
Synthesis of Novel β-Keto-Enol Derivatives Tethered Pyrazole, Pyridine and Furan as New Potential Antifungal and Anti-Breast Cancer Agents

 ,
, 
Abstract
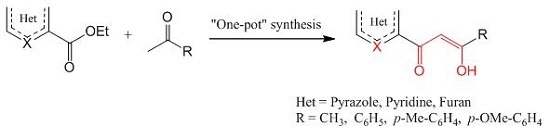
:
1. Introduction

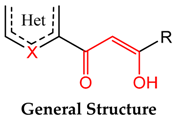
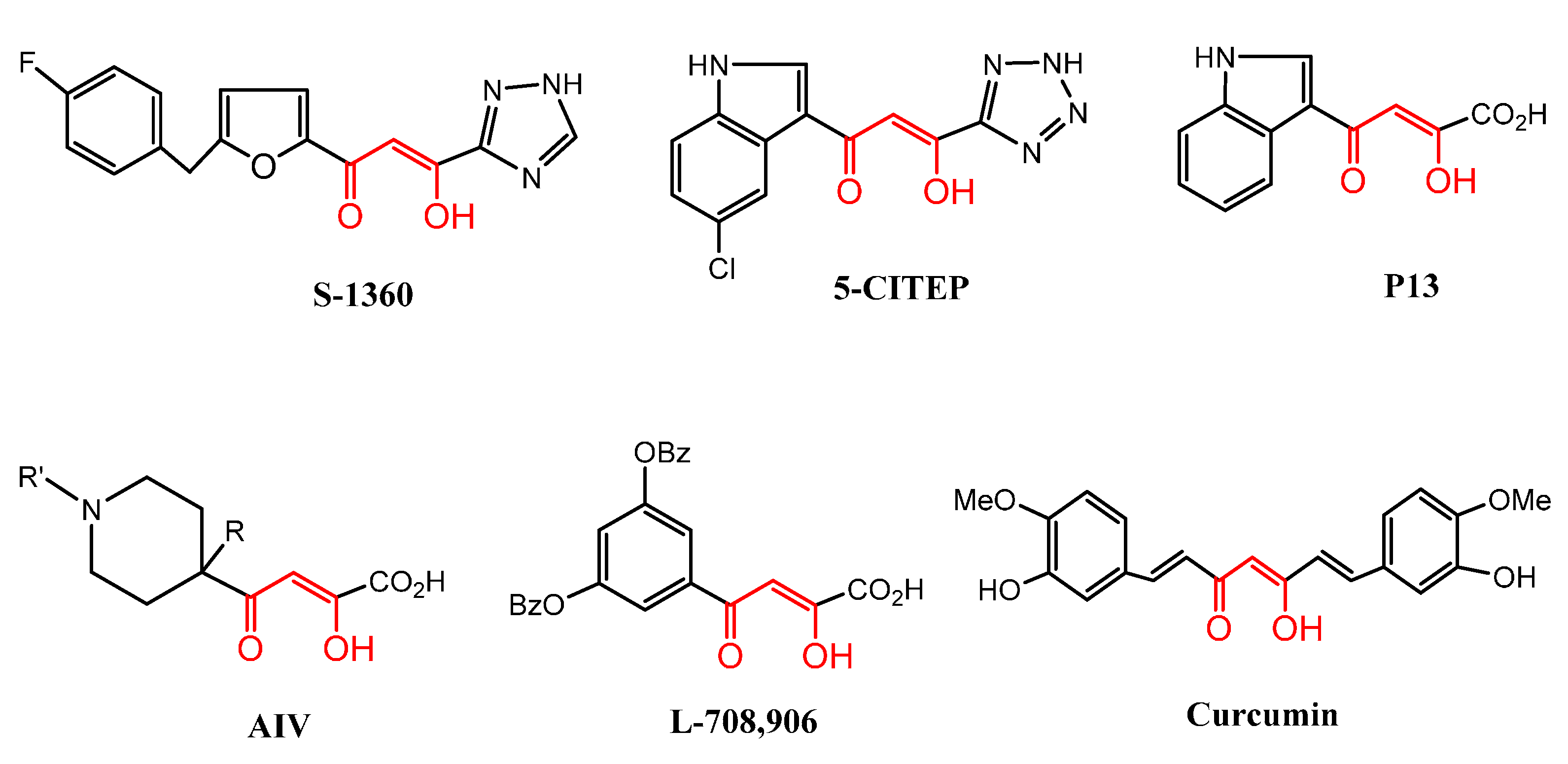
2. Results and Discussion
2.1. Chemistry


2.2. Biological Activities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Products | MDA-MB241 | Fusarium Oxysporum f.sp Albedinis | ||||
|---|---|---|---|---|---|---|
| No. | Heterocycles | R | IC50 (μg/mL) | IC50 (μM) | IC50 (μg/mL) | IC50 (μM) |
| 1 |  | CH3 | 46.20 | 256.38 | 0.01 | 0.055 |
| 2 |  | C6H5 | 44.33 | 183.00 | 12.83 | 53.39 |
| 3 | 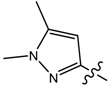 | p-Me-C6H4 | 21.95 | 85.65 | 142 | 554.03 |
| 4 | 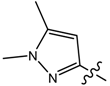 | p-MeO-C6H4 | 34.93 | 128.30 | 150 | 550.86 |
| 5 |  | CH3 | 47.00 | 288.04 | 0.013 | 0.079 |
| 6 | 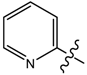 | C6H5 | 17.62 | 78.23 | 16.43 | 72.94 |
| 7 | 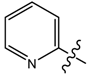 | p-Me-C6H4 | 128.67 | 537.8 | 35.80 | 149.62 |
| 8 |  | p-MeO-C6H4 | 28.97 | 113.50 | N/A * | N/A * |
| 9 | 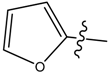 | CH3 | N/A * | N/A * | 0.014 | 0.092 |
| 10 |  | C6H5 | 18.79 | 87.72 | 68 .45 | 319.53 |
3. Experimental
3.1. General Information
3.2. General Procedure for the Synthesis of β-Keto-Enol Heterocycles
3.3. Anticancer Assays
3.4. Antibacterial and Antifungal Tests
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pommier, Y.; Johnson, A.A.; Marchand, C. Integrase inhibitors to treat HIV/AIDS. Nat. Rev. Drug Discov. 2005, 4, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Egbertson, S.S.; Anthony, N.J.; Summa, V. HIV integrase inhibitors: From diketo acids to heterocyclic templates: History of HIV integrase medicinal chemistry at Merck West Point and Merck Rome (IRBM) leading to discovery of raltegravir. In Pharmaceutical & Medicinal Chemistry; Neamati, N., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011; Chapter 14; pp. 197–230. [Google Scholar]
- Goldgur, Y.; Craigir, R.; Cohen, G.H.; Fujiwara, T.; Yoshinaga, T.; Fujishita, T.; Sugimoto, H.; Endo, T.; Murai, H.; Davies, D.R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: A platform for antiviral drug design. Proc. Natl. Acad. Sci. USA 1999, 96, 13040–13043. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Pluymers, W.; Pais, G.; Maele, B.V.; Pannecouque, C.; Fikkert, V.; Burke, J.T.R.; de Clercq, E.; Witvrouw, M.; Neamati, N.; Debyser, Z. Inhibition of human immunodeficiency virus type 1 integration by diketo derivatives. Antimicrob. Agents Chemother. 