
New Non-Toxic Semi-Synthetic Derivatives from Natural Diterpenes Displaying Anti-Tuberculosis Activity

Abstract
: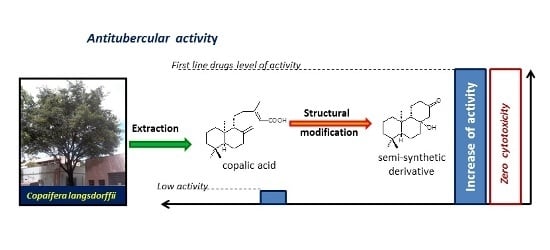
1. Introduction

2. Results and Discussion
2.1. Diterpenes
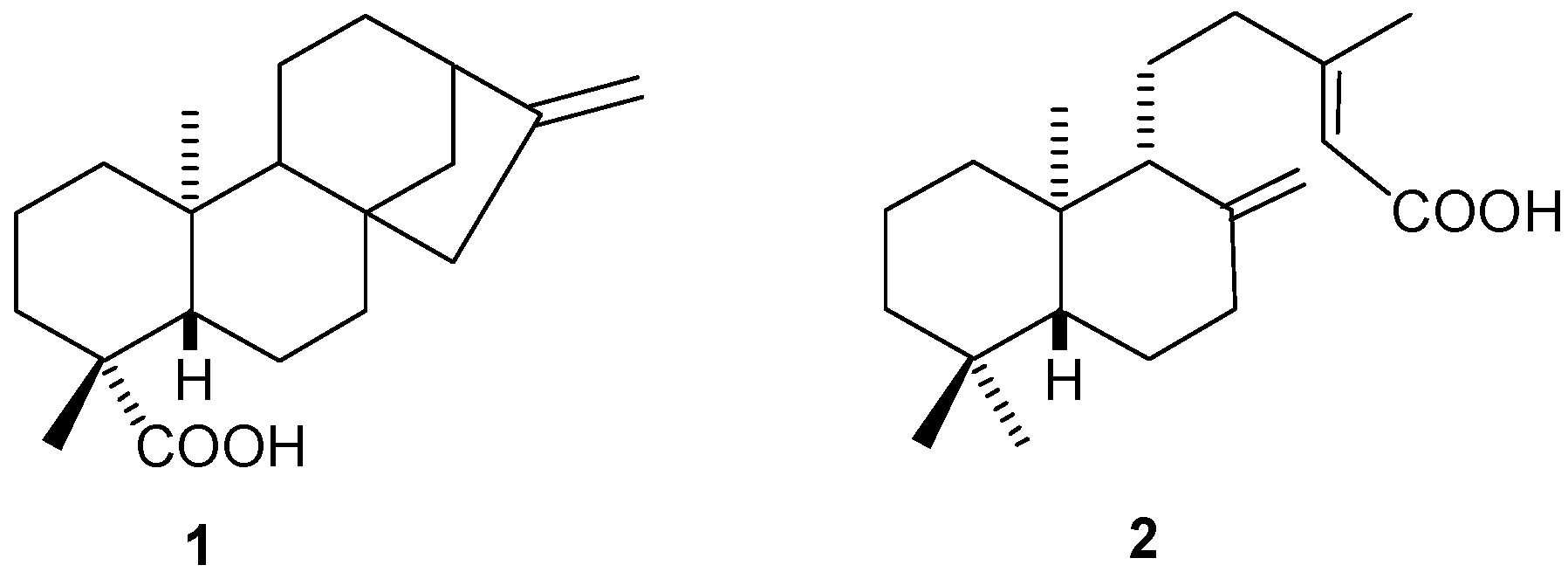
2.1.1. Kaurenoic Acid (1)
2.1.2. Copalic Acid (2)
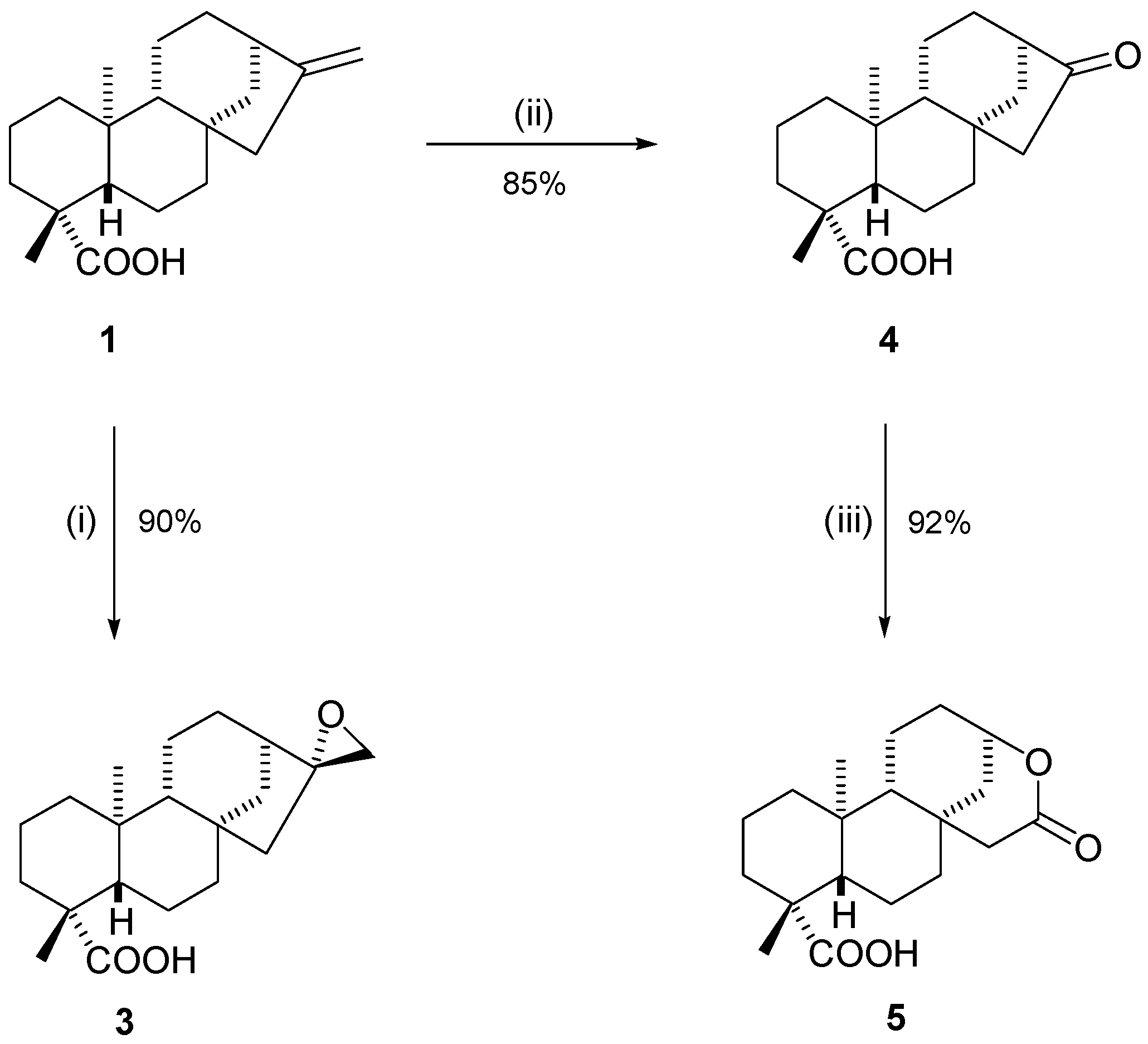
2.2. Synthetic Modifications


2.3. Biological Assays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity Level | Range of MIC Values (μg·mL−1) |
|---|---|
| Inactive | 125 and higher |
| Weak | 80 to 100 |
| Moderate | 31.25 to 64 |
| Significant | 12.5 to 25 |
| Promising | 10 or lower |
| Compound | Obtention | MIC (μg·mL−1) | MIC (μM) | Toxicity ** (Cell Viability) | |
|---|---|---|---|---|---|
| Origin Compound | Process | ||||
| 1 | ----- | Isolation | 125 | --- | ne |
| 2 | ----- | Isolation | 125 | --- | ne |
| 3 | 1 | Epoxidation | 100 | --- | ne |
