
An Improved Helferich Method for the α/β-Stereoselective Synthesis of 4-Methylumbelliferyl Glycosides for the Detection of Microorganisms

Abstract
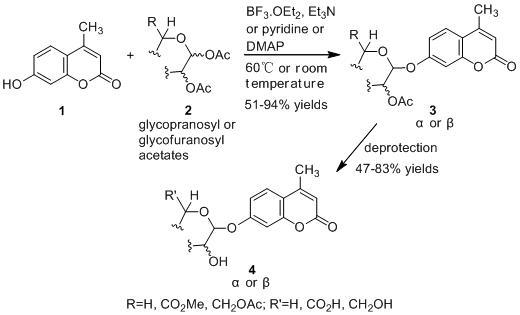
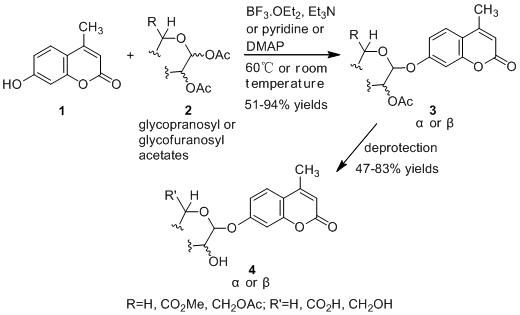
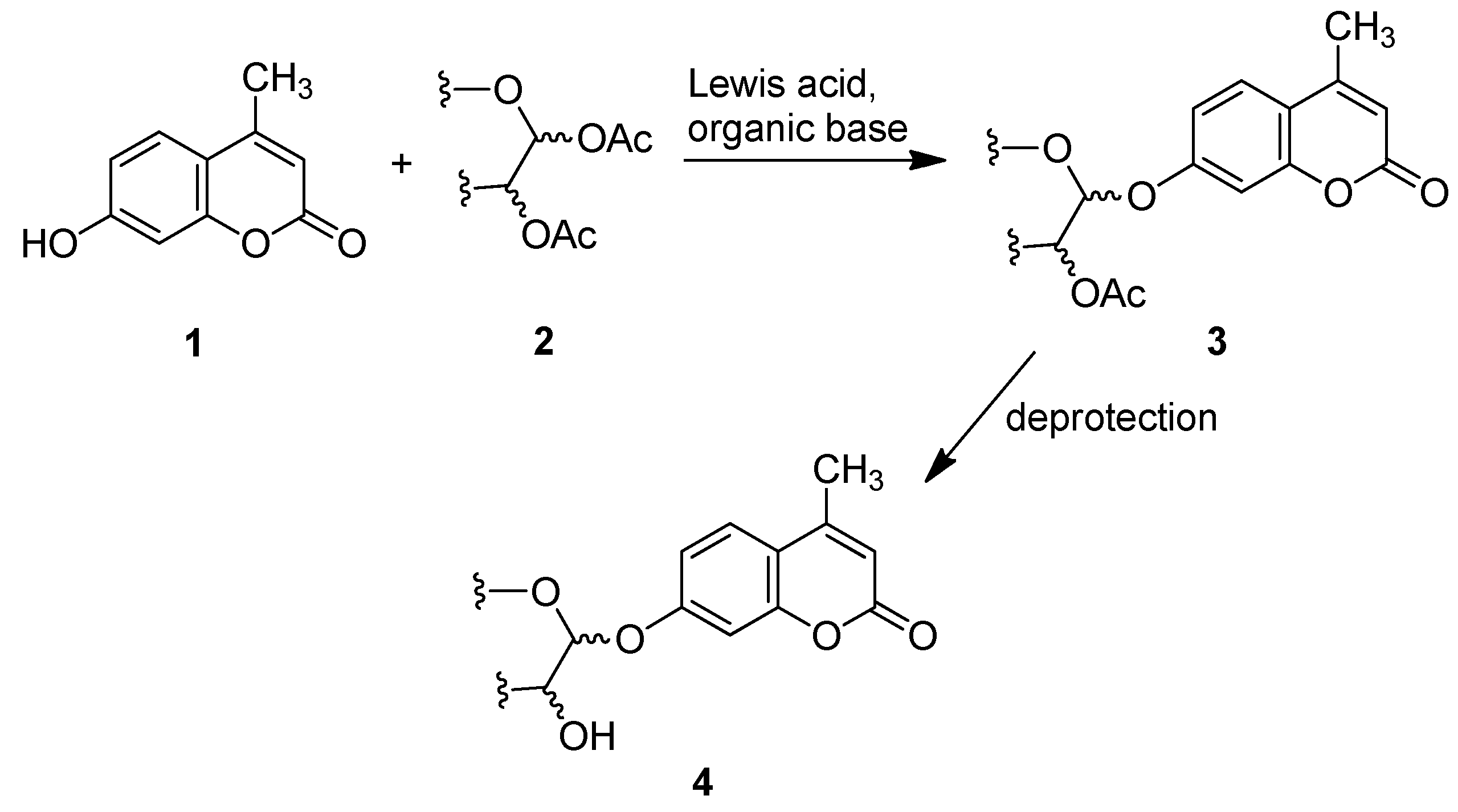
:
1. Introduction

2. Results and Discussion
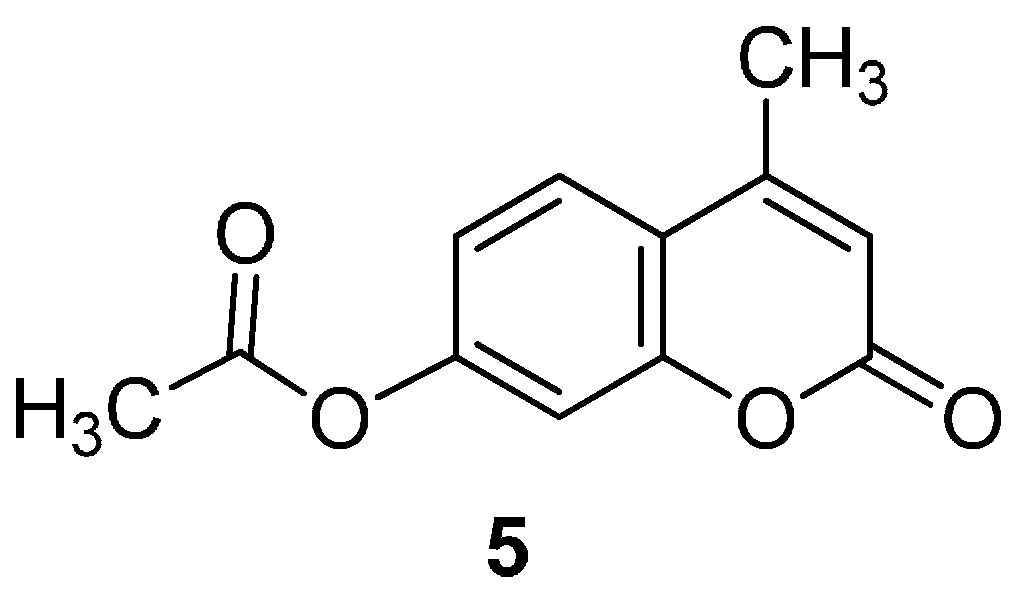
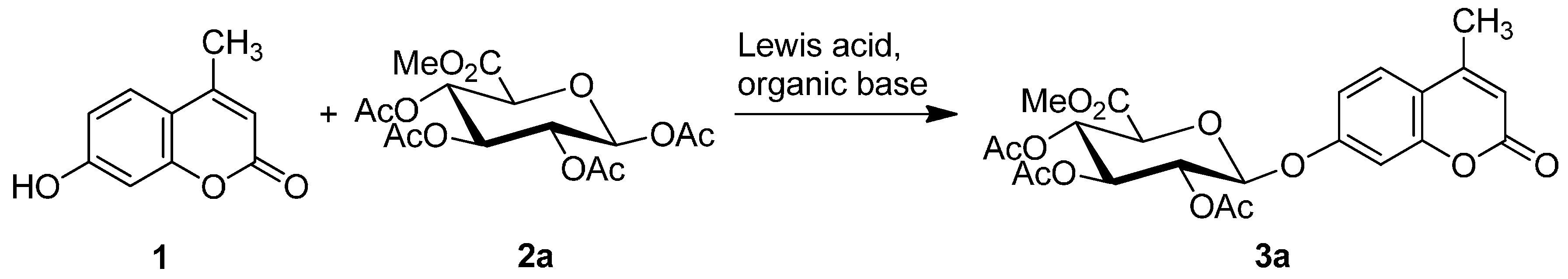
2.1. Synthesis of the Protected β-d-Glucopyranuronide 3a


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
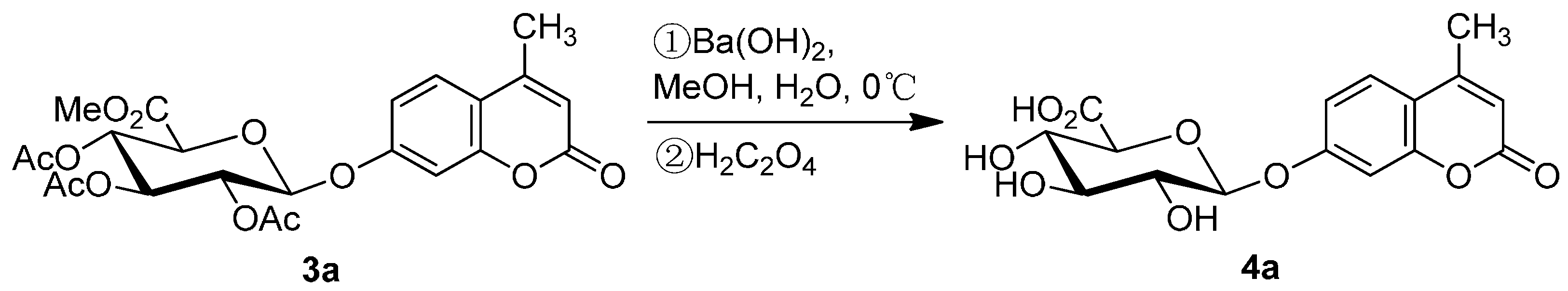{kind=link}
{kind=link}
| Entry | 1 (Equiv.) | 2a (Equiv.) | Organic Base (Equiv.) | Lewis Acid (Equiv.) | Reaction Conditions | Yield a of 3a | The Main Anomer b |
|---|---|---|---|---|---|---|---|
| 1 | 1.5 | 1.0 | TEA (3.75) | BF3·OEt2 (10.0) | CH2Cl2, 25–29 °C, 72 h | 17% | β |
| 2 | 1.5 | 1.0 | TEA (3.75) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 17% | β |
| 3 | 1.5 | 1.0 | TEA (3.75) | BF3·OEt2 (15.0) | ClCH2CH2Cl, 60 °C, 5 h | 19% | β |
| 4 | 1.5 | 1.0 | — c | BF3·OEt2 (15.0) | ClCH2CH2Cl, 60 °C, 10 h | — | — |
