
Energy and Molecules from Photochemical/Photocatalytic Reactions. An Overview

Abstract
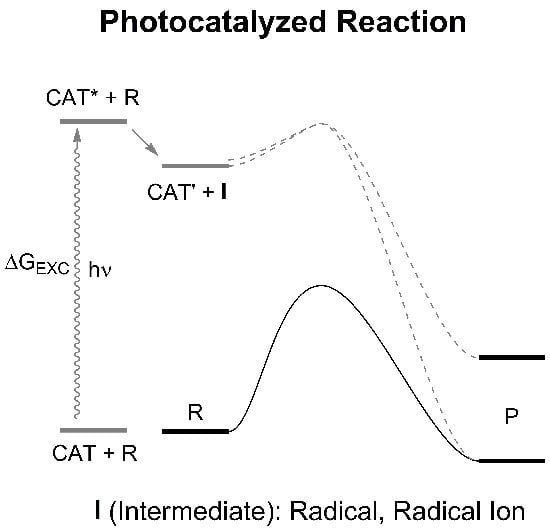
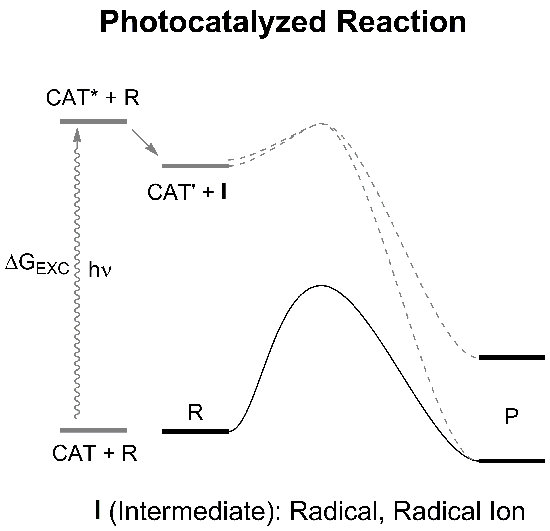
:
1. Introduction: Photochemistry for Synthesis and Energy










2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
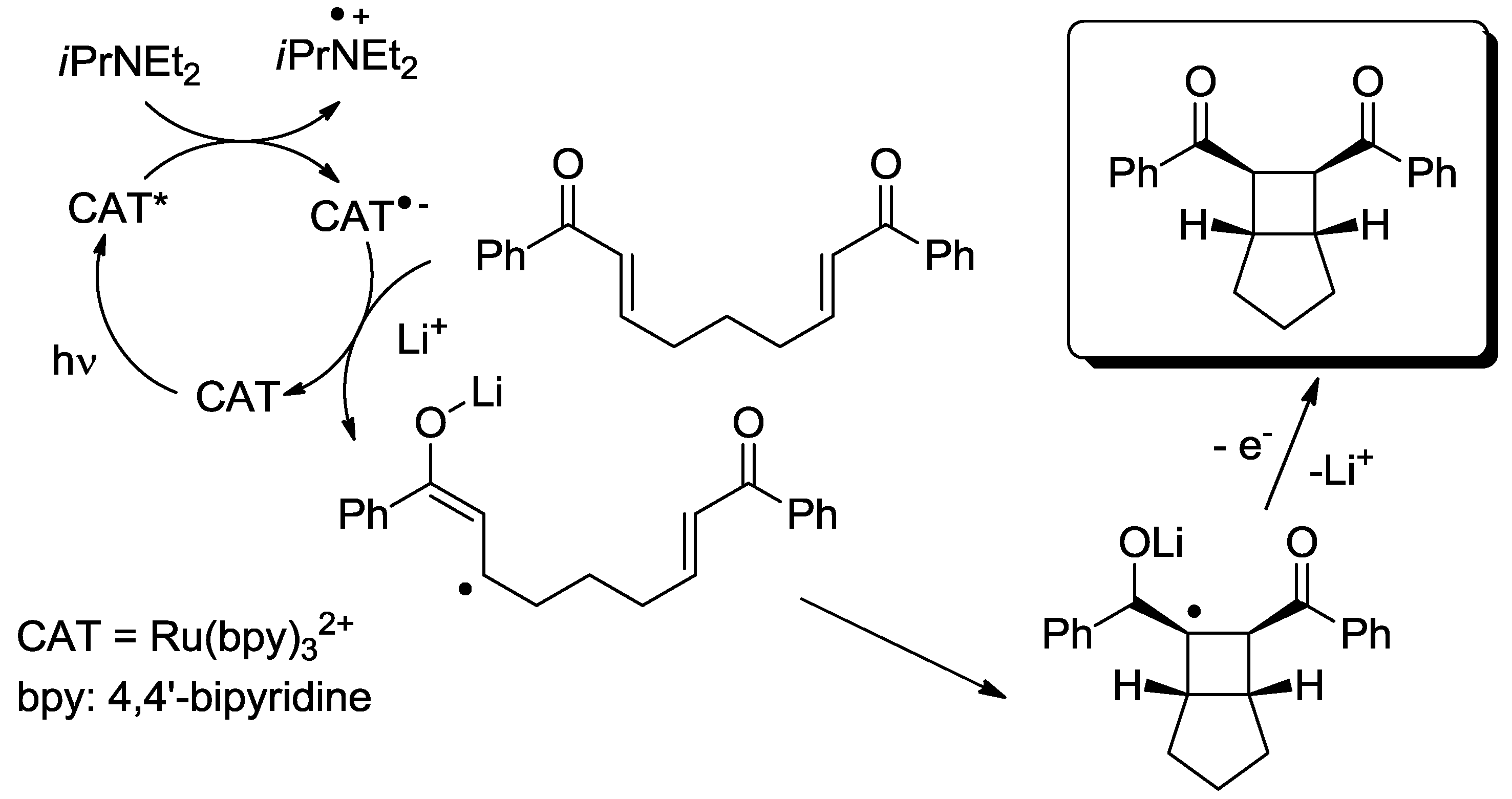{kind=link}
{kind=link}
| Entry | Reaction | Class of Reaction | ΔGEXC a | ΔG a |
|---|---|---|---|---|
| (λEXC) b | ( ΔH) a | |||
| 1 [28] |  | PS/PC | 53 (540) | −80.24 (−80.74) |
| 2 [30] |  | PS | 53 (540) | −11.52 (−25.16) |
| 3 [31] |  | P | 94 (304) | −3.05 (−13.99) |
| 4 [32] |  | PC | 73 (392) | −11.22 (−22.05) |
| 5 [15,33] |  | PC | 73 (392) | −17.00 (−28.89) |
| 6 [34] |  | PS | 92 (311) | −18.34 (−31.13) |
| 7 [35] |  | P | 95 (301) | −19.31 (−32.89) |
| 8 [36] |  | P | 77 (371) | −18.55 (−32.75) |
| 9 [37] |  | P | 95 (301) | −17.91 (−6.15) |
| 10 [38] |  | PC c | 58 (493) | −6.91 (−20.86) |
| 11 [39] |  | P | 119 (240) | 3.74 (0.27) |
| 12 [40] |  | PS | 87 (330) | 24.63 (24.28) |
| 13 [41] |  | PS | 92 (311) | 19.86 (18.43) |
| 14 [42] |  | PC | 89 (321) | −8.72 (−12.90) |
| 15 [43] |  | P | − | −17.92 (−31.43) |
| 16 [44] |  | P | 94 (304) | 20.75 (7.53) |
| 17 [45] |  | PS | 84 (340) | 8.71 (8.16) |
| 18 [46] |  | PS | 92 (311) | 9.60 (9.57) |
| 19 [47] |  | P | − | −24.25 (−24.71) |
| 20 |  | - | − | −107.2 (−113.5) |
| 21 |  | - | − | −193.6 (−193.9) |
| 22 [48] |  | PC | 67 (427) | −5.77 (1.40) |


3. Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ciamician, G. Actions chimiques de la lumière. Bull. Soc. Chim. Fr. 1908, 3–4, i–xxvii. [Google Scholar]
- Albini, A.; Fagnoni, M. Green chemistry and photochemistry were born at the same time. Green Chem. 2004, 6, 1–6. [Google Scholar]
- Albini, A.; Fagnoni, M. 1908: Giacomo Ciamician and the concept of green chemistry. ChemSusChem 2008, 1, 63–66. [Google Scholar]
- Albini, A.; Dichiarante, V. The belle époque of photochemistry. Photochem. Photobiol. Sci. 2009, 8, 248–254. [Google Scholar]
- Paternò, E. Sintesi in chimica organica per mezzo della luce. Nota I. Introduzione. Gazz. Chim. Ital. 1914, 44, 31. [Google Scholar]
- D’Auria, M. Una polemica fra Paternò e Ciamician. Chim. Ind. (Milan) 2009, 106–109. [Google Scholar]
