Abstract
Two new ent-kaurane diterpene glycosides, ranunculosides A (1) and B (2), and a new benzophenone, ranunculone C (3), were isolated from the aerial part of Ranunculus muricatus Linn. The chemical structures of compounds 1–3 were established to be (2S)-ent-kauran-2β-ol-15-en-14-O-β-d-glucopyranoside, (2S,4S)-ent-kauran-2β,18-diol-15-en-14-O-β-d-glucopyranoside, and (R)-3-[2-(3,4-dihydroxybenzoyl)-4,5-dihydroxy-phenyl]-2-hydroxylpropanoic acid, respectively, by spectroscopic data and chemical methods. The absolute configuration of 1 was determined by the combinational application of RP-HPLC analysis and Mosher’s method.
1. Introduction
Ranunculus muricatus Linn. (Ranunculaceae) is widely distributed in Asia, South America, Australia, and Europe. This plant mainly grows in the lower Yangtze River regions of China. It is used in folklore medicines in India for the therapy of tonsillitis diseases [1]. Although there is little documentation for folk and clinical use of the titled plant in China, other Ranunculus plants such as R. sceleratus Linn., R. japonicus Thunb., and R. termatus Thunb. have diverse clinical uses with a long history in traditional Chinese medicines and folk medicines, including those used for the treatment of lymphatic tuberculosis, malaria, swollen hemorrhoids, scrofula, and arthritis [2,3,4]. A series of simple lactone derivatives with 5- or 6-member rings, some steroids, and phenols was found in these plants, some of which indicated various biological activities, and they are responsible for their traditional or folk uses and clinical efficacies [5,6]. Our interest is in finding out the chemical characteristics and biological activities of the widely distributed plant R. muricatus. Wang et al.’s research led to the isolation and identification of eight known constituents from this plant, which are stigmasta-4-ene-3,6-dione, stigmasterol, anemonin, sitosterol, protocatechuic aldehyde, protocatechuic acid, and luteoin, and most of which belong to common compounds in plants [7]. Our previous study has revealed the presence of rich flavonoid glycosides in the plant [8]. Now, our further phytochemical investigation on the ethanolic extract from the aerial part of R. muricatus Linn. led to the isolation and identification of three new compounds (Figure 1), ranunculoside A (1), ranunculoside B (2), and ranunculone C (3). It is notable that ranunculosides A (1) and B (2) are the first two ent-kaurane diterpenoids from the genus plants and are present in glycoside. Herein we describe the isolation and structure elucidation of these compounds through spectroscopic analysis and chemical methods.
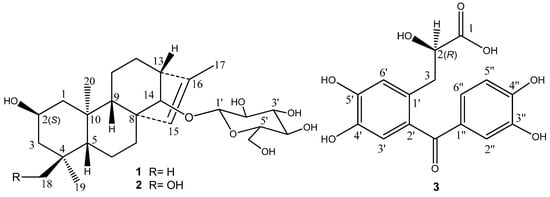
Figure 1.
Structures of compounds 1–3.
2. Results and Discussion
The ethanolic extract from the aerial part of R. muricatus Linn. was successively partitioned in an H2O suspension solution with petroleum ether, EtOAc, and n-BuOH, then applied to various column chromatography with silica gel, Octadecylsilyl (ODS), and Sephadex LH20, following by preparative reversed phase HPLC purification to yield compounds 1–3.
