
Algae Undaria pinnatifida Protects Hypothalamic Neurons against Endoplasmic Reticulum Stress through Akt/mTOR Signaling


Abstract
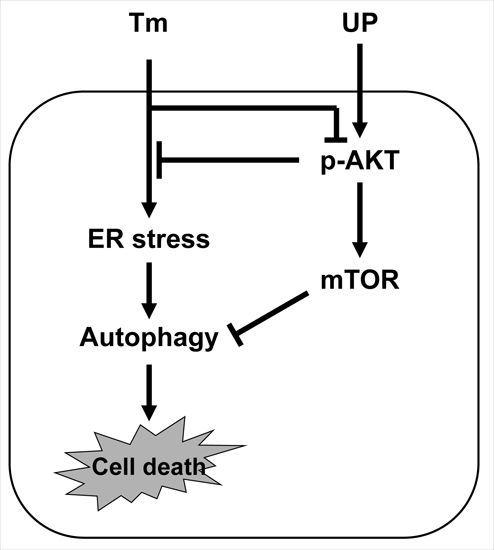
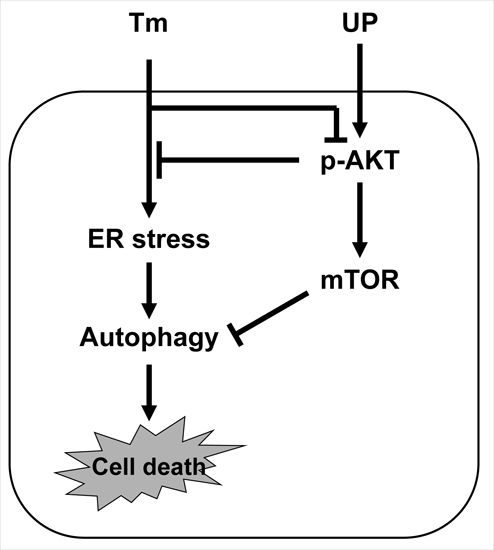
:
1. Introduction
2. Results
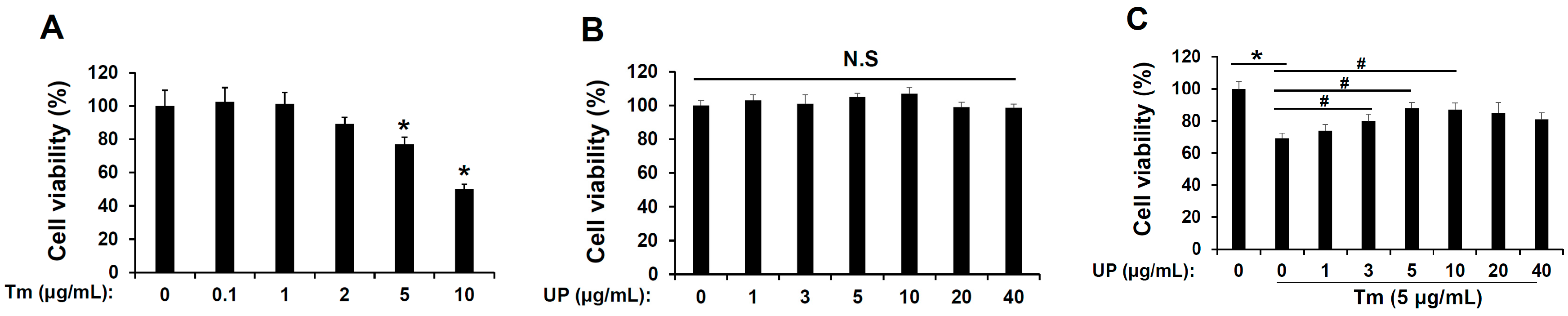
2.1. UP Has a Protective Effect in Tunicamycin-Induced Cell Death

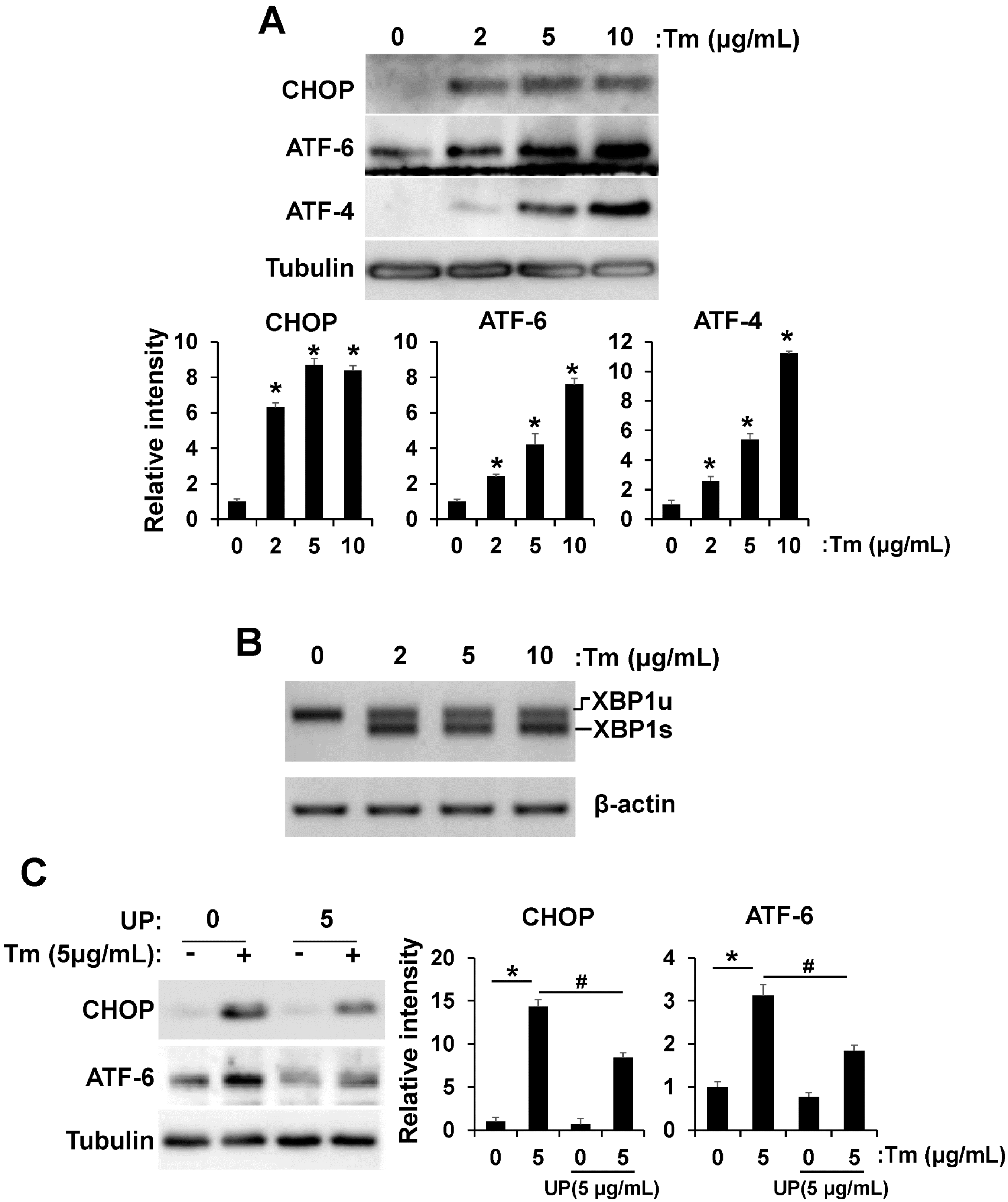
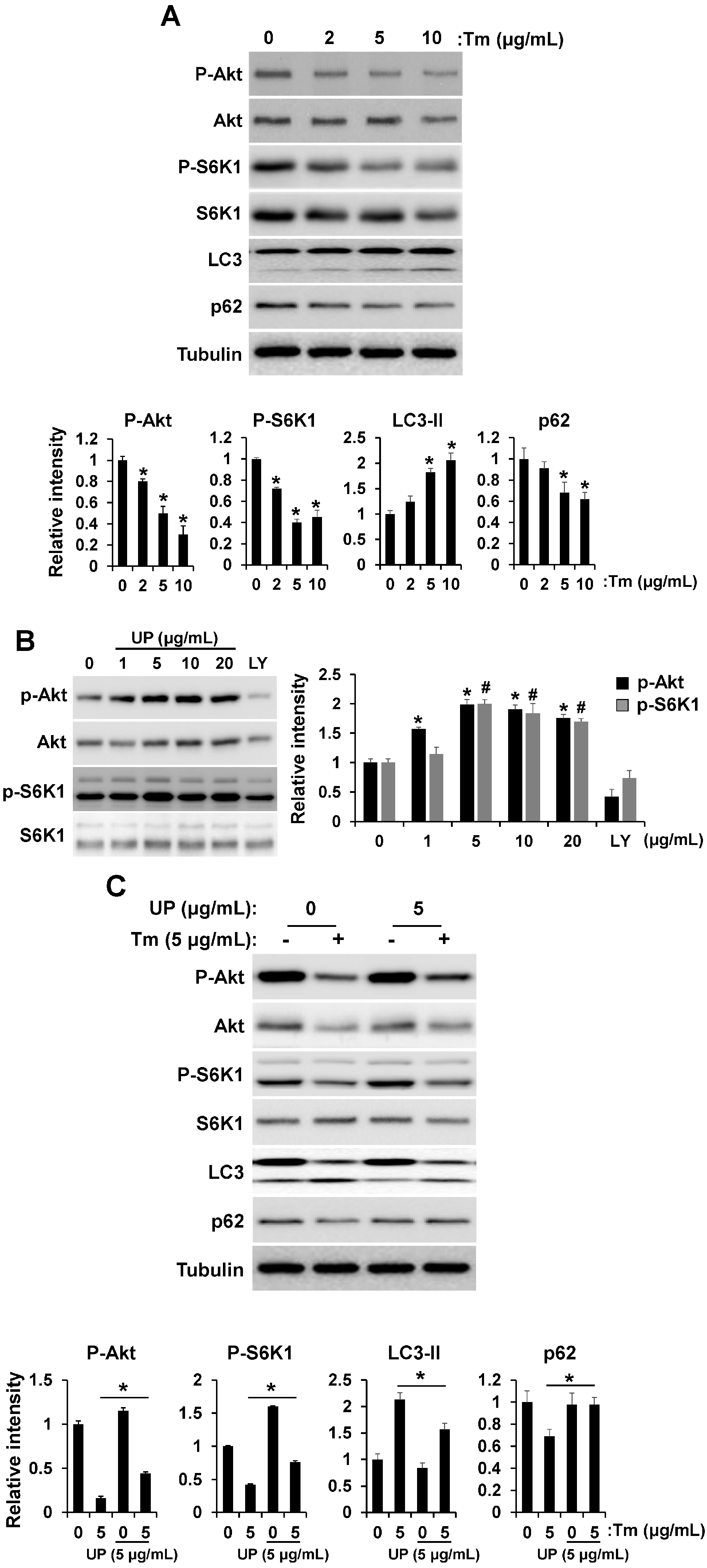
2.2. UP Attenuates ER Stress

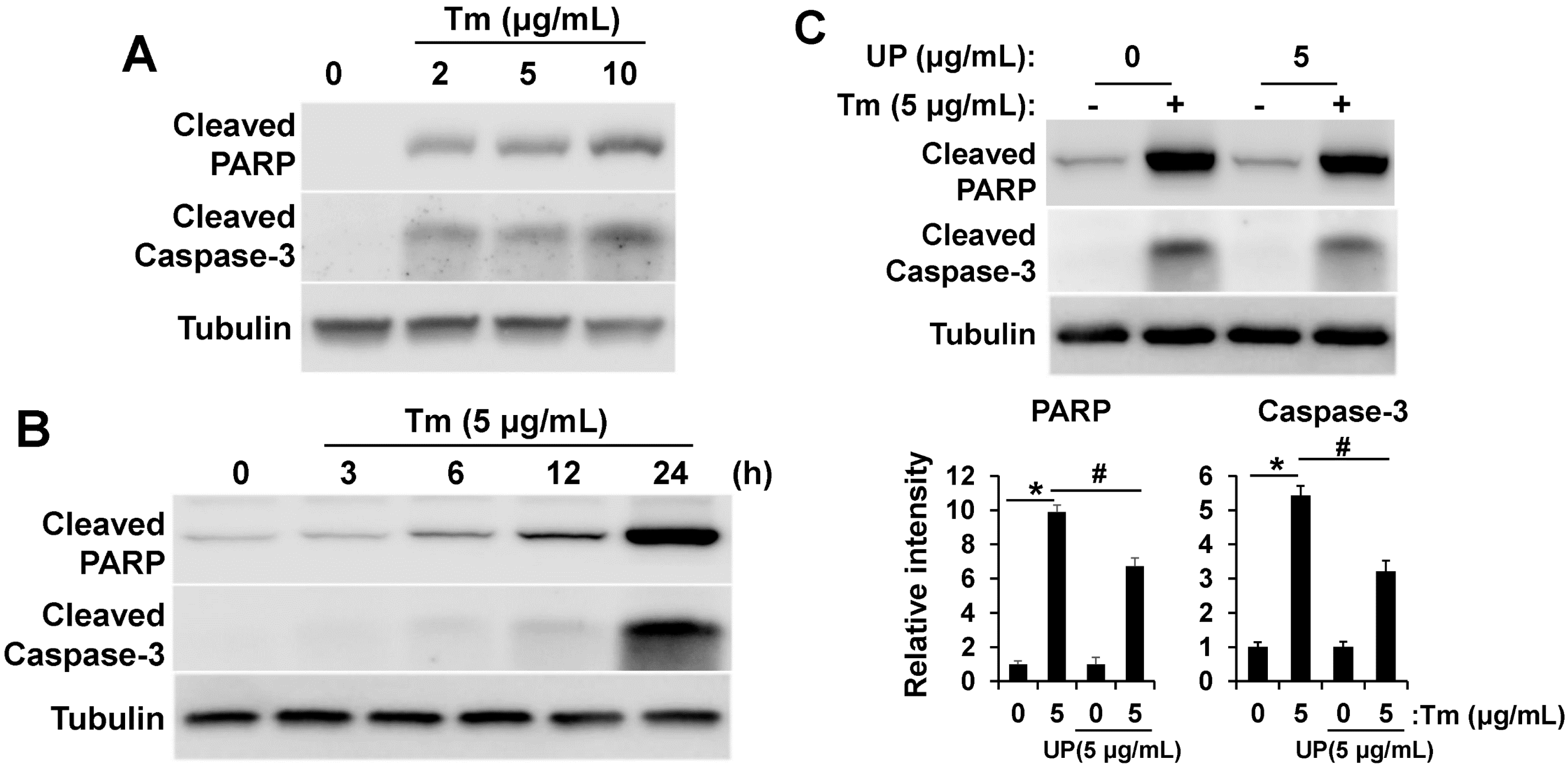
2.3. UP Attenuates ER Stress Induced Apoptotic Cell Death

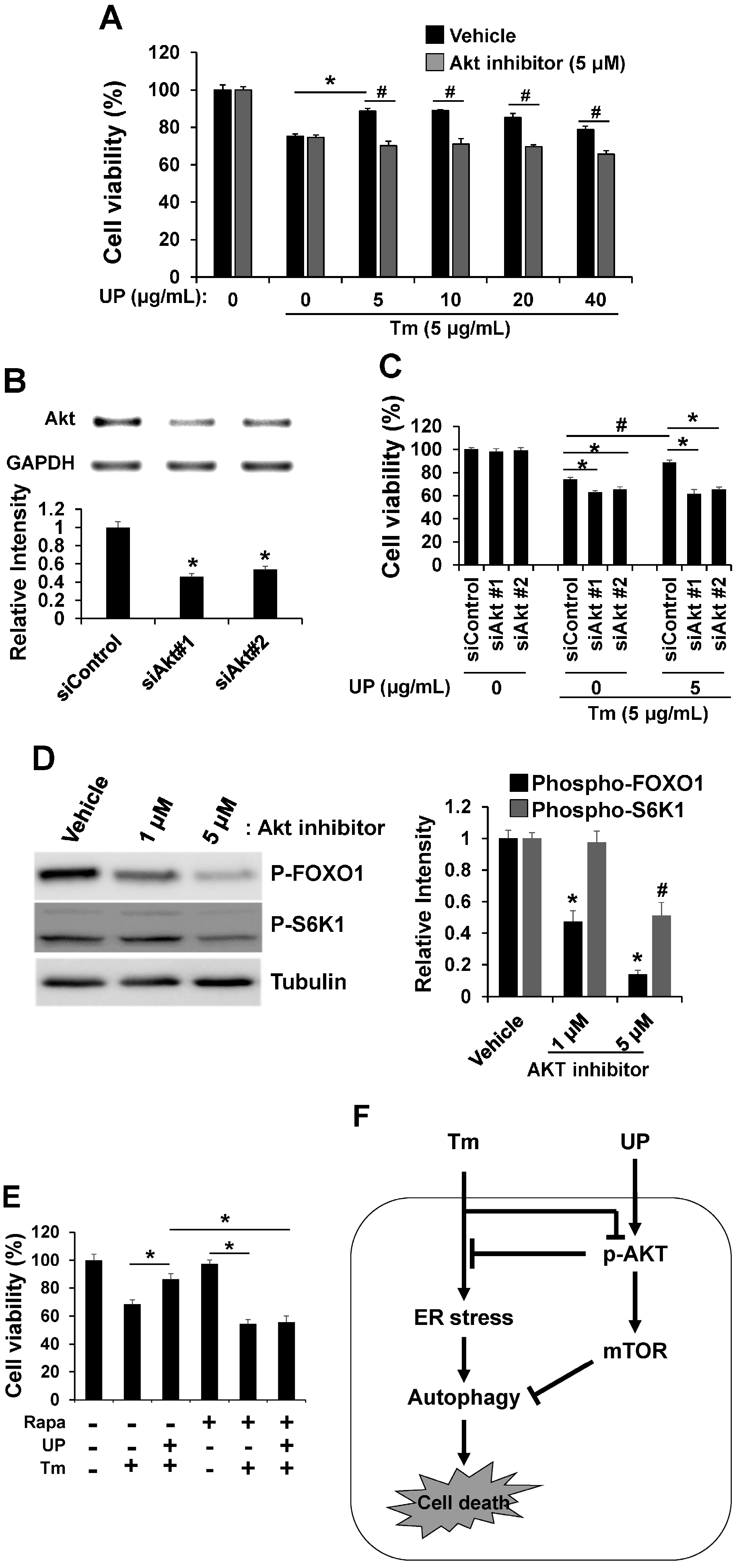
2.4. UP Increases Cell Viability via Akt/mTOR Signaling


3. Discussion
4. Experimental Section
4.1. Reagents and Cells
4.2. Preparation of Undaria pinnatifida Extract
4.3. Cell Viability Test
4.4. Transient Transfection with siRNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Genes | Symbols | GenBank No. | siRNA Sequences | |
|---|---|---|---|---|
| Control siRNA | Forward, 5’-ACGUGACACGUUCGGAGAAUU-3’ Reverse, 5’-UUCUCCGAACGUGUCACGUUU-3’ | |||
| Thymoma viral proto-oncogene (Akt) | ||||
| Thymoma viral proto-oncogene | akt | NM_009652 | #1 | Forward, 5’-GAACGAUGGCACCUUUAUUUU-3’ Reverse, 5’-AAUAAAGGUGCCAUCGUUCUU-3’ |
| #2 | Forward,5’-GGUUCUUUGCCAACAUCGUUU-3’ Reverse, 5’-ACGAUGUUGGCAAAGAACCUU-3’ | |||