2002, 46, 3292–3297. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Fujii, S. Binding Mode prediction and inhibitor design of anti-influenza virus diketo acids targeting metalloenzyme RNA polymerase by molecular docking. Bioinformation 2011, 6, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, A.; Yamakoshi, H.; Hongo, S.; Kanoh, N.; Shibata, H.; Iwabuchi, Y. Structure-activity relationships of the antitumor C5-curcuminoid GO-Y030. Molecules 2015, 20, 15374–15391. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Thomas, S.G.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Sung, B.; Tharakan, S.T.; Misra, K.; Priyadarsini, I.K.; Rajasekharan, K.N.; et al. Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol. 2008, 76, 1590–1611. [Google Scholar] [CrossRef] [PubMed]
- Minassi, A.; Sanchez-Duffhues, G.; Collado, J.A.; Munoz, E.; Appendino, G. Dissecting the pharmacophore of curcumin. Which structural element is critical for which action? J. Nat. Prod. 2013, 76, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Cai, J.; Armstrong, J.; Herold, M.; Lu, Y.J.; Sun, A.; Snyder, J.P.; Liotta, D.C.; Jones, D.P.; Shoji, M. EF24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anti-Cancer Drug. 2005, 16, 263–275. [Google Scholar] [CrossRef]
- Tan, K.L.; Ali, A.; Du, Y.; Fu, H.; Jin, H.X.; Chin, T.M.; Khan, M.; Go, M.L. Synthesis and evaluation of bisbenzylidenedioxotetrahydrothiopranones as activators of endoplasmic reticulum (ER) stress signaling pathways and apoptotic cell death in acute promyelocytic leukemic cells. J. Med. Chem. 2014, 57, 5904–5918. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg. Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Li, X.; Chen, L.; Yang, S.; Wu, X.; Studer, E.; Gurley, E.; Hylemon, P.B.; Ye, F.; Li, Y.; et al. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Bioorg. Med. Chem. Lett. 2008, 18, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.-G.; Zhao, Y.; Ma, C.; Xu, X.-M.; Zhang, X.-M.; Huang, N.-Y.; He, H.-Q. Synthesis and anti-integrase evaluation of novel calix[4]arene derivatives containing the triazolyl 1,3-diketo moiety. Chin. Chem. Lett. 2014, 25, 737–740. [Google Scholar] [CrossRef]
- Song, W.-H.; Liu, M.-M.; Zhong, D.-W.; Zhu, Y.-L.; Bosscher, M.; Ye, D.-Y.; Yuan, Z.-H.; Zhou, L. Tetrazole and triazole as bioisosteres of carboxylic acid: Discovery of diketo tetrazoles and diketo triazoles as anti-HCV agents. Bioorg. Med. Chem. Lett. 2013, 23, 4528–4531. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Karthikeyan, C.; Solomon, V.R.; Moorthy, N.S.H.N.; Lee, H.; Sahu, K.; Deora, G.S.; Trivedi, P. Synthesis of some coumarinyl chalcones and their antiproliferative activity against breast cancer cell lines. Lett. Drug Des. Discov. 2011, 8, 308–311. [Google Scholar] [CrossRef]