Compound 1 was obtained as an amorphous solid. Its HR-ESI-MS in positive ion mode showed a pseudomolecular ion peak at m/z 489.28287 [M + Na]+, suggesting its molecular formula as C26H42O7. In the 1H-NMR spectrum of 1 (Table 1), there were the signals at δH 0.83 (s, 3H), 0.86 (s, 3H), 0.87 (s, 3H), and 1.66 (d, J = 1.3 Hz, 3H) for four methyl groups. The 1H-NMR spectrum displayed signals for the protons of a sugar moiety including a β-oriented anomeric proton signal at δH 4.88 (d, J = 7.8 Hz, 1H), and two other oxygenated methine protons at δH 4.12 (m, 1H) and 3.78 (br s, 1H) in the range of δH 3.90–5.00. In addition, there was an olefinic proton signal that appeared at δH 5.34 (br s, 1H). The 13C-NMR spectrum of 1 (Table 2) displayed 26 resonances, including six resonances ascribable to a sugar unit. The distortionless enhancement by polarization transfer (DEPT) experiment revealed that the 26 carbons were comprised of four methyls, seven methylenes, eleven methines, and four quaternary carbons, including a pair of cyclic olefinic carbons at δC 127.3 and 138.1. The heteronuclear multiple-bond coherence (HMBC) spectrum showed the correlations of protons at δH 5.34 (br s, H-C(15)) with carbons at δC 52.8 (s, C(8))/48.8 (d, C(13))/97.7 (d, C(14))/138.1 (s, C(16)), and protons at δH 3.78 (br s, H-C(14)) with carbons at δC 32.7 (t, C(7))/53.3 (d, C(9))/127.3 (d, C(15)), suggesting that the olefinic group was located between C(15) and C(16). The OH group at the C(2) position was evident from the correlations between the proton at δH 4.12 (m, Hα-C(2)) and protons at δH (2.20/1.02) (m, CH2(1))/(2.03/1.43) (m, CH2(3)) in the 1H,1H-COSY spectrum of 1 (Figure 2). The above data suggested that the aglycone of 1 may be a diterpene (Figure 1). Meanwhile, the correlations of protons at δH 0.86 (s, Me(18)) with carbons at δC 52.3 (t, C(3))/35.2 (s, C(4))/55.1 (d, C(5))/23.4 (q, C(19)), and protons at δ(H) 0.83 (s, Me(20)) with carbons at δC 49.8 (q, C(1))/55.1 (d, C(5))/53.3 (d, C(9))/39.4 (s, C(10)) were observed in the HMBC spectrum, suggesting that the diterpene has an ent-kaurane-type skeleton, which was further confirmed to have connections with CH2(1)/H-C(2)/CH2(3), H-C(5)/CH2(6)/CH2(7), and H-C(9)/CH2(11)/CH2(12)/H-C(13)/H-C(14) spin-coupling systems (Figure 2) by the 1H, 1H-COSY. In the ROESY spectrum of 1, the explicit nuclear Overhauser effects (NOE) correlations (Figure 3) of protons at δH 4.12 (m, Hα-C(2)) with protons at δH 0.86 (s, Me(19))/0.83 (s, Me(20)) determined the OH group at C(2) to be in β-orientation. Similarly, the NOEs of protons at δH 3.78 (br s, H-C(14)) with protons at δH 0.97 (m, Hβ-C(9))/4.88 (d, J = 7.8 Hz, Hβ-C(1′))/2.72 (br s, Hβ-C(13)) suggested H-C(14) to be β-orientational. Furthermore, the NOEs described in Figure 3 offered full relative stereochemical information of the diterpene skeleton. The sugar moiety was identified as d-glucopyranose by its 1H and 13C data and the HPLC analysis of the sugar derivative after the acid hydrolysis of 1 and the successive chemical derivatization of monosaccharide. The connection of the sugar group at C(14) was established by the HMBC correlation between protons at δH 4.88 (d, J = 7.8 Hz, H-C(1′)) and carbons at δC 97.7 (d, C(14)), and the comparison with the NMR data of pterokaurane M2, a very similar molecule with an aglycone in its structure [9]. The absolute configuration of the aglycone (1a) in 1 was confirmed by Mosher’s method [10]. Based on the differences of the chemical shifts of diagnosing protons (for Δδ(S-R) values, see Figure 4) between the (R)-MTPA ester and (S)-MTPA ester and the model for determining R or S configuration, the absolute configuration of acyloxylated carbon (C(2)) in 1a, then that of 1, was assigned as S by using the diagnostic MTPA ester model. Therefore, the structure of compound 1 was clarified to be (2S)-ent-kauran-2β-ol-15-en-14-O-β-d-glucopyranoside, and named as ranunculoside A.

Table 1.
1H-NMR Data of 1, 1a, and 2 (at 300 MHz, in CD5N5; δ in ppm, J in Hz).