4.5. Reverse Transcription-PCR
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001, 3, E255–E263. [Google Scholar] [CrossRef] [PubMed]
- Lupachyk, S.; Watcho, P.; Obrosov, A.A.; Stavniichuk, R.; Obrosova, I.G. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp. Neurol. 2013, 247, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Liu, T. Inflammatory cause of metabolic syndrome via brain stress and nf-kappab. Aging 2012, 4, 98–115. [Google Scholar] [PubMed]
- Denis, R.G.; Arruda, A.P.; Romanatto, T.; Milanski, M.; Coope, A.; Solon, C.; Razolli, D.S.; Velloso, L.A. Tnf-α transiently induces endoplasmic reticulum stress and an incomplete unfolded protein response in the hypothalamus. Neuroscience 2010, 170, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Won, J.C.; Jang, P.G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.Y.; Lee, K.U.; Kim, M.S. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Boo, H.J.; Hong, J.Y.; Kim, S.C.; Kang, J.I.; Kim, M.K.; Kim, E.J.; Hyun, J.W.; Koh, Y.S.; Yoo, E.S.; Kwon, J.M.; et al. The anticancer effect of fucoidan in pc-3 prostate cancer cells. Mar. Drugs 2013, 11, 2982–2999. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, J.; Chang, A.K.; Liu, B.; Yang, L.; Li, Q.; Wang, P.; Zou, X. Fucoidan extract derived from undaria pinnatifida inhibits angiogenesis by human umbilical vein endothelial cells. Phytomedicine 2012, 19, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Hannan, A.; Kang, J.Y.; Hong, Y.K.; Lee, H.; Choi, J.S.; Choi, I.S.; Moon, I.S. The marine alga gelidium amansii promotes the development and complexity of neuronal cytoarchitecture. Phytother. Res. 2013, 27, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kitamura, A.; Machida, H.; Watanabe, M.; Negishi, H.; Hiraoka, J.; Nakano, T. Effect of undaria pinnatifida (wakame) on the development of cerebrovascular diseases in stroke-prone spontaneously hypertensive rats. Clin. Exp. Pharmacol. Physiol. 2003, 30, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Michinaga, S.; Hisatsune, A.; Isohama, Y.; Katsuki, H. Orexin neurons in hypothalamic slice cultures are vulnerable to endoplasmic reticulum stress. Neuroscience 2011, 190, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. Er stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Schmelzle, T.; Hall, M.N. Tor, a central controller of cell growth. Cell 2000, 103, 253–262. [Google Scholar] [CrossRef]