- Radi, S.; Tighadouini, S.; Ben Hadda, T.; Akkurt, M.; Özdemir, N.; Sirajuddin, M.; Mabkhot, Y.N. Crystal structure of (2Z)-3-hydroxy-1-(1,5-dimethyl-1H-pyrazol-3-yl)but-2-en-1-one, C9H12N2O2. Z. Kristallogr. 2015. submitted. [Google Scholar]
- Riahi, A.; Wurster, M.; Lalk, M.; Lindequist, U.; Langer, P. Synthesis and antimicrobial activity of 4-hydroxy-4-(pyridyl)alk-3-en-2-ones. Bioorg. Med. Chem. 2009, 17, 4323–4326. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.E.; Borisov, E.V.; Lindon, J.C. Determination of the tautomeric equilibria of pyridoyl benzoyl β-diketones in the liquid and solid state through the use of deuterium isotope effects on 1H and 13C-NMR chemical shifts and spin coupling constants. Spectrochim. Acta A Mol. Spectrosc. 2015, 136, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Defresne, F.; Bouzin, C.; Guilbaud, C.; Dieu, M.; Delaive, E.; Michiels, C.; Raes, M.; Feron, O. PP 82 Pleiotropic influences of radio- and chemotherapy on auto-antibodies warrant caution for their use as biomarkers of tumor response: The anti-GRP78 paradigmatic example. Eur. J. Cancer 2011, 47, S16–S16. [Google Scholar] [CrossRef]
- Carrod, L.P.; Grady, F.D. Antibiotics and Chemotherapy, 3rd ed.; Churchill Livingstone: Edinburgh, UK, 1972; p. 477. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radi, S.; Tighadouini, S.; Feron, O.; Riant, O.; Bouakka, M.; Benabbes, R.; Mabkhot, Y.N. Synthesis of Novel β-Keto-Enol Derivatives Tethered Pyrazole, Pyridine and Furan as New Potential Antifungal and Anti-Breast Cancer Agents. Molecules 2015, 20, 20186-20194. https://doi.org/10.3390/molecules201119684
Radi S, Tighadouini S, Feron O, Riant O, Bouakka M, Benabbes R, Mabkhot YN. Synthesis of Novel β-Keto-Enol Derivatives Tethered Pyrazole, Pyridine and Furan as New Potential Antifungal and Anti-Breast Cancer Agents. Molecules. 2015; 20(11):20186-20194. https://doi.org/10.3390/molecules201119684
Chicago/Turabian StyleRadi, Smaail, Said Tighadouini, Olivier Feron, Olivier Riant, Mohammed Bouakka, Redouane Benabbes, and Yahia N. Mabkhot. 2015. "Synthesis of Novel β-Keto-Enol Derivatives Tethered Pyrazole, Pyridine and Furan as New Potential Antifungal and Anti-Breast Cancer Agents" Molecules 20, no. 11: 20186-20194. https://doi.org/10.3390/molecules201119684