3.4. Direct acylation of Barbituric Acid Templates
General procedure: To a solution of carboxilic acid (1.0 eq) in dichloromethane were added DCC (1.1 eq), barbituric acid (1.0 eq) and DMAP (1.2 eq) at room temperature. The mixture was stirred overnight at room temperature. The crude reaction mixture was filtered with dichloromethane. Concentration in vacuo followed by flash column chromatography gave metal-chelated barbituric acid. The crude product was dissolved in dichloromethane and washed with 1M HCl. The organic layer was dried with MgSO4 and concentrated in vacuo to give 3-acylbarbituric acid.
3.4.1. Synthesis of Compound 3
Yield; 51%; M.P.; 92 °C; 1H-NMR (400 MHz, CDCl3); 3.73 (t, 2H, J = 6.4 Hz, C11), 3.36 (t, 2H, J = 6.4 Hz, C10), 3.35 (s, 3H, C7), 3.31 (s, 3H, C8), 1.16 (s, 9H, C13–C15). 13C-NMR (100 MHz, CDCl3); 197.5 (C9), 169.7 (C2), 160.8 (C4), 150.3 (C6), 95.7 (C3), 73.2 (C12), 57.5 (C11), 38.3 (C10), 28.0 (N-CH3), 27.8 (N-CH3), 27.4 (C13–C15). MS (ES−); 283.14 (M−H), MS (ES+); 307.14 (M+Na), HRMS (M+Na); calcd for C13H20N2Na1O5; 307.1264; found; 307.1269.
3.4.2. Synthesis of Compound 4
Yield; 57% (oil); 1H-NMR (400 MHz, CDCl3); 3.98–3.89 (m, 4H, C7 and C9), 3.14 (s, 2H, C12), 1.22–1.13 (m, 6H, C8 and C10), 1.02 (s, 9H, C14–C16). 13C-NMR (100 MHz, CDCl3); 198.5 (C11), 169.5 (C2), 160.6 (C4), 149.3 (C6), 96.8 (C3), 46.8 (C12), 36.5 (C7 and C9), 33.4 (C13), 29.9 (C14–C16), 13.1 (C8 and C10). MS (ES−); 281.15 (M−H); HRMS (M−H); calcd for C14H21N2O4; 281.1507; found; 281.1505.
3.4.3. Synthesis of Compound (±)-5b
Yield; 71% (oil); 1H-NMR (400 MHz, CDCl3); 4.03–3.94 (m, 4H, C7 and C9), 3.08 (d, 2H, J = 7.2 Hz, C12), 2.22–2.12 (m, 1H, C13), 1.35 (dd, 1H, J1 = 14.0 Hz, J2 = 3.2 Hz, C14), 1.26–1.15 (m, 7H, C8, C10 and C14), 1.01 (d, 3H, J = 6.8 Hz, C19), 0.90 (s, 9H, C16-C18). 13C-NMR (100 MHz, CDCl3); 199.3 (C11), 169.6 (C2), 160.5 (C4), 149.7 (C6), 96.0 (C3), 50.7 (C14), 45.5 (C12), 36.6 (NCH2), 36.5 (NCH2), 31.1 (C15), 29.9 (C16–C18), 27.7 (C13), 22.7 (C19), 13.2 (C8 and C10). MS (ES−); 323.20 (M−H); HRMS (M−H); calcd for C17H27N2O4; 323.1976; found; 323.1980.
3.4.4. Synthesis of Compound (±)-5c
Yield; 42% (oil); 1H-NMR (400 MHz, CDCl3); 4.17–4.12 (m, 2H, C9), 4.00–3.92 (m, 2H, C7), 3.62–3.55 (m, 2H, C10), 3.33 and 3.32 (2 of s, 3H, C11), 3.06 (d, 2H, J = 7.2 Hz, C13), 2.19–2.12 (m, 1H, C14), 1.35–1.30 (m, 1H, C15), 1.24–1.12 (m, 4H, C8 and C15), 0.99 and 0.98 (2 of d, 3H, J = 6.8 Hz, C20), 0.87 (s, 9H, C17–C19). 13C-NMR (100 MHz, CDCl3); 199.3 and 199.2 (C12), 169.7 and 169.5 (C2), 160.6 and 160.3 (C4), 149.7 and 149.6 (C6), 95.9 and 95.8 (C3), 69.3 and 69.2 (C10), 58.7 and 58.6 (C11), 50.6 (C15), 45.4 and 45.3 (C13), 40.1 and 40.0 (C9), 36.7 and 36.6 (C7), 31.0 (C16), 29.8 (C17–C19), 27.7 (C14), 22.7 (C20), 13.1 (C8). MS (ES−); 353.22 (M−H); MS (ES+); 355.23 (M+H), 377.20 (M+Na); HRMS (M+Na); calcd for C18H30N2Na1O5; 377.2047; found; 377.2040.