| 5 | 1.5 | 1.0 | TEA (2.0) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 16% | β |
| 6 | 2.0 | 1.0 | TEA (2.0) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 18% | β |
| 7 | 1.0 | 1.5 | TEA (2.0) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 23% | β |
| 8 | 1.0 | 1.5 | TEA (2.0) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 4A MS, 60 °C, 5 h | 13% | β |
| 9 | 1.0 | 1.5 | DIPEA (2.0) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 20% | β |
| 10 | 1.0 | 1.5 | Pyridine (2.0) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 23% | β |
| 11 | 1.0 | 1.5 | TMU (2.0) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 6% | β |
| 12 | 1.0 | 1.5 | DMAP (1.5) | BF3·OEt2 (10.0) | ClCH2CH2Cl, 60 °C, 5 h | 28% | β |
| 13 | 1.0 | 1.5 | Pyridine (3.0) | BF3·OEt2 (15.0) | ClCH2CH2Cl, 60 °C, 10 h | 19% | β |
| 14 | 1.0 | 1.5 | Pyridine (3.0) | BF3·OEt2 (15.0) | THF, 60 °C, 10 h | — | — |
| 15 | 1.0 | 1.5 | Pyridine (3.0) | BF3·OEt2 (15.0) | CH3CN, 60 °C, 10 h | 2% | β |