| H-Atom | 1a (Multiplet) | 1 (Multiplet) | 2 (Multiplet) |
|---|---|---|---|
| CH2(1) | α 2.27 (ddd, J = 12.3, 3.8, 2.1) | α 2.20 (brd, J = 12.3) | α 2.25 o |
| β 1.13 (m) | β 1.02 (m) | β 1.10 (m) | |
| H-C(2) | α 4.16 (m) | α 4.12 (m) | α 4.27 (m) |
| CH2(3) | α 2.05 (ddd, J = 12.3, 3.8, 2.1) | α 2.03 (brd, J = 12.3) | α 2.16 (m) |
| β 1.46 (m) | β 1.43 (m) | β 2.06 (m) | |
| H-C(5) | β 0.88 o | β 0.71 (dd, J = 10.6, 1.2) | β 1.52 o |
| CH2(6) | α 1.36 o | α 1.26 o | α 1.50 o |
| β 1.61 o | β 1.47 o | β 1.70 o | |
| CH2(7) | α 2.33 (dd, J = 10.7, 3.2) | α 2.21 (brd, J = 10.2) | α 2.29 o |
| β 1.62 (m) | β 1.57 (m) | β 1.73 (m) | |
| CH2(9) | β 1.09 (m) | β 0.97 (m) | β 1.03 o |
| CH2(11) | α 1.45 o | α 1.38 o | α 1.40 o |
| β 1.24 o | β 1.18 o | β 1.23 o | |
| CH2(12) | α 1.50 o | α 1.51 o | α 1.52 o |
| β 1.32 o | β 1.30 o | β 1.28 o | |
| H-C(13) | β 2.44 (br s) | β 2.72 (br s) | β 2.73 (br s) |
| H-C(14) | β 3.50 (br s) | β 3.78 (br s) | β 3.75 (br s) |
| H-C(15) | 5.38 (br s) | 5.34 (br s) | 5.38 (br s) |
| Me(17) | 1.70 (d, J = 1.1) | 1.66 (d, J = 1.3) | 1.67 (d, J = 1.3) |
| Me(18) | 0.94 (s) | 0.87 (s) | a 3.64 (brd, J = 10.5) |
| CH2(18) | b 3.43 (brd, J = 10.5) | ||
| Me(19) | 0.92 (s) | 0.86 (s) | 0.98 (s) |
| Me(20) | 0.90 (s) | 0.83 (s) | 0.94 (s) |
| H-C(1′) | β 4.88 (d, J = 7.8) | β 4.88 (d, J = 7.8) | |
| H-C(2′) | 3.95 (m) | 3.95 (m) | |
| H-C(3′) | 3.98 (m) | 3.99 (m) | |
| H-C(4′) | 4.28 o | 4.26 o | |
| H-C(5′) | 4.28 o | 4.27 o | |
| CH2(6′) | a 4.43 (dd, J = 11.7, 5.3) | a 44.4 (dd, J = 11.8, 5.3) | |
| b 4.61 (dd, J = 11.7, 2.3) | b 4.59 (dd, J = 11.7, 2.4) |
Note: (i) o Overlapped; (ii) Multiplicity: s-singlet, br s-broad singlet; d-doublet, dd-double doublet, ddd-double double doublet, m-multiplet; (iii) α and β mean the orientations of protons; (iv) a and b mean the two protons of a methylene; (v) Assignments were performed by two-dimensional (2D) NMR.
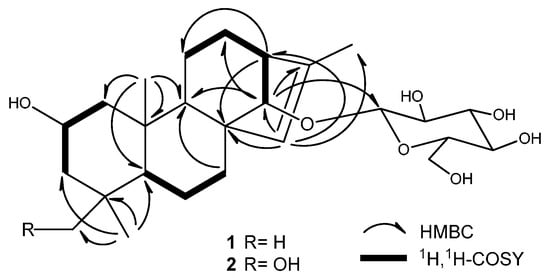
Figure 2.
Key HMBC and 1H,1H-COSY correlations of compounds 1 and 2.
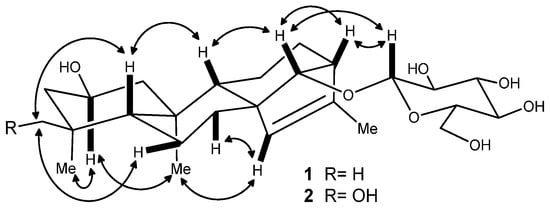
Figure 3.
Key NOE correlations of compounds 1 and 2.
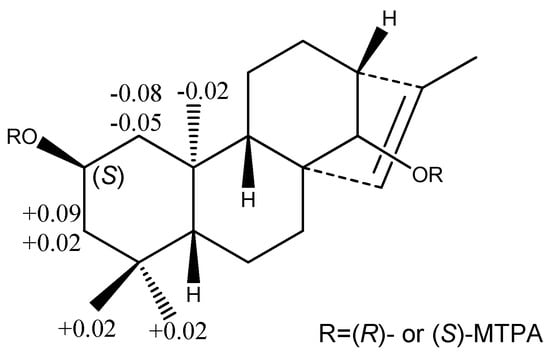
Figure 4.
The Δδ(S-R) values of some protons (in C5D5N, in ppm) obtained from (S)- and (R)-MTPA esters of compound 1a.
Table 2.