- Hwang, H.J.; Jung, T.W.; Ryu, J.Y.; Hong, H.C.; Choi, H.Y.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, K.M.; Choi, D.S.; et al. Dipeptidyl petidase-iv inhibitor (gemigliptin) inhibits tunicamycin-induced endoplasmic reticulum stress, apoptosis and inflammation in h9c2 cardiomyocytes. Mol. Cell Endocrinol. 2014, 392, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mtor signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Liu, T. Hypothalamic inflammation: A double-edged sword to nutritional diseases. Ann. N. Y. Acad. Sci. 2011, 1243, E1–E39. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, S.; Zhang, H.; Zhang, G.; Ahmed, Z.; Wang, Y.; Cai, D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc. Natl. Acad. Sci. USA 2011, 108, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Han, Z.; Couvillon, A.D.; Exton, J.H. Critical role of endogenous akt/iaps and mek1/erk pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 2004, 279, 49420–49429. [Google Scholar] [CrossRef] [PubMed]
- Marte, B.M.; Downward, J. Pkb/akt: Connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem. Sci. 1997, 22, 355–358. [Google Scholar] [CrossRef]
- Hosoi, T.; Hyoda, K.; Okuma, Y.; Nomura, Y.; Ozawa, K. Akt up- and down-regulation in response to endoplasmic reticulum stress. Brain Res. 2007, 1152, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Wendel, H.G.; de