| 16 | 1.0 | 1.5 | Pyridine (2.0) | TMSOTf (4.0) | ClCH2CH2Cl, 60 °C, 16 h | <1% | β |
| 17 | 1.5 | 1.0 | DIPEA (3.0) | SnCl4 (5.0) | CH2Cl2, 25–29 °C, 48 h | — | — |

| Entry a | 1 (mmol) | 2a (mmol) | DMAP (mmol) | BF3·OEt2 (mmol) | Yield of Recovered 1 b | Yield c | Main Anomer d | |
|---|---|---|---|---|---|---|---|---|
| 3a | 5 | |||||||
| 1 | 1.0 | 1.5 | 3.0 | 10.0 | 21% | 30% | 14% | β |
| 2 | 1.0 | 1.5 | 4.5 | 10.0 | 28% | 32% | 11% | β |
| 3 | 1.0 | 1.5 | 6.0 | 10.0 | 44% | 17% | 9% | β |
| 4 | 1.0 | 1.5 | 5.0 | 10.0 | 37% | 23% | 10% | β |
| 5 | 1.0 | 1.5 | 4.0 | 10.0 | 24% | 34% | 12% | β |
| 6 | 1.0 | 1.5 | 3.5 | 10.0 | 26% | 33% | 13% | β |
| 7 | 1.0 | 3.0 | 8.0 | 20.0 | 22% | 36% | 14% | β |
| 8 | 1.0 | 2.0 | 5.0 | 12.5 | 11% | 49% | 12% | β |
| 9 | 1.0 | 2.0 | 4.0 | 12.5 | 9% | 51% | 15% | β |
| 10 | 1.0 | 2.0 | 4.0 | 15.0 | 10% | 41% | 17% | β |
| 11 | 1.0 | 2.0 | 4.0 | 12.5 | 15% | 26% | 26% | β |
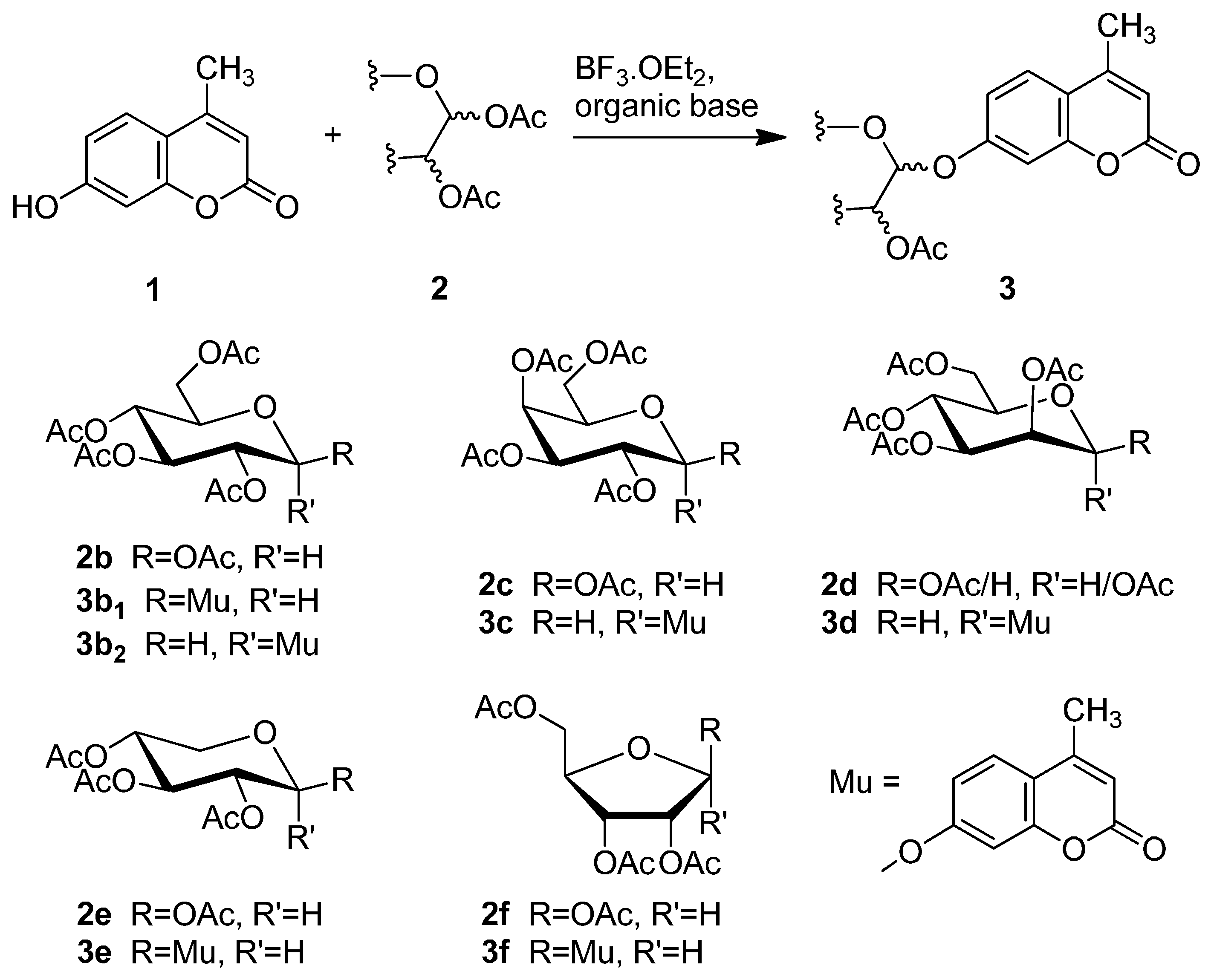
2.2. Synthesis of Other Protected Glycosides 3b–3f

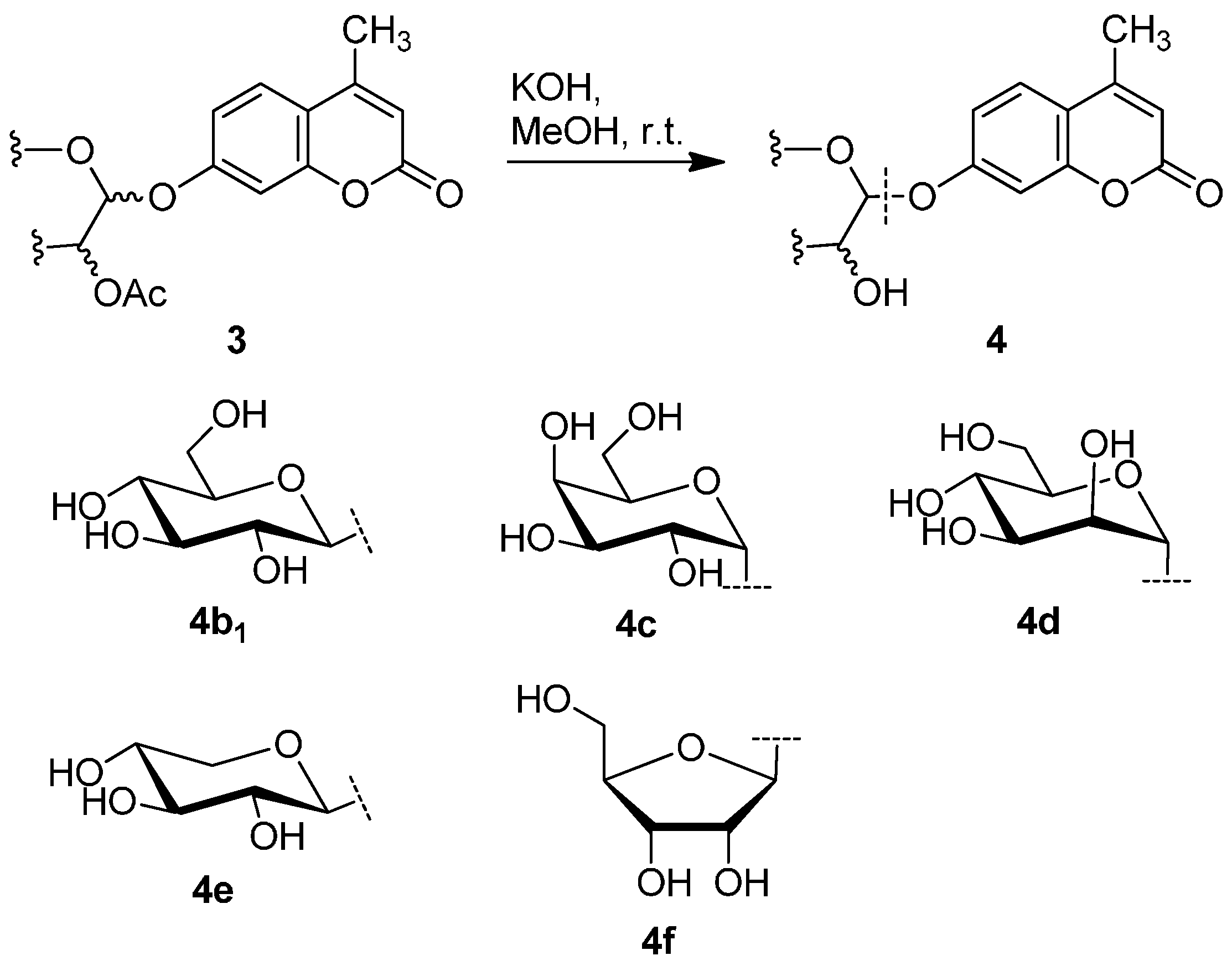