13C-NMR Data of 1, 1a, and 2 (at 75 MHz, in CD5N5; δ in ppm).
| C-Atom | 1a | 1 | 2 |
|---|---|---|---|
| C(1) | 50.1 (t) | 49.8 (t) | 49.7 (t) |
| C(2) | 64.3 (d) | 64.2 (d) | 64.5 (d) |
| C(3) | 52.4 (t) | 52.3 (t) | 46.5 (t) |
| C(4) | 35.3 (s) | 35.2 (s) | 40.0 (s) |
| C(5) | 55.7 (d) | 55.1 (d) | 48.7 (d) |
| C(6) | 20.3 (t) | 20.0 (t) | 20.0 (t) |
| C(7) | 32.8 (t) | 32.7 (t) | 32.5 (t) |
| C(8) | 54.0 (s) | 52.8 (s) | 53.0 (s) |
| C(9) | 53.7 (d) | 53.3 (d) | 53.3 (d) |
| C(10) | 39.5 (s) | 39.4 (s) | 39.4 (s) |
| C(11) | 19.5 (t) | 19.4 (t) | 19.5 a (t) |
| C(12) | 24.0 (t) | 23.5 (t) | 23.6 (t) |
| C(13) | 52.9 (d) | 48.8 (d) | 48.6 (d) |
| C(14) | 92.2 (d) | 97.7 (d) | 97.6 (d) |
| C(15) | 127.4 (d) | 127.3 (d) | 127.5 (d) |
| C(16) | 138.5 (s) | 138.1 (s) | 138.0 (s) |
| C(17) | 16.3 (q) | 16.2 (q) | 16.2 (q) |
| C(18) | 34.3 (q) | 34.1 (q) | 71.9 (t) |
| C(19) | 23.5 (q) | 23.4 (q) | 19.5 a (q) |
| C(20) | 17.0 (q) | 16.9 (q) | 17.6 (q) |
| C(1′) | 102.9 (d) | 102.8 (d) | |
| C(2′) | 75.2 (d) | 75.2 (d) | |
| C(3′) | 78.9 a (d) | 79.0 a (d) | |
| C(4′) | 72.1 (d) | 72.2 (d) | |
| C(5′) | 78.9 a (d) | 79.0 a (d) | |
| C(6′) | 63.2 (t) | 63.3 (t) |
Note: a Overlapped; d indicates CH, q indicates CH3, s indicates quaternary carbon, t indicates CH2.
Compound 2 was obtained as an amorphous powder, and had a molecular formula of C26H42O8 according to the pseudomolecular ion peak at m/z 505.27707 [M + Na]+ in HR-ESI-MS (positive mode), 16 mass units higher than that of 1, implying the presence of an additional oxygen atom in 2. Comparison of the NMR data of 2 with those of 1 indicated that they were a pair of compounds extremely analogous in structure. The mere difference was that the methyl group (Me(18)) in 1 was replaced by a hydroxymethyl group in 2, because the hydroxymethyl signals at δH 3.43/3.64 (dd, J = 10.5 Hz, 1H each) and δC 71.9 (t) were observed in the 1H- and 13C-NMR spectra of 2. The complete structure of 2 was confirmed by HMBC and NOE correlations, which were similar to those of 1 (Figure 2 and Figure 3). The acid hydrolysis of 2 under the same conditions mentioned in 1 also afforded the same sugar, d-glucose, which was also determined by the comparative HPLC analysis of the sugar derivative with that of standard sugars. Based on the consideration of the same biogeneric pathway, the absolute configurations of all the chiral centers except C-4 in the aglycone part of 2 should be identical to those of 1. The absolute configuration of the newly present chiral carbon C(4) due to the hydroxyl substitution was determined by careful analysis of its NOESY. Therefore, compound 2 was determined to be (2S,4S)-ent-kauran-2β,18-diol-15-en-14-O-β-d-glucopyranoside, which was named ranunculoside B.