- Warburg, E. The transformation of energy in photochemical processes in gases. II. Sitzb. Preuss. Akad. Wiss. 1912, 746, 216–225. [Google Scholar]
- Benrath, A. On pure and combined photochemical reactions. Z. Phys. Chem. 1910, 74, 115–124. [Google Scholar]
- IUPAC. Compendium of Chemical Terminology. The Gold Book. Available online: http://goldbook.iupac.org/ (accessed on 5 November 2014).
- Prignano, A.L.; Trogler, W.C. Silica-Supported Bis(trialky1phosphine)platinum Oxalates. Photogenerated Catalysts for Hydrosilation of Olefins. J. Am. Chem. Soc. 1987, 109, 3586–3595. [Google Scholar]
- Arceo, E.; Jurberg, I.D.; Alvarez-Fernàndez, A.; Melchiorre, P. Photochemical activity of a key donor-acceptor complex can drive stereoselective catalytic α-alkylation of aldehydes. Nat. Chem. 2013, 3, 750–756. [Google Scholar]
- Minero, C.; Catozzo, F.; Pelizzetti, E. Role of Adsorption in Photocatalyzed Reactions of Organic Molecules in Aqueous TiO2 Suspensions. Langmuir 1992, 8, 481–486. [Google Scholar]
- Kamat, P.V. Photochemistry on Nonreactive and Reactive (Semiconductor) Surfaces. Chem. Rev. 1992, 93, 267–300. [Google Scholar]
- Gerischer, H. Photochemistry of adsorbed species. Faraday Discuss. Chem. Soc. 1974, 58, 219–236. [Google Scholar]
- Protti, S.; Ravelli, D.; Fagnoni, M.; Albini, A. Solar light-driven photocatalyzed alkylations. Chemistry on the window ledge. Chem. Commun. 2009, 7351–7353. [Google Scholar]
- Ravelli, D.; Protti, S.; Fagnoni, M.; Albini, A. Visible Light Photocatalysis. A Green Choice? Curr. Org. Chem. 2013, 17, 2366–2373. [Google Scholar]
- Buckel, W. Radical and Electron Recycling in Catalysis. Angew. Chem. Int. Ed. 2009, 48, 6779–6787. [Google Scholar]
- Zeitler, K. Photoredox Catalysis with Visible Light. Angew. Chem. Int. Ed. 2009, 48, 9785–9789. [Google Scholar]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar]
- Narayanam, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar]
- Teplý, F. Photoredox catalysis by [Ru(bpy)3]2+ to trigger transformations of organic molecules. Organic synthesis using visible-light photocatalysis and its 20th century roots. Collect. Czech. Chem. Commun. 2011, 76, 859–917. [Google Scholar]
- Xuan, J.; Xiao, W.-J. Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2012, 51, 6828–6838. [Google Scholar]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar]