2.3. Deprotection of the Protected Glycosides 3a–3f
| Entry a | 1 (Equiv.) | 2 (Equiv.) | Organic Base (Equiv.) | BF3·OEt2 (Equiv.) | Reaction Conditions | Yield b | The Main Anomer c |
|---|---|---|---|---|---|---|---|
| 1 | 1.0 | 2b (2.0) | DMAP (1.5) | 10.0 | ClCH2CH2Cl, 60 °C, 5 h | 3b1 (36%) | β |
| 2 | 1.0 | 2b (2.0) | Pyridine (4.5) | 12.5 | ClCH2CH2Cl, 60 °C, 5 h | 3b1 (42%) | β |
| 3 d | 1.0 | 2b (2.0) | DMAP (4.0) | 12.5 | ClCH2CH2Cl, 60 °C, 5 h | 3b1 (45%) | β |
| 4 | 1.0 | 2b (2.0) | — e | 12.5 | ClCH2CH2Cl, 60–70 °C, 10 h | — | — |
| 5 | 1.0 | 2c (2.0) | TEA (2.5) | 12.5 | ClCH2CH2Cl, 60 °C, 5 h | 3c (54%) | α |
| 6 | 1.0 | 2c (2.0) | TEA (2.5) | 12.5 | ClCH2CH2Cl, 0 °C, 48 h | 3c (17%) | α |
| 7 f | 1.0 | 2c (2.0) | DMAP (4.0) | 12.5 | ClCH2CH2Cl, 60 °C, 5 h | 3c (56%) | α |
| 8 | 2.0 | 2d (1.0) | Pyridine (3.0) | 15.0 | ClCH2CH2Cl, 60 °C, 5 h | 3d (59%) | α |
| 9 g | 1.0 | 2d (2.0) | DMAP (4.0) | 12.5 | ClCH2CH2Cl, 60 °C, 5 h | 3d (62%) | α |
| 10 | 1.0 | 2e (2.0) | TEA (2.5) | 12.5 | CH2Cl2, 20–21 °C, 5 h | 3e (73%) | β |