Compound 3 showed a sodiated molecular ion peak at m/z 357.05848 ([M + Na]+) in HR-ESI-MS, corresponding to the molecular formula C16H14O8. The 13C-NMR spectrum displayed 16 carbon signals, ascribable to two aromatic rings at δC 115–155 (Table 3), one carbonyl at δC 199.3, a carboxyl carbon at δC 177.5, an oxygenated methine at δC 73.2, and a methylene at δC 38.0. The 1H-NMR spectrum (Table 3) of 3 displayed a AA′X spin system with protons at δH 2.92 (dd, J = 13.8, 8.0 Hz, 1H), 3.12 (dd, J = 13.7, 3.8 Hz, 1H), and 4.28 (dd, J = 7.8, 3.9 Hz, 1H), which was verified by 1H,1H-COSY, and five aromatic protons. An ABX-type aromatic proton spin system at δH 7.30 (d, J = 1.8 Hz, H–C(2″), 1H), 7.20 (dd, J = 8.3, 1.8 Hz, H-C(6″), 1H), and 6.83 (d, J = 8.3 Hz, H-C(5″), 1H) was assigned to one tri-substituted benzene ring. Additionally, the other two aromatic protons at δH 6.81 (br s, H-C(3′), 1H) and 6.88 (br s, H-C(6′), 1H) belonged to the second tetra-substituted benzene ring. The methylene at δH 2.92/3.12 and C(3) (t, δC 38.0) was directly connected to oxygenated methine at δH 4.28/(d, δC 73.2, C(2)), a chiral center. The HMBC correlations between protons at δH 2.92/3.12 (CH2(3)) and carbon at δC 177.5 (s, C(1)) suggested that 3 had a 2-hydroxypropanoic acid group. The correlations of protons at δH 2.92/3.12 (CH2(3)) with carbons at δC 177.5 (C(1))/131.6 (s, C(2′))/119.3 (d, C(6′)) suggested that the 2-hydroxypropanoic acid group connected to the aromatic ring have an AB-type spin system. The key correlations of carbon at δC 199.3 (s, C=O) with protons at δH 6.81 (s, H-C(3′))/7.30 (s, H-C(2″))/7.20 (s, H-C(6″)) in the HMBC spectrum of 3 (Figure 5) implied its skeleton of diphenylketone. Importantly, other correlations (Figure 5) of H-C(2) with C(1′), H-C(6′) with C(2′), and H-C(5″) with C(1″) also confirmed the above conclusion. Structurally, compound 3 was nearly identical to the reported compound ethyl (S)-3-[2-(3,4-dihydroxybenzoyl)-4,5-dihydroxyphenyl]-2-hydroxypropanoate except for the ethanolic esterification at its C(1) in the latter, which had a negative specific rotation and S absolute configuration [11]. Noticeably, the specific rotation of 3 is positive, opposite to the reported one above, but consistent with the compound salvianic acid A [12] with R absolute configuration. Therefore, the only chiral center (d, C(2)) in 3 was discriminated as R. According to all the above evidence, compound 3 was identified to be (R)-3-[2-(3,4-dihydroxybenzoyl)-4,5-dihydroxyphenyl]-2-hydroxylpropanoic acid, designated as ranunculone C.
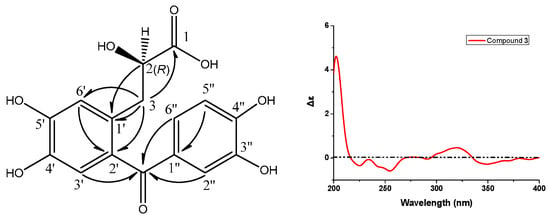
Figure 5.
Key HMBC (H → C) correlations and CD spectrum of compound 3.
Table 3.
1H- and 13C-NMR Data of 3 (at 300/75 MHz for 1H/13C, in CD3OD; δ in ppm, J in Hz).
| C-Atom | δ(C) | H-Atom | δ(H) Multiplet |
|---|---|---|---|
| C(1) | 177.5 (s) | - | |
| C(2) | 73.2 (d) | H-C(2) | 4.28 (dd, J = 7.8, 3.9) |
| C(3) | 38.0 (t) | CH2(3) | a 2.92 (dd, J = 13.8, 8.0) |
| b 3.12 (dd, J = 13.7, 3.8) | |||
| C=O | 199.3 (s) | - | |
| C(1′) | 131.0 (s) | - | |
| C(2′) | 131.6 (s) | - | |
| C(3′) | 118.6 (d) | H-C(3′) | 6.81 (br s) |
| C(4′) | 143.9 (s) | - | |
| C(5′) | 149.0 (s) | - | |
| C(6′) | 119.3 (d) | H-C(6′) | 6.88 (br s) |
| C(1″) | 131.4 (s) | - | |
| C(2″) | 118.3 (d) | H-C(2″) | 7.30 (d, J = 1.8) |
| C(3″) | 146.1 (s) | - | |
| C(4″) | 152.2 (s) | - | |
| C(5″) | 115.6 (d) | H-C(5″) | 6.83 (d, J = 8.3) |
| C(6″) | 125.9 (d) | H-C(6″) | 7.20 (dd, J = 8.3, 1.8) |
Note: For δ(C), d indicates CH, s indicates quaternary carbon, t indicates CH2. - means no correlation to be observed.
All the new compounds were found to be inactive in evaluation for their cytotoxicity against HEp-2 and HeLa cell lines at a test concentration of 50 μg/mL by MTT assay. The procedure was similar to the one in the literature [13], and the data were not presented here.