| 4 | 1 | Ozonolisis | 100 | --- | ne |
| 5 | 4 | Baeyer-Villiger | 200 | --- | ne |
| 6 | 2 | Epoxidation | 25 | 78 | 97% |
| 7 | 2 | Ozonolisis | 12.5 | 47.3 | 96.7% |
| 8 | 7 | Aldol reaction | 6.25 | 25.36 | 100% |
| Ethambutol * [31] | ----- | --- | 1.64 | 7.22 | --- |
| Pyrazinamide [32] | ----- | --- | 3.12 | 25.34 | --- |
| Streptomycin [32] | ----- | --- | 6.25 | 10.75 | --- |
| Rifampicin [32] | ----- | --- | 0.12 | 0.15 | --- |
| Isoniazid | ----- | --- | 0.06 | 0.44 | --- |
3. Experimental Section
3.1. General Methods
3.2. General Methods for Substance Analysis
3.3. Anti-Tuberculosis Activity Assay
3.4. Natural Products Extraction
3.4.1. Kaurenoic Acid
3.4.2. Copalic Acid
3.5. Individual Experiments
3.6. Cytotoxic Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO) 2013 global tuberculosis report. Available online: http://www.who.int/tb/publications/factsheet_global.pdf?ua=1 (accessed on 12 May 2015).