3.4.5. Synthesis of Compound 6a
Yield; 90%; M.P.; 133 °C; 1H-NMR (500 MHz, CDCl3); 3.35 (s, 3H, C7), 3.31 (s, 3H, C8), 3.06 (s, 2H, C10), 1.99 (brs, 3H, C18–C20), 1.65–1.34 (m, 12H, C12–C17). 13C-NMR (125 MHz, CDCl3); 197.8 (C9), 169.7 (C2), 161.1 (C4), 150.2 (C6), 97.1 (C3), 47.9 (C10), 42.6 (CH2), 36.6 (CH2), 36.3 (C11), 28.8 (N-CH3), 28.1 (CH), 27.8 (CH). MS (ES−); 331.18 (M−H), HRMS (M−H); calcd for C18H23N2O4; 331.1663; found; 331.1657.
3.4.6. Synthesis of Compound 6b
Yield; 54% (oil); 1H-NMR (400 MHz, CDCl3); 3.36 (s, 3H, C7), 3.31 (s, 3H, C8), 3.08 (s, 2H, C10), 1.99 (brs, 2H, C18 and C19), 1.65–1.34 (m, 12H, C12-C17), 0.78 (s, 3H, C21). 13C-NMR (100 MHz, CDCl3); 197.8 (C9), 169.7 (C2), 161.1 (C4), 150.2 (C6), 97.1 (C3), 49.6 (CH2), 47.6 (C10), 43.6 (CH2), 41.8 (CH2), 37.0 (C11), 35.8 (CH2), 30.9 (C21), 30.8 (C20), 29.3 (C18 and C19), 28.1 (N-CH3), 27.8 (N-CH3). MS (ES−); 345.20 (M−H), HRMS (M−H); calcd for C19H25N2O4; 345.1820; found; 345.1811.
3.4.7. Synthesis of Compound 6c
Yield; 39%; M.P.; 77 °C; 1H-NMR (400 MHz, CDCl3); 3.37 (s, 3H, C7), 3.32 (s, 3H, C8), 3.10 (s, 2H, C10), 2.05–2.02 (m, 1H, C20), 1.52–1.07 (m, 12H, C12–C17), 0.79 (s, 6H, C21 and C22). 13C-NMR (100 MHz, CDCl3); 197.8 (C9), 169.7 (C2), 161.1 (C4), 150.2 (C6), 97.1 (C3), 50.8 (CH2), 48.9 (CH2), 47.3 (C10), 42.9 (CH2), 41.0 (CH2), 37.7 (C18 and C19), 31.5 (C11), 30.5 (C21 and C22), 29.8 (C20), 28.2 (N-CH3), 27.8 (N-CH3). MS (ES−); 359.18 (M−H), HRMS (M-H); calcd for C20H27N2O4; 359.1976; found; 359.1965.
3.4.8. Synthesis of Compound 7a
Yield; 50%; M.P.; 130 °C; 1H-NMR (400 MHz, CDCl3); 3.36 (s, 3H, C7), 3.33 (s, 3H, C8), 3.11–3.07 (m, 2H, C10), 1.98 (brs, 3H, C19–C21), 1.73–1.54 (m, 12H, C13–C18), 1.45–1.41 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 201.2 (C9), 169.7 (C2), 160.8 (C4), 150.4 (C6), 95.2 (C3), 42.0 (C10), 40.0 (CH2), 37.0 (CH2), 32.4 (C12), 30.8 (CH2), 28.6 (N-CH3), 28.0 (CH), 27.8 (CH). MS (ES−); 345.19 (M−H), HRMS (M−H); calcd for C19H25N2O4; 345.1820; found; 345.1815.