- Ohtani, B. Photocatalysis by inorganic solid materials: Revisiting its definition, concepts, and experimental procedures. In Advances in Inorganic Chemistry, Inorganic Photochemistry; van Eldik, R., Stochel, G., Eds.; Academic Press, Elsevier Ltd.: London, UK, 2011; Volume 63, Chapter 10. [Google Scholar]
- Bethke, S.; Drandm, S.; Treptow, B.; Geiter, R. Strained hydrocarbons from cyclic diynes, preparation and reactivity. J. Phys. Org. Chem. 2002, 15, 484–489. [Google Scholar]
- Mills, A.; Le Hunte, S. An overview of semiconductor photocatalysis. J. Photochem. Photobiol. A Chem. 1997, 108, 1–35. [Google Scholar]
- Kisch, H. Semiconductor Photocatalysis for Organic Synthesis. In Advances in Photochemistry; Neckers, D.C., von Bünau, G., Jenks, V.S., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2007; Volume 26, p. 93. [Google Scholar]
- Oelgemöller, M.; Jung, C.; Ortner, J.; Mattay, J.; Zimmermann, E. Green photochemistry: Solar photooxygenations with medium concentrated sunlight. Green Chem. 2005, 7, 35–38. [Google Scholar]
- Oelgemöller, M.; Healy, N.; de Oliveira, L.; Jung, C.; Mattay, J. Green photochemistry: Solar-chemical synthesis of Juglone with medium concentrated sunlight. Green Chem. 2006, 8, 831–834. [Google Scholar]
- Wootton, R.C.R.; Fortt, R.; de Mello, A.J. A microfabricated nanoreactor for safe, continuous generation and use of singlet oxygen. Org. Proc. Res. Dev. 2002, 6, 187–189. [Google Scholar]
- Li, J.-T.; Yang, J.-H.; Han, J.-F.; Li, T.-S. Reductive coupling of aromatic aldehydes and ketones in sunlight. Green Chem. 2003, 5, 433–435. [Google Scholar]
- Dondi, D.; Fagnoni, M.; Albini, A. Tetrabutylammonium Decatungstate-Photosensitized Alkylation of Electrophilic Alkenes: Convenient Functionalization of Aliphatic C-H Bonds. Chem. Eur. J. 2006, 12, 4153–4163. [Google Scholar]
- Esposti, S.; Dondi, D.; Fagnoni, M.; Albini, A. Acylation of Electrophilic Olefins through Decatungstate-Photocatalyzed Activation of Aldehydes. Angew. Chem. Int. Ed. 2007, 46, 2531–2534. [Google Scholar]
- Elad, D.; Rokach, J. The Light-Induced Amidation of Terminal Olefins. J. Org. Chem. 1964, 29, 1855–1859. [Google Scholar]
- Lazzaroni, S.; Protti, S.; Fagnoni, M.; Albini, A. Photoinduced Three-Component Reaction: A Convenient Access to 3-Arylacetals or 3-Arylketals. Org. Lett. 2009, 11, 349–352. [Google Scholar]