Stanchina, E.; Fridman, J.S.; Malina, A.; Ray, S.; Kogan, S.; Cordon-Cardo, C.; Pelletier, J.; Lowe, S.W. Survival signalling by akt and eif4e in oncogenesis and cancer therapy. Nature 2004, 428, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Bachelder, R.E.; Lipscomb, E.A.; Shaw, L.M.; Mercurio, A.M. Integrin (alpha 6 beta 4) regulation of eif-4e activity and vegf translation: A survival mechanism for carcinoma cells. J. Cell. Biol. 2002, 158, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. Mtorc1 serves er stress-triggered apoptosis via selective activation of the ire1-jnk pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Kramvis, I.; Cho, N.; Sadowski, A.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mtor-dependent manner. J. Neurosci. 2009, 29, 5926–5937. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Hiramatsu, N.; Hayakawa, K.; Tagawa, Y.; Okamura, M.; Ogata, R.; Huang, T.; Nakajima, S.; Yao, J.; Paton, A.W.; et al. Activation of the akt-nf-kappab pathway by subtilase cytotoxin through the atf6 branch of the unfolded protein response. J. Immunol. 2009, 183, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Hiramatsu, N.; Hayakawa, K.; Saito, Y.; Kato, H.; Huang, T.; Yao, J.; Paton, A.W.; Paton, J.C.; Kitamura, M. Selective abrogation of bip/grp78 blunts