| 11 h | 1.0 | 2e (2.0) | DMAP (4.0) | 12.5 | ClCH2CH2Cl, 26–27 °C, 5 h | 3e (69%) | β |
| 12 | 1.0 | 2f (2.0) | TEA (2.5) | 12.5 | CH2Cl2, 20–21 °C, 3 h | 3f (94%) | β |
| 13 i | 1.0 | 2f (2.0) | DMAP (4.0) | 12.5 | ClCH2CH2Cl, 26–27 °C, 5 h | 3f (77%) | β |


3. Experimental Section
3.1. General Information



3.2. General Procedure for the Glycosylation Step














4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yang, X.; Wu, Q.; Zhang, J.; Huang, J.; Chen, L.; Liu, S.; Yu, S.; Cai, S. Prevalence, enumeration, and characterization of Salmonella isolated from aquatic food products from retail markets in China. Food Control 2015, 57, 308–313. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, X.; Wu, Q.; Zhang, J.; Xu, X.; Li, H. Prevalence and characterization of Escherichia coli O157 and O157:H7 in retail fresh raw meat in South China. Ann. Microbiol. 2015. [Google Scholar] [CrossRef]
- Food Safety in Fact Sheets. Available online: www.who.int/mediacentre/factsheets/fs399/en/ (accessed on 28 November 2015).