- Schiel, C.; Oelgemöller, M.; Ortner, J.; Mattay, J. Green photochemistry: The solar-chemical “Photo-Friedel-Crafts acylation” of quinones. Green Chem. 2001, 3, 224–228. [Google Scholar]
- Dichiarante, V.; Fagnoni, M.; Albini, A. Eco-friendly hydrodehalogenation of electron-rich aryl chlorides and fluorides by photochemical reaction. Green Chem. 2009, 11, 942–945. [Google Scholar]
- Du, J.; Yoon, T.P. Crossed Intermolecular [2+2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis. J. Am. Chem. Soc. 2009, 131, 14604–14605. [Google Scholar]
- Büchi, G.; Goldman, I.M. Photochemical Reactions. VII. The Intramolecular Cyclization of Carvone to Carvonecamphor. J. Am. Chem. Soc. 1957, 79, 4741–4748. [Google Scholar]
- Grutsch, P.A.; Kutal, C. Use of copper(I) phosphine compounds to photosensitize the valence isomerization of norbornadiene. J. Am. Chem. Soc. 1977, 99, 6460–6463. [Google Scholar]
- Eaton, P.E.; Or, Y.S.; Branca, S.J. Pentaprismane. J. Am. Chem. Soc. 1981, 103, 2134–2136. [Google Scholar]
- Langer, K.; Mattay, J. Stereoselective Intramolecular Copper(I)-Catalyzed [2+2]-Photocycloadditions. Enantioselective Synthesis of (+)- and (−)-Grandisol. J. Org. Chem. 1996, 60, 7256–7266. [Google Scholar]
- Hook, B.D.A.; Dohle, W.; Hirst, P.R.; Pickworth, M.; Berry, M.B.; Booker-Milburn, K.I. A Practical Flow Reactor for Continuous Organic Photochemistry. J. Org. Chem. 2005, 70, 7558–7564. [Google Scholar]
- D’Auria, M.; Racioppi, R.; Viggiani, L. Paternò-Büchi reaction between furan and heterocyclic aldehydes: Oxetane formation vs. metathesis. Photochem. Photobiol. Sci. 2010, 9, 1134–1138. [Google Scholar]
- Armesto, D.; Ortiz, M.J.; Agarrabeitia, A.R.; El-Boulifi, N. Efficient photochemical synthesis of 2-vinylcyclopropanecarbaldehydes, precursors of cyclopropane components present in pyrethroids, by using the oxa-di-π-methane rearrangement. Tetrahedron 2010, 66, 8690–8697. [Google Scholar]
- Hsu, D.-S.; Chou, Y.-Y.; Tung, Y.-S.; Liao, C.-C. Photochemistry of Tricyclo[5.2.2.02,6]undeca-4,10-dien-8-ones: An Efficient General Route to Substituted Linear Triquinanes from 2-Methoxyphenols. Total Synthesis of (±)-Δ9(12)-Capnellene. Chem. Eur. J. 2010, 16, 3121–3131. [Google Scholar]