activation of nf-kappab through the atf6 branch of the upr: Involvement of c/ebpbeta and mtor-dependent dephosphorylation of akt. Mol. Cell. Biol. 2011, 31, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Fonseca, S.G.; Oslowski, C.M.; Jurczyk, A.; Shearstone, J.R.; Zhu, L.J.; Permutt, M.A.; Greiner, D.L.; Bortell, R.; Urano, F. Aatf mediates an antiapoptotic effect of the unfolded protein response through transcriptional regulation of akt1. Cell Death Differ. 2010, 17, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Brule, S.; Hee Um, S.; Li, Y.; Masuda, K.; Roden, M.; Sun, X.J.; Krebs, M.; Polakiewicz, R.D.; Thomas, G.; et al. Identification of irs-1 ser-1101 as a target of s6k1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad Sci. USA 2007, 104, 14056–14061. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Huang, Q.; Ong, C.N.; Shen, H.M. Activation of the pi3k-akt-mtor signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy 2009, 5, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [PubMed]
- Knapik, J.; Meredith, C.; Jones, B.; Fielding, R.; Young, V.; Evans, W. Leucine metabolism during fasting and exercise. J. Appl. Physiol. 1991, 70, 43–47. [Google Scholar] [PubMed]
- Bhuiyan, M.M.H.; Mohibbullah, M.; Hannan, M.A.; Hong, Y.K.; Choi, J.S.; Choi, I.S.; Moon, I.S. Undaria pinnatifida Promotes Spinogenesis and Synaptogenesis and Potentiates Functional Presynaptic Plasticity in Hippocampal Neurons. Am. J. Chin. Med. 2015, 43, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Moon, I.S.; Goo, T.-W.; Moon, S.-S.; Seo, M. Algae Undaria pinnatifida Protects Hypothalamic Neurons against Endoplasmic Reticulum Stress through Akt/mTOR Signaling. Molecules 2015, 20, 20998-21009. https://doi.org/10.3390/molecules201219744
Kim J, Moon IS, Goo T-W, Moon S-S, Seo M. Algae Undaria pinnatifida Protects Hypothalamic Neurons against Endoplasmic Reticulum Stress through Akt/mTOR Signaling. Molecules. 2015; 20(12):20998-21009. https://doi.org/10.3390/molecules201219744
Chicago/Turabian StyleKim, Jongwan, Il Soo Moon, Tae-Won Goo, Seong-Su Moon, and Minchul Seo. 2015. "Algae Undaria pinnatifida Protects Hypothalamic Neurons against Endoplasmic Reticulum Stress through Akt/mTOR Signaling" Molecules 20, no. 12: 20998-21009. https://doi.org/10.3390/molecules201219744
APA StyleKim, J., Moon, I. S., Goo, T. -W., Moon, S. -S., & Seo, M. (2015). Algae Undaria pinnatifida Protects Hypothalamic Neurons against Endoplasmic Reticulum Stress through Akt/mTOR Signaling. Molecules, 20(12), 20998-21009. https://doi.org/10.3390/molecules201219744