Scholar] [CrossRef]
- Amiri-Attou, O.; Terme, T.; Vanelle, P. Functionalization of 6-nitrobenzo[1,3]dioxole with carbonyl compounds via TDAE methodology. Molecules 2005, 10, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Montana, M.; Crozet, M.D.; Castera-Ducros, C.; Terme, T.; Vanelle, P. Rapid synthesis of new azaheterocyclic hydroxymalonate derivatives using TDAE approach. Heterocycles 2008, 75, 925–932. [Google Scholar] [CrossRef]
- Nadji-Boukrouche, A.R.; Khoumeri, O.; Terme, T.; Liacha, M.; Vanelle, P. Original TDAE reactivity in benzoxa- and benzothiazolone series. ARKIVOC 2010, 10, 358–370. [Google Scholar] [CrossRef]
- Montana, M.; Terme, T.; Vanelle, P. Original synthesis of oxiranes via TDAE methodology: reaction of 2-(dibromomethyl)quinoxaline with aromatic aldehydes. Tetrahedron Lett. 2005, 46, 8373–8376. [Google Scholar] [CrossRef]
- Montana, M.; Terme, T.; Vanelle, P. Original synthesis of α-chloro ketones in aza heterocyclic series using TDAE approach. Tetrahedron Lett. 2006, 47, 6573–6576. [Google Scholar] [CrossRef]
- Khoumeri, O.; Montana, M.; Terme, T.; Vanelle, P. First TDAE approach in quinonic series: Synthesis of new 2-substituted 1,4-dimethoxy-9,10-anthraquinones. Tetrahedron 2008, 64, 11237–11242. [Google Scholar] [CrossRef]
- Vanelle, P.; Maldonado, J.; Madadi, N.; Gueiffier, A.; Chapat, J.-P.; Crozet, M.P. SRN1 reactions in imidazo[1,2-a]pyridine series. Tetrahedron Lett. 1990, 31, 3013–3016. [Google Scholar] [CrossRef]
- Delmas, F.; Gasquet, M.; Timon-David, P.; Madadi, N.; Vanelle, P.; Vaille, A.; Maldonado, J. Synthesis and in vitro anti-protozoan activity of new 5-nitrothiophene oxime ether derivatives. Eur. J. Med. Chem. 1993, 28, 23–27. [Google Scholar] [CrossRef]
- Gellis, A.; Vanelle, P.; Kaafarani, M.; Benakli, K.; Crozet, M.P. Synthesis and SRN1 reactions of nitrothiazoles. Tetrahedron 1997, 53, 5471–5484. [Google Scholar] [CrossRef]
- Crozet, M.D.; Botta, C.; Gasquet, M.; Curti, C.; Remusat, V.; Hutter, S.; Chapelle, O.; Azas, N.; de Méo, M.; Vanelle, P. Lowering of 5-nitroimidazole’s mutagenicity: Towards optimal antiparasitic pharmacophore. Eur. J. Med. Chem. 2009, 44, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.A.; Burgess, A.G.; Krauer, K.G.; Eckmann, L.; Vanelle, P.; Crozet, M.D.; Gillin, F.D.; Upcroft, P.; Upcroft, J.A. A new-generation 5-nitroimidazole can induce highly metronidazole-resistant Giardia lamblia in vitro. Int. J. Antimicrob. Agents 2010, 36, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Khoumeri, O.; Crozet, M.D.; Terme, T.; Vanelle, P. Original TDAE application: Synthesis of 2-substituted-4,11-dimethoxy-anthra[2,3-b]furan-5,10-diones via intramolecular Buchwald reaction. Tetrahedron Lett. 2009, 50, 6372–6376. [Google Scholar] [CrossRef]
- Suzuki, F.; Trenbeath, S.; Gleim, R.D.; Sih, C.S. Total synthesis of anthracyclinones via intramolecular base-catalyzed cyclizations. J. Org. Chem. 1978, 43, 4159–4169. [Google Scholar] [CrossRef]
- Gökhan, N.; Köksal, M.; Küpeli, E.; Yeşilada, E.; Erdoğan, H. Some new Mannich bases of 5-methyl-2-benzoxazolinones with analgesic and anti-inflammatory activities. Turk. J. Chem. 2005, 29, 445–454. [Google Scholar]
- Amiri-Attou, O.; Terme, T.; Médebielle, M.; Vanelle, P. Original formation of benzyl benzoates by TDAE strategy. Tetrahedron Lett. 2008, 49, 1016–1020. [Google Scholar] [CrossRef]
- Juspin, T.; Laget, M.; Terme, T.; Azas, N.; Vanelle, P. TDAE assisted synthesis of new imidazo[2,1-b]thiazole derivatives as anti-infectious agents. Eur. J. Med. Chem. 2010, 45, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Montana, M.; Correard, F.; Khoumeri, O.; Esteve, M.-A.; Terme, T.; Vanelle, P. Synthesis of new quinoxalines containing an oxirane ring by the TDAE strategy and in vitro evaluation in neuroblastoma cell lines. Molecules 2014, 19, 14987–14998. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1, 2, 3, 5a–j, 6 and 7a–j are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadji-Boukrouche, A.R.; Khoumeri, O.; Terme, T.; Liacha, M.; Vanelle, P. TDAE Strategy in the Benzoxazolone Series: Synthesis and Reactivity of a New Benzoxazolinonic Anion. Molecules 2015, 20, 1262-1276. https://doi.org/10.3390/molecules20011262
Nadji-Boukrouche AR, Khoumeri O, Terme T, Liacha M, Vanelle P. TDAE Strategy in the Benzoxazolone Series: Synthesis and Reactivity of a New Benzoxazolinonic Anion. Molecules. 2015; 20(1):1262-1276. https://doi.org/10.3390/molecules20011262
Chicago/Turabian StyleNadji-Boukrouche, Aïda R., Omar Khoumeri, Thierry Terme, Messaoud Liacha, and Patrice Vanelle. 2015. "TDAE Strategy in the Benzoxazolone Series: Synthesis and Reactivity of a New Benzoxazolinonic Anion" Molecules 20, no. 1: 1262-1276. https://doi.org/10.3390/molecules20011262