3. Experimental Section
3.1. General
TLC: precoated aluminum gel plates (Yantai Chemical Industry Research Institute, Yantai, China). Column Chromatography (CC): silica gel (SiO2; 200–300, and 300–400 mesh; Qingdao Marine Chemical Factory, Qingdao, China); D101 macroporous resin (Tianjin Agricultural Pesticide Factory, Tianjin, China); ODS (50 µm, YMC, Tokyo, Japan); Sephadex LH-20 (GE Healthcare Co. Uppsala, Sweden). M.p.: X-5 micro-melting point apparatus, uncorrected. Optical rotations: Jasco P-1030 automatic digital polarimeter (JASCO Corporation, Tokyo, Japan). UV spectra: Jasco V-550 UV/VIS spectrometer (JASCO Corporation), in anh. MeOH; λmax (log ε) in nm. IR spectra: Jasco FT/IR-480 plus spectrometer (JASCO Corporation), as KBr pellets; in cm−1. HR-ESI-MS spectra: Agilent 6210 LC/MSD TOF mass spectrometer (Agilent Technologies, Santa Clara, CA, USA); in m/z (rel. %). NMR spectra: Bruker AV-300, AV-400 (for data of HMBC), AV-500 (for data of ROESY) instruments (Bruker Corporation, Fallanden, Switzerland), TMS as internal standard, δ in ppm, J in Hz. HPLC: Agilent series 1200 (Agilent Technologies) with quaternary pump, multiple wavelength detector and autosampler, and Ultimate™ XB-C18 column (5 µm, 250 × 4.6 mm, or 21.2 × 250 mm, Welch Materials, Inc., Shanghai, China).
3.2. Plant Material
The experimental material was gathered in Jiujiang County, Jiangxi Province, China, in July 2010 and authenticated as Ranunculus muricatus Linn. by Prof. X.-Z. Zhang, College of Pharmacy, Guangdong Pharmaceutical University. A voucher specimen (No. RM-200927) was deposited in the Department of Pharmacognosy, College of Pharmacy, Jinan University.
3.3. Extraction and Isolation
Air-dried and powdered herbal materials (aerial part) of Ranunculus muricatus Linn. (10.0 kg) were soaked in 95% alcohol for 24 h and filtered each time, and the process repeated three times. The filtered solution was gathered and evaporated off the alcoholic solvent in vacuo to get an extract in about 1.0 kg. The alcoholic extract was suspended in distilled water and then partitioned successively with petroleum ether, ethyl acetate and n-BuOH to afford three fractions: petroleum ether fraction (80.0 g), EtOAc fraction (27.1 g), and BuOH fraction (65.2 g), respectively. The EtOAc fraction (27.0 g) was subjected to CC (SiO2; PE/EtOAc 30:1 → 0:1), to afford seven pooled fractions (FrA–G) according to the TLC pattern. FrF (700.0 mg) was applied to CC (SiO2; CHCl3/MeOH 15:1 → 5:1) to get three subfractions (SFr 1–3). SFr 2 (50.0 mg) was chromatographed on CC (SiO2; CHCl3/MeOH 8:1) to get SFr 2-1, and it was then purified by CC (sephadex LH-20; CHCl3/MeOH 1:1) to afford 1 (30.3 mg) as an amorphous white powder. The n-BuOH fraction (65.0 g) was suspended in water, then applied to D101 macroporous resin (EtOH/H2O 0:1 → 90:10) to obtain SFr H-1 (12.2 g), SFr H-2 (6.2 g), and SFr H-3 (35.1 g). SFr H-1 (12.0 g) was applied to CC (ODS; MeOH/H2O 1:5 → 3:5) to get SFr H-1-8 (400.0 mg). This fraction was further chromatographed on CC (SiO2; CHCl3/MeOH 10:1 → 5:1) to get SFr H-1-8-1, which was finally purified by CC (SephadexLH-20; CHCl3/MeOH 1:1) to afford 2 (25.1 mg) as an amorphous white powder. The fraction SFr H-1-2 (300.0 mg) was applied to CC (ODS; MeOH/H2O 1:5 → 3:5), and then purified on CC (Sephadex LH-20; CHCl3/MeOH 1:1) to afford SFr H-1-2-1 (25.1 mg), which was further purified by preparative HPLC to afford 3 (20.0 mg).