- World Health Organization (WHO) health topics (tuberculosis). Available online: http://www.who.int/topics/tuberculosis/en/ (accessed on 12 May 2015).
- Zwerling, A; Behr, M.A.; Verma, A.; Brewer, T.; Menzies, D.; Pai, M. The BCG world atlas: A database of global BCG vaccination policies and practices. PLoS Med. 2011, 83, e1001012. [Google Scholar] [CrossRef] [PubMed]
- Rawat, D.S. Antituberculosis drug research: A critical overview. Med. Res. Rev. 2013, 33, 693–764. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Montanari, C.A.; Bolzani, V.S. Drug design based on natrural products. Quim. Nova. 2001, 24, 105–111. [Google Scholar]
- Rates, S.M.K. Plants as source of drugs. Toxicon 2001, 39, 603–613. [Google Scholar] [CrossRef]
- Souza, A.B.; Martins, C.H.; Souza, M.G.; Furtado, N.A.; Heleno, V.C.; Sousa, J.P.; Rocha, E.M.; Bastos, J.K.; Cunha, W.R.; Veneziani, R.C.; et al. Antimicrobial activity of terpenoids from Copaifera langsdorffii Desf. against cariogenic bacteria. Phytoher. Res. 2011, 25, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.B.; Moreira, M.R.; Ambrósio, S.R.; Furtado, N.A.; Cunha, W.R.; Heleno, V.C.; Silva, A.N.; Simão, M.R.; Rocha, E.M.; Marttins, C.H.; et al. Evaluation of ent-kaurenoic acid derivatives for their anticariogenic activity. Nat. Prod. Comm. 2011, 6, 777–780. [Google Scholar]
- Carvalho, T.C.; Simão, M.R.; Ambrósio, S.R.; Furtado, N.A.; Veneziani, R.C.; Heleno, V.C.; Da Costa, F.B.; Gomes, B.P.; Souza, M.G.; Reis, E.B.; et al. Antimicrobial activity of diterpenes from Viguiera arenaria against endodontic bacteria. Molecules 2011, 16, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.B.; Souza, M.G.; Moreira, M.A.; Moreira, M.R.; Furtado, N.A.; Martins, C.H.; Bastos, J.K.; Santos, R.A.; Heleno, V.C.; Ambrósio, S.R.; et al. Antimicrobial evaluation of diterpenes from Copaifera langsdorffii oleoresin against periodontal anaerobic bacteria. Molecules 2011, 16, 9611–9619. [Google Scholar] [CrossRef] [PubMed]
- Traves, P.G.; Pimentel-Santillana, M.; Rico, D.; Rodriguez, N.; Miethke, T.; Castrillo, A.; Theodorakis, E.A.; Martin-Sanz, P.; Palladino, M.A.; Bosca, L. Anti-inflammatory actions of acanthoic acid-related diterpenes involve activation of the Pl3K p110 γ/δ subunits and inhibition of NF-κB. Chem. Biol. 2014, 21, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Antonisamy, P.; Dhanasekaran, M.; Ignacimuthu, S.; Duraipandivan, V.; Balthazar, J.D.; Agastian, P; Kim, J.H. Gastroprotective effect of epoxy clerodane diterepene isolated from Tinospora cordifolia Miers (Guduchi) on indomethacin-induced gastric ulcer in rats. Phytomedicine 2014, 21, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Bohgaki, T.; Matsubara, T.; Shibuya, H. Indonesian medicinal plants XXIII. Chemical structures of two new migrated pimarane-type diterpenes, neoosthosiphols A and B, and suppressive effects on rat thoracic aorta of chemical constituents isolated from the leaves of Orthosiphon aristatus (Lamiaceae). Chem. Pharm. Bull. 2000, 48, 433–435. [Google Scholar] [PubMed]
- Ghisalberti, E.L. The biological activity of naturally occurring kaurane diterpenes. Fitoterapia 1997, 68, 303–325. [Google Scholar]