- Manafi, M.; Kneifel, W.; Bascomb, S. Fluorogenic and chromogenic substrates used in bacterial diagnostics. Microbiol. Mol. Biol. Rev. 1991, 55, 335–348. [Google Scholar]
- Manafi, M. Fluorogenic and chromogenic enzyme substrates in culture media and identification tests. Int. J. Food Microbiol. 1996, 31, 45–58. [Google Scholar] [CrossRef]
- Manafi, M. New developments in chromogenic and fluorogenic culture media. Int. J. Food Microbiol. 2000, 60, 205–218. [Google Scholar] [CrossRef]
- Jokerst, J.C.; Adkins, J.A.; Bisha, B.; Mentele, M.M.; Goodridge, L.D.; Henry, C.S. Development of a paper-based analytical device for colorimetric detection of select foodborne pathogens. Anal. Chem. 2012, 84, 2900–2907. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wei, X.; Zhang, J.; Guo, W. Research progress of syntheses and applications in detecting microorganisms of indoxyl chromogenic substrates. Chemistry 2013, 76, 580–589. [Google Scholar]
- Orenga, S.; James, A.L.; Manafi, M.; Perry, J.D.; Pincus, D.H. Enzymatic substrates in microbiology. J. Microbiol. Methods 2009, 79, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Nilssen, Ø.; Stensland, H.M.F.R.; Malm, D. Clinical utility gene card for: α-Mannosidosis. Eur. J. Hum. Genet. 2011, 19. [Google Scholar] [CrossRef] [PubMed]
- Marsden, D.; Levy, H. Newborn screening of lysosomal storage disorders. Clin. Chem. 2010, 56, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Hattori, K.; Endo, F. Newborn screening for lysosomal storage disorders. Am. J. Med. Genet. Part C 2011, 157, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Oemardien, L.F.; Boer, A.M.; Ruijter, G.J.; van der Ploeg, A.T.; de Klerk, J.B.; Reuser, A.J.; Verheijen, F.W. Hemoglobin precipitation greatly improves 4-methylumbelliferone-based diagnostic assays for lysosomal storage diseases in dried blood spots. Mol. Genet. Metab. 2011, 102, 44–48. [Google Scholar] [CrossRef] [PubMed]
- O'Riordan, N.; Kane, M.; Joshi, L.; Hickey, R.M. Glycosidase activities in bovine milk over lactation. Int. Dairy J. 2014, 35, 116–121. [Google Scholar] [CrossRef]
- Oguri, S.; Nakaoka, A.; Amano, Y.; Ito, Y. Application of glycosidase activity as a marker for characterizing and identifying vegetables. J. Food Compos. Anal. 2012, 25, 39–46. [Google Scholar] [CrossRef]
- Carrasco, L.C.; Romar, R.; Avilés, M.; Gadea, J.; Coy, P. Determination of glycosidase activity in porcine oviductal fluid at the different phases of the estrous cycle. Reproduction 2008, 136, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Gold, H.; Munneke, S.; Dinkelaar, J.; Overkleeft, H.S.; Aerts, J.M.; Codée, J.D.; van der Marel, G.A. A practical synthesis of capped 4-methylumbelliferyl hyaluronan disaccharides and tetrasaccharides as potential hyaluronidase substrates. Carbohydr. Res. 2011, 346, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.D.; James, A.L.; Morris, K.A.; Oliver, M.; Chilvers, K.F.; Reed, R.H.; Gould, F.K. Evaluation of novel fluorogenic substrates for the detection of glycosidases in Escherichia coli and enterococci. J. Appl. Microbiol. 2006, 101, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.J. O-Glycosylations under neutral or basic conditions. J. Chem. Soc. Perkin Trans. 1 2002, 2219–2233. [Google Scholar] [CrossRef]
- Zhu, J.; Dong, J.; Yang, Z. Synthesis and application of Mugal, an enzyme-substrate rapid determination reagent. Huaxue Shiji 1997, 19, 210–212. [Google Scholar]