- Podgorsêk, A.; Stavber, S.; Zupan, M.; Iskra, J. Visible light induced “on water” benzylic bromination with N-bromosuccinimide. Tetrahedron Lett. 2006, 47, 1097–1099. [Google Scholar]
- Zhang, D.; Wu, L.-Z.; Zhou, L.; Han, X.; Yang, Q.-Z.; Zhang, L.-P.; Tung, C.-H. Photocatalytic Hydrogen Production from Hantzsch 1,4-Dihydropyridines by Platinum(II) Terpyridyl Complexes in Homogeneous Solution. J. Am. Chem. Soc. 2004, 126, 3440–3441. [Google Scholar]
- Fagnoni, M.; Albini, A. Photochemically-Generated Intermediates in Synthesis. Wiley: Hoboken, NJ, USA, 2013; Chapter 8. [Google Scholar]
- Giese, B.; Gonzàlez-Gòmez, J.; Witzel, A. The Scope of Radical CC-Coupling by the “Tin Method”. Angew. Chem. Int. Ed. 1984, 23, 69–70. [Google Scholar]
- Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A. Photocatalysis for the Formation of the C-C Bond. Chem. Rev. 2007, 107, 2725–2756. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Version D.01; Gaussian, Inc.: Wallingford CT, USA, 2009. [Google Scholar]
- Hoffmann, N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar]
- Dichiarante, V.; Protti, S. Photochemistry in ecosustanaible syntheses. Recent advances. In CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed.; Griesbeck, A.G., Oelgemöller, M., Ghetti, A., Eds.; Taylor and Francis: Boca Raton, FL, USA, 2012; Chapter 9. [Google Scholar]
- Protti, S.; Albini, A.; Serpone, N. Photocatalytic generation of solar fuels from the reduction of H2O and CO2: A look at the patent literature. Phys. Chem. Chem. Phys. 2014, 16, 19790–19827, and references therein. [Google Scholar]
- Sample Availability: Not apply.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravelli, D.; Protti, S.; Albini, A. Energy and Molecules from Photochemical/Photocatalytic Reactions. An Overview. Molecules 2015, 20, 1527-1542. https://doi.org/10.3390/molecules20011527
Ravelli D, Protti S, Albini A. Energy and Molecules from Photochemical/Photocatalytic Reactions. An Overview. Molecules. 2015; 20(1):1527-1542. https://doi.org/10.3390/molecules20011527
Chicago/Turabian StyleRavelli, Davide, Stefano Protti, and Angelo Albini. 2015. "Energy and Molecules from Photochemical/Photocatalytic Reactions. An Overview" Molecules 20, no. 1: 1527-1542. https://doi.org/10.3390/molecules20011527