Acid hydrolysis of 1 and identification of absolute configuration of 1a by Mosher’s method [9,10]. Compound 1 (9.0 mg) was hydrolyzed with 10 mL of 2 M HCl for 2 h at 90 °C; after cooling down in the air, the mixture was extracted with EtOAc (20 mL) three times. The EtOAc portion was dried in vacuo, and applied to CC (SiO2; CHCl3/acetone 20:1), and finally purified by CC (Sephadex LH-20; CHCl3/MeOH 1:1) to yield the aglycone of 1 (1a, 7 mg) as an amorphous white powder. The aglycone 1a (1.4 mg) was dissolved in 500 μL of C5D5N, and then we added (R)-(−)-α-methoxy-α-(trifluoromethyl) phenylacetyl chloride (10 μL; Sigma, St. Louis, MO, USA) into it. The Mosher reaction process was protected by nitrogen. The reaction mixture containing the (R)-MTPA ester (1ar) of 1a was directly analyzed by NMR. The (S)-MTPA ester (1as) preparation and analysis was in the same procedure as that of 1ar. On the basis of the proton chemical shift between 1ar and 1as, the differences of the δ values of protons at the same positions were calculated (Figure 4). So the absolute configuration of C(2) was determined.
Determination of absolute configuration of sugar by RP-HPLC [14]. The absolute configuration of sugar unit of 1 was discriminated by reversed-phase (RP) HPLC method. Compound 1 (2.0 mg) was hydrolyzed with 2 mL of 1 M HCl for 2 h at 90 °C, and the mixture was dried in vacuo. The residue was dissolved in pyridine (1.5 mL) containing l-cysteine methyl ester hydrochloride (1.5 mg; Adamas-beta, Shanghai, China) and heated at 60 °C for 1 h, and then we added O-tolylisothiocyanate (5 μL; Sigma) into the mixture and heated it at 60 °C for 1 h. The final reaction mixture was directly analyzed by RP-HPLC. Analytical HPLC was performed on the column at 30 °C with isocratic elution of 25% CH3CN (0.8‰ formic acid) for 40 min and subsequently washing the column with 90% CH3CN at a flow rate of 0.8 mL/min. The peak signals were detected by a UV detector at 250 nm. The peak of the derivative from 1 was clearly observed at 22.62 min. Standard sugars (Sigma) d-glucose and l-glucose were applied to the same analytical procedure. The peaks of control sugar derivatives were recorded at 22.63 (d-glucose), and 20.75 (l-glucose) min, respectively. The key peak of the derivative from 1 corresponded with the derivative of control d-glucose (tR 22.63 min), which confirmed the D configuration of the sugar moiety.
The absolute configuration of the sugar moiety of 2 was determined by the same method and analytical procedure as those of 1. The peak of the derivative from 2 was observed at 21.73 min, which coincided with the derivative of control D-glucose (tR 21.77 min).
Ranunculoside A (=(2S)-ent-kauran-2β-ol-15-en-14-O-β-d-glucopyranoside; 1): Amorphous white powder; M.p. 230–232 °C; = −31.0 (c 0.01, MeOH); UV (MeOH) λmax 206 nm; IR (KBr) νmax: 3371, 2925, 2852, 1161, 1036 cm−1; 1H- and 13C-NMR data (in C5D5N), see Table 1 and Table 2; HR-ESI-MS: 489.28287 ([M + Na]+, calcd. for C26H42O7Na, 489.28227).
The aglycone (1a) of 1(=(2S)-ent-2β,14α-diol-15-en-kauran; 1a). Amorphous white powder; M.p. 246–248 °C; = −17.2 (c 0.05, MeOH); UV (MeOH) λmax 204 nm; IR (KBr) νmax: 3367, 2925, 2854, 2360, 1383, 1036 cm−1; 1H- and 13C-NMR data (in C5D5N), see Table 1 and Table 2; HR-ESI-MS: 327.22969 ([M + Na]+, C20H32NaO2+; calcd. 327.22945).
Ranunculoside B (=(2S,4S)-ent-kauran-2β,18-diol-15-en-14-O-β-d-glucopyranoside; 2). Amorphous white powder; M.p. 198–200 °C; = −20.4 (c 0.05, MeOH); UV (MeOH) λmax 206 nm; IR (KBr) νmax: 3371, 2932, 2856, 1169, 1033 cm−1; 1H- and 13C-NMR data (in C5D5N), see Table 1 and Table 2; HR-ESI-MS: 505.27707 ([M + Na]+, C26H42NaO8+; calcd. 505.27719).