- Robinson, D. The fluorimetric determination of β-glucosidase: Its occurrence in the tissues of animals, including insects. Biochem. J. 1956, 63, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Leaback, D.H.; Walker, P.G. Studies on glucosaminidase. 4. The fluorimetric assay of N-acetyl-β-glucosaminidase. Biochem. J. 1961, 78, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Strachan, R.; Wood, J.; Hirschmann, R. Synthesis and properties of 4-methyl-2-oxo-1,2-benzopyran-7-yl β-d-galactoside (galactoside of 4-methylumbelliferone). J. Org. Chem. 1962, 27, 1074–1075. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, Q.; Zhang, J.; Guo, W.; Wei, X. Synthesis and application of Mugal which is the specific fluorescent substrate for detection of coliform. Xiandai Shipin Keji 2014, 30, 251–257. [Google Scholar]
- Carriere, D.; Meunier, S.J.; Tropper, F.D.; Cao, S.; Roy, R. Phase transfer catalysis toward the synthesis of O-, S-, Se- and C-glycosides. J. Mol. Catal. A Chem. 2000, 154, 9–22. [Google Scholar] [CrossRef]
- Walsh, J.S.; Patanella, J.E.; Halm, K.A.; Facchine, K.L. An improved HPLC assay for the assessment of liver slice metabolic viability using 7-ethoxycoumarin. Drug Metab. Dispos. 1995, 23, 869–874. [Google Scholar] [PubMed]
- Courtin-Duchateau, M.C.; Veyrières, A. Synthesis of 4-methylumbelliferyl 1,2-cis-glycosides. Carbohydr. Res. 1978, 65, 23–33. [Google Scholar] [CrossRef]
- Vervoort, A.; de Bruyne, C.K. Synthesis of substituted phenyl α-d-mannopyranosides. Carbohydr. Res. 1970, 12, 277–280. [Google Scholar] [CrossRef]
- Woollen, J.W.; Walker, P.G. The fluorimetric estimation of β-glucuronidase in blood plasma. Clin. Chim. Acta 1965, 12, 659–670. [Google Scholar] [CrossRef]
- Szweda, R.; Spohr, U.; Lemieux, R.U.; Schindler, D.; Bishop, D.F.; Desnick, R.J. Synthesis of 4-methylumbelliferyl glycosides for the detection of α- and β-d-galactopyranosaminidases. Can. J. Chem. 1989, 67, 1388–1391. [Google Scholar] [CrossRef]
- Chilvers, K.F.; Perry, J.D.; James, A.L.; Reed, R.H. Synthesis and evaluation of novel fluorogenic substrates for the detection of bacterial beta-galactosidase. J. Appl. Microbiol. 2001, 91, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Marsh, C.A.; Levvy, G.A. Synthesis of 4-methylumbelliferone β-d-glucuronide, a substrate for the fluorimetric assay of β-glucuronidase. Nature 1956, 178, 589–590. [Google Scholar] [CrossRef]
- Lopez-Lopez, M.A.; Balbuzano-Deus, A.; Rodriguez-Dominguez, J.C.; Mesa Hernandez, M.; Fernandez-Villalobo, A.; Reyes, Y.I.; Kirsch, G. A new synthetic route to 4-methylumbelliferyl-β-d-glucopyranosiduronic acid (MUG). Synlett 2007, 2007, 649–651. [Google Scholar]
- Brown, R.T.; Stachulski, A.V. Intermediates for glucuronide synthesis: 7-Hydroxycoumarin glucuronide. J. Chem. Res. Synop. 1997, 10, 370–371. [Google Scholar] [CrossRef]
- Pearson, A.G.; Kiefel, M.J.; Ferro, V.; von Itzstein, M. Towards the synthesis of aryl glucuronides as potential heparanase probes. An interesting outcome in the glycosidation of glucuronic acid with 4-hydroxycinnamic acid. Carbohydr. Res. 2005, 340, 2077–2085. [Google Scholar] [CrossRef] [PubMed]
- Stachulski, A.V.; Jenkins, G.V. The synthesis of O-glucuronides. Nat. Prod. Rep. 1998, 15, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Rho, E.S.; Min, Y.K.; Kim, B.T.; Kim, K.H. Practical beta-stereoselective O-glycosylation of phenols with penta-O-acetyl-beta-d-glucopyranose. J. Carbohydr. Chem. 2001, 20, 503–506. [Google Scholar] [CrossRef]
- Zhu, X.R.; Zheng, Y.; Wang, P.; Yang, S.B.; Chen, R.; Zhu, Y.Q. Synthesis of two metabolites of edaravone. J. Chin. Pharm. Sci. 2010, 19, 307–311. [Google Scholar] [CrossRef]
- Toshima, K.; Tatsuta, K. Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem. Rev. 1993, 93, 1503–1531. [Google Scholar] [CrossRef]
- Andersson, M.; Oscarson, S. Synthesis of O-glycopyranosyl-N-hydroxysuccinimides of glucose and lactose and their opening by nucleophiles into prespacer glycosides. Glycoconj. J. 1992, 9, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Hosoya, T.; Suzuki, K. Improvement in O→C-glycoside rearrangement approach to C-aryl glycosides: Use of 1-O-acetyl sugar as stable but efficient glycosyl donor. Tetrahedron Lett. 1990, 31, 4629–4632. [Google Scholar] [CrossRef]