- Sass, D.C.; Heleno, V.C.; Lopes, J.L.; Constantino, M.G. One-step biomimetic conversion of a furanoheliangolide into an eremantholide using Stryker’s reagent. Tetrahedron Lett. 2008, 49, 3877–3880. [Google Scholar] [CrossRef]
- Sass, D.C.; Heleno, V.C.; Morais, G.O.; Lopes, J.L.; Lopes, N.P.; Constantino, M.G. Selectivity in reduction of natural furanoheliangolides with Striker’s reagent. Org. Biomol. Chem. 2011, 9, 6148–6153. [Google Scholar] [CrossRef] [PubMed]
- Sass, D.C.; Morais, G.O.; Miranda, R.A.; Magalhães, L.G.; Cunha, W.R.; Santos, R.A.; Arakawa, N.S.; Da Costa, F.B.; Constantino, M.G.; Heleno, V.C. Structurally modified natural sesquitepene lactones constitute effective and less toxic schistosomicidal compounds. Org. Biomol. Chem. 2014, 12, 7957–7964. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.B.; O’Brien, T.E.; Moore, J.T.; Anderson, D.E.; Foss, M.H.; Weibel, D.B.; Ames, J.B.; Shaw, J.T. The synthesis and antimicrobial activity of heterocyclic derivatives of totarol. ACS Med. Chem. Lett. 2012, 3, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.G.; Ni, T.F.; Gao, W.; He, Y.; Wang, Y.Y.; Cui, H.W.; Yang, C.G.; Qiu, W.W. The synthesis and antibacterial activity of pyrazole-fused tricyclic diterpenes derivatives. Eur. J. Med. Chem. 2015, 90, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Mang, C.; Jakupovic, S.; Schunk, S.; Ambrosi, H.D.; Schwarz, O.; Jakupovic, J. Natural products in combinatorial chemistry: An andrographolide-based Library. J. Comb. Chem. 2005, 8, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, O.; Jakupovic, S.; Ambrosi, H.D.; Haustedt, L.O.; Mang, C.; Müller-Kuhrt, L. Natural Products in parallel chemistry—Novel 5-lipoxygenase inhibitors from BIOS-based libraries starting from α-santonin. J. Comb. Chem. 2007, 9, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Batista, R.; García, P.A.; Castro, M.A.; del Corral, J.M.; Speziali, N.L.; Varotti, F.P.; de Paula, R.C.; García-Fernández, L.F.; Francesch, A.; Feliciano, A.S.; Oliveira, A.B. Synthesis, cytotoxicity and antiplasmodial activity of novel ent-kaurane derivatives. Eur. J. Med. Chem. 2013, 62, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Hueso-Falcón, I.; Girón, N.; Velasco, P.; Amaro-Luis, J.M.; Ravelo, A.G.; Heras, B.; Hortelano, S.; Estevez-Braun, A. Synthesis and induction of apoptosis signaling pathway of ent-kaurane derivatives. Bioorg. Med. Chem. 2010, 18, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- van Wyk, A.W.; Lobb, K.A.; Caira, M.R.; Hoppe, H.C.; Davies-Coleman, M.T. Transformations of manool. Try- and tetracyclic norditerpenoids with in vitro activity against Plasmodium falciparum. J. Nat. Prod. 2007, 70, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, B.L.; Burfitt, I.R.; Felkin, H.; Joly-Goudket, M.; Naemura, K.; Salomon, M.F.; Wenkert, E.; Wovkulich, P.M. Stereoselective conversión of keto groups into methyl vinyl quaternary carbón centers. J. Amer. Chem. Soc. 1978, 100, 6445–6450. [Google Scholar] [CrossRef]