Ranunculone C (=(R)-3-[2-(3,4-dihydroxybenzoyl)-4,5-dihydroxyphenyl]-2-hydroxylpropanoic acid; 3). Amorphous yellow powder; M.p. 203–205 °C; = +7.5 (c 0.05, MeOH); UV (MeOH) λmax (log ε): 204 (4.29), 236 (4.00), 287 (3.80), 322 (3.84) nm; CD: 219 nm (Δε-0.17); IR (KBr) νmax: 3422 (br s), 1715, 1592, 1521, 1298, 889, 832, 784, 635 cm−1; 1H- and 13C-NMR data (in CD3OD), see Table 3; HR-ESI-MS: 357.05848 ([M + Na]+, C16H14NaO8+; calcd. 357.05809).
4. Conclusions
Ranunculosides A (1) and B (2) are the first two ent-kaurane diterpenoids present in glycoside from the genus plants. Up until now, with the exception of R. ternatus [11], R. muricatus is the second Ranunculus plant to have benzophenone-type compounds isolated out. All the compounds showed no cytotoxicity against HEp-2 and HeLa cell lines at a test concentration of 50 μg/mL by MTT assay.
Acknowledgments
This project was partly supported by the Foundation (No. 8351063201000003) of Guangdong Natural Sciences Committee, China. All the spectroscopic data were measured in the Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine and New Drug Research at Jinan University.
Author Contributions
Bi-Ling Wu and Guangxiong Zhou conceived and designed the experiments and wrote the paper. Bi-Ling Wu and Fang-Min Qin performed the experiments. Bi-Ling Wu and Guangxiong Zhou analyzed the spectroscopic data and determined the chemical structures of the natural products. Hui-Liang Zou and Hong-yu Li contributed to bioassay of compounds.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lal, S.D.; Yadav, B.K. Folk medicines of Kurukshetra district (Haryana), India. Econ. Bot. 1983, 37, 299–305. [Google Scholar] [CrossRef]
- Wang, A.W.; Wang, M.; Yuan, J.-R.; Tian, J.-K.; Wu, L.-M.; Geng, H. The study on antitumour effects in vitro of different extracts in Radix Ranunculus Ternati. J. Nat. Prod. Res. Dev. 2004, 16, 529–531. [Google Scholar]
- Xie, J.P.; He, Y.; Yue, J.; Hu, C.H.; Wang, H.-H. Identification of differential expression proteins of Mycobacterium tuberculosis strain isolated from clinical species treated with Radix Ranuncoli Ternati Extracts by comparative proteomics. Chin. Biochem. Mol. Biol. 2006, 22, 63–69. [Google Scholar]
- Qasem, J.R. Fungicidal activity of Ranunculus asiaticus and other weeds against Fusarium oxysporum f. sp. lycopersici. Ann. Appl. Biol. 1996, 128, 533–540. [Google Scholar] [CrossRef]
- Zhong, Y.M.; Feng, Y.-F. Advances in studies on flavonoids and lactones in plants of Ranunculus Linn. Chin. Tradit. Herb. Drug 2011, 42, 825–828. [Google Scholar]
- Xiong, Y.; Deng, K.Z.; Guo, Y.Q.; Gao, W.Y. Studies on chemical constituents of flavonoids and glycosides in Ranunculus ternatus. Chin. Tradit. Herb. Drug 2008, 39, 1449–1452. [Google Scholar]
- Wang, L.J.; Gao, X.Z. Studies on the chemical constituents in Ranunculus muricatus L. Chin. JMAP 2009, 26, 460–462. [Google Scholar]
- Wu, B.L.; Qin, F.M.; Zhou, G.X. Studies on chemical constituents of Ranunculus muricatus Linn. Nat. Prod. Res. Dev. 2013, 25, 736–741. [Google Scholar]
- Ge, X.; Ye, G.; Li, P.; Tang, W.J.; Gao, J.L.; Zhao, W.M. Cytotoxic diterpenoids and sesquiterpenoids from Pteris multifida. J. Nat. Prod. 2008, 71, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Ye, Q.; Oberlies, N.H.; Shi, G.; Gu, Z.M.; He, K.; Mclaughlin, J.L. Recent advances in Annonaceous acet-ogenins. Nat. Prod. Rep. 1996, 13, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Deng, K.Z.; Gao, W.Y.; Guo, Y.Q.; Zhang, T.J. A novel alkenoic acid ester and a new benzophenone from Ranunculus ternatus. Chin. Chem. Lett. 2007, 18, 1364–1366. [Google Scholar] [CrossRef]
- Zhang, D.C.; Wu, W.L.; Liu, X.X.; Pan, D.J.; Yang, C.Y.; Zhang, B.T. Study on the water-soluble constituents of Salvia miltiorrhiza. Chin. Tradit. Pat. Med. 1982, 1, 9, 24–25. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).