- Zeng, S.; Chen, L.; Liu, D.; Li, H.; Wu, G.; Liao, Y. Synthesis of p-nitrophenyl α-d-xylopyranoside. Huaxue Shiji 2013, 35, 599–600. [Google Scholar]
- De Bruyne, C.K.; Loontiens, F.G. A fluorigenic substrate for β-d-xylosidase. Naturwissenschaften 1965, 52, 661. [Google Scholar] [CrossRef]
- Schramm, V.L.; Furneaux, R.H.; Tyler, P.C.; Clinch, K. Enzyme Detection/Assay Method and Substrates. Patent Cooperation Treaty International Publication Number WO/1997/031008A1, 28 August 1997. [Google Scholar]
- Yamaguchi, M.; Horiguchi, A.; Fukuda, A.; Minami, T. Novel synthesis of aryl 2,3,4,6-tetra-O-acetyl-d-glucopyranosides. J. Chem. Soc. Perkin Trans. 1 1990, 1079–1082. [Google Scholar] [CrossRef]
- Demchenko, A.V. 1,2-cis O-Glycosylation: Methods, strategies, principles. Curr. Org. Chem. 2003, 7, 35–79. [Google Scholar] [CrossRef]
- Zhu, X.; Schmidt, R. New principles for glycoside-bond formation. Angew. Chem. Int. Ed. 2009, 48, 1900–1934. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Ogawa, T. A highly efficient, practical, and stereoselective approach to the synthesis of α 1→4 linked galactooligosaccharides. Tetrahedron Lett. 1987, 28, 2731–2734. [Google Scholar] [CrossRef]
- Demchenko, A.V.; Rousson, E.; Boons, G.J. Stereoselective 1,2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40, 6523–6526. [Google Scholar] [CrossRef]
- Stachulski, A.V.; Meng, X. Glucuronides from metabolites to medicines: A survey of the in vivo generation, chemical synthesis and properties of glucuronides. Nat. Prod. Rep. 2013, 30, 806–848. [Google Scholar] [CrossRef] [PubMed]
- Needs, P.W.; Williamson, G. Syntheses of daidzein-7-yl beta-d-glucopyranosiduronic acid and daidzein-4′,7-yl di-beta-d-glucopyranosiduronic acid. Carbohydr. Res. 2001, 330, 511–515. [Google Scholar] [CrossRef]
- Khan, M.K.; Rakotomanomana, N.; Loonis, M.; Dangles, O. Chemical synthesis of citrus flavanone glucuronides. J. Agric. Food Chem. 2010, 58, 8437–8443. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Alshafie, G.; Nieves, N.; Ahrens, J.; Clagett-Dame, M.; Abou-Issa, H.; Curley, R.W., Jr. Synthesis and preliminary chemotherapeutic evaluation of the fully C-linked glucuronide of N-(4-hydroxyphenyl)retinamide. Bioorg. Med. Chem. 2006, 14, 3038–3048. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Alcantara, D.; Morales, J.C. A concise synthesis of glucuronide metabolites of urolithin-B, resveratrol, and hydroxytyrosol. Carbohydr. Res. 2009, 344, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.; Ma, Y.; Wu, Q.; Zhang, J.; Cai, Z.; Lu, M. An Improved Helferich Method for the α/β-Stereoselective Synthesis of 4-Methylumbelliferyl Glycosides for the Detection of Microorganisms. Molecules 2015, 20, 21681-21699. https://doi.org/10.3390/molecules201219789
Wei X, Ma Y, Wu Q, Zhang J, Cai Z, Lu M. An Improved Helferich Method for the α/β-Stereoselective Synthesis of 4-Methylumbelliferyl Glycosides for the Detection of Microorganisms. Molecules. 2015; 20(12):21681-21699. https://doi.org/10.3390/molecules201219789
Chicago/Turabian StyleWei, Xianhu, Yanxia Ma, Qingping Wu, Jumei Zhang, Zhihe Cai, and Mianfei Lu. 2015. "An Improved Helferich Method for the α/β-Stereoselective Synthesis of 4-Methylumbelliferyl Glycosides for the Detection of Microorganisms" Molecules 20, no. 12: 21681-21699. https://doi.org/10.3390/molecules201219789
APA StyleWei, X., Ma, Y., Wu, Q., Zhang, J., Cai, Z., & Lu, M. (2015). An Improved Helferich Method for the α/β-Stereoselective Synthesis of 4-Methylumbelliferyl Glycosides for the Detection of Microorganisms. Molecules, 20(12), 21681-21699. https://doi.org/10.3390/molecules201219789