- Rangaka, M.X.; Wilkinson, R.J.; Boulle, A.; Glynn, J.R.; Fielding, K.; van Cutsem, G.; Wilkinson, K.A.; Goliath, R.; Mathee, S.; Goemaere, E.; et al. Isoniazid plus antiretroviral therapy to prevent tuberculosis: A randomised double-blind, placebo-controlled trial. Lancet 2014, 384, 682–690. [Google Scholar] [CrossRef]
- Parikh, R.; Dalwadi, S. Preparation and characterization of controlled release poly-ɛ-caprolactonemicroparticles of isoniazid for drug delivery through pulmonary route. Powder Technol. 2014, 264, 158–165. [Google Scholar] [CrossRef]
- Cantrell, C.L; Franzblau, S.G.; Fischer, N.H. Antimycobacterial plant terpenoids. Planta Med. 2001, 67, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Stavrakov, G.; Valcheva, V.; Philipova, I.; Doytchinova, I. Novel camphane-based anti-tuberculosis agents with nanomolar activity. Eur. J. Med. Chem. 2013, 70, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Kalalbandi, V.K.; Seetharamappa, J.; Katrahalli, U.; Bhat, K.G. Synthesis, cristal studies, anti-tuberculosis and cytotoxic studies of 1-[(2E)-3-phenylprop-2-enoyl]-1H-benzimidazole derivatives. Eur. J. Med. Chem. 2014, 79, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Gültekin, Z.; Grainger, R.S.; Adams, H.; Spargo, P.L. (1R,3R)-2-Methylene-1,3-dithiolane 1,3-dioxide: A highly reactive and highly selective chiral ketene equivalent towards a broad range of dienes. J. Chem. Soc. Perkin Trans. 1998, 1, 2771–2781. [Google Scholar] [CrossRef]
- Palomino, J.C.; Martin, A.; Camacho, M.; Guerra, H.; Swings, J.; Portaels, F. Resazurin microtiter assay plate: Simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. [Google Scholar] [CrossRef]
- Pelletier, S.W.; Chokshi, H.P.; Desai, H.K. Separation of diterpenoid alkaloid mixtures using vacuum liquid chromatography. J. Nat. Prod. 1986, 49, 892–900. [Google Scholar] [CrossRef]
- Sample Availability: Samples are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matos, P.M.; Mahoney, B.; Chan, Y.; Day, D.P.; Cabral, M.M.W.; Martins, C.H.G.; Santos, R.A.; Bastos, J.K.; Page, P.C.B.; Heleno, V.C.G. New Non-Toxic Semi-Synthetic Derivatives from Natural Diterpenes Displaying Anti-Tuberculosis Activity. Molecules 2015, 20, 18264-18278. https://doi.org/10.3390/molecules201018264
Matos PM, Mahoney B, Chan Y, Day DP, Cabral MMW, Martins CHG, Santos RA, Bastos JK, Page PCB, Heleno VCG. New Non-Toxic Semi-Synthetic Derivatives from Natural Diterpenes Displaying Anti-Tuberculosis Activity. Molecules. 2015; 20(10):18264-18278. https://doi.org/10.3390/molecules201018264
Chicago/Turabian StyleMatos, Priscilla M., Brian Mahoney, Yohan Chan, David P. Day, Mirela M. W. Cabral, Carlos H. G. Martins, Raquel A. Santos, Jairo K. Bastos, Philip C. Bulman Page, and Vladimir C. G. Heleno. 2015. "New Non-Toxic Semi-Synthetic Derivatives from Natural Diterpenes Displaying Anti-Tuberculosis Activity" Molecules 20, no. 10: 18264-18278. https://doi.org/10.3390/molecules201018264
APA StyleMatos, P. M., Mahoney, B., Chan, Y., Day, D. P., Cabral, M. M. W., Martins, C. H. G., Santos, R. A., Bastos, J. K., Page, P. C. B., & Heleno, V. C. G. (2015). New Non-Toxic Semi-Synthetic Derivatives from Natural Diterpenes Displaying Anti-Tuberculosis Activity. Molecules, 20(10), 18264-18278. https://doi.org/10.3390/molecules201018264