3.4.9. Synthesis of Compound 7b
Yield; 44% (oil); 1H-NMR (400 MHz, CDCl3); 4.18–4.14 (m, 2H, C9), 4.02–3.95 (m, 2H, C7), 3.63–3.57 (m, 2H, C10), 3.34 and 3.33 (2 of s, 3H, C11), 3.11–3.07 (m, 2H, C13), 1.96 (brs, 3H, C22–C24), 1.71–1.61 (m, 6H, C16, C17 and C20), 1.52 (brs, 6H, C18, C19 and C21), 1.44–1.40 (m, 2H, C14), 1.26–1.18 (m, 3H, C8). 13C-NMR (100 MHz, CDCl3); 201.3 and 201.2 (C12), 169.7 and 169.5 (C2), 160.5 and 160.3 (C4), 149.8 and 149.7 (C6), 95.3 and 95.2 (C3), 69.4 and 69.2 (C10), 58.7 and 58.6 (C11), 42.0 (C16, C17 and C20), 40.1 and 40.0 (C9), 39.8 and 39.7 (C13), 37.0 (C18, C19 and C21), 36.7 and 36.6 (C7), 32.3 (C15), 30.8 and 30.7 (C14), 28.6 (C22–C24), 13.2 and 13.1 (C8). MS (ES−); 403.23 (M-H); MS (ES+); 427.21 (M+Na); HRMS (M+Na); calcd for C22H32N2Na1O5; 427.2203; found; 427.2191.
3.4.10. Synthesis of Compound 8a
Yield; 25%; M.P.; 55 °C; 1H-NMR (400 MHz, CDCl3); 3.34 (s, 3H, C7), 3.31 (s, 3H, C8), 3.14–3.10 (m, 2H, C10), 1.76–1.62 (m, 5H, CH2), 1.58–1.52 (m, 2H, C11), 1.37–1.27 (m, 1H, C12), 1.23–1.11 (m, 3H, CH2), 0.98–0.89 (m, 2H, CH2). 13C-NMR (100 MHz, CDCl3); 200.3 (C9), 169.7 (C2), 160.7 (C4), 150.3 (C6), 95.1 (C3), 37.6 (C12), 34.4 (C10), 33.1 (C11), 32.9 (CH2), 28.0 (N-CH3), 27.7 (N-CH3), 26.5 (CH2), 26.2 (CH2). MS (ES−); 293.16 (M−H), HRMS (M−H); calcd for C15H21N2O4; 293.1507; found; 293.1510.
3.4.11. Synthesis of Compound 8b
Yield; 53% (oil); 1H-NMR (400 MHz, CDCl3); 3.34 (s, 3H, C7), 3.30 (s, 3H, C8), 3.09 (t, 2H, J = 7.6 Hz, C10), 1.71–1.60 (m, 7H, C13 and CH2), 1.30–1.09 (m, 6H, CH2), 0.91–0.81 (m, 2H, CH2). 13C-NMR (100 MHz, CDCl3); 199.9 (C9), 169.7 (C2), 160.7 (C4), 150.3 (C6), 95.1 (C3), 37.3 (C13), 37.1 (CH2), 36.9 (CH2), 33.1 (CH2), 27.9 (N-CH3), 27.7 (N-CH3), 26.6 (CH2), 26.2 (CH2), 23.1 (CH2). MS (ES−); 307.16 (M−H); HRMS (M−H); calcd for C16H23N2O4; 307.1663; found; 307.1661.
3.4.12. Synthesis of Compound 9
Yield; 84%; M.P.; 67 °C; 1H-NMR (400 MHz, CDCl3); 5.00 (d, 1H, J = 20.4 Hz, C10), 4.87 (d, 1H, J = 20.4 Hz, C10), 3.33 (s, 3H, C7), 3.27 (s, 3H, C8), 3.21–3.15 (m, 1H, C11), 2.29–2.21 (m, 1H, C15), 2.09–2.05 (m, 1H, C16), 1.62–1.57 (m, 2H, CH2), 1.35–1.27 (m, 2H, C12 and C18), 1.00–0.80 (m, 11H, C13, C14, C16, C19 and C20), 0.74 (d, 3H, J = 6.8 Hz, C17). 13C-NMR (100 MHz, CDCl3); 197.1 (C9), 169.4 (C2), 160.5 (C4), 150.1 (C6), 93.5 (C3), 80.5 (C11), 69.3 (C10), 47.9 (C12), 39.9 (C16), 34.3 (C14), 31.4 (N-CH3), 27.7 (N-CH3), 25.5 (C15), 23.2 (C13), 22.1 (C18), 20.8 (C19 and C20), 16.2 (C17). MS (ES−); 351.19 (M-H); MS (ES+); 375.22 (M+H); HRMS (M−H); calcd for C18H27N2O5; 351.1925; found; 351.1922.
3.4.13. Synthesis of Compound (±)-10
Yield; 51% (oil); 1H-NMR (500 MHz, CDCl3); 3.32 (s, 3H, C7), 3.28 (s, 3H, C8), 3.15–3.05 (m, 2H, C10), 2.25–2.21 (m, 1H, C13), 2.18 (d, 1H, J = 12.5 Hz, C16), 2.07 (d, 1H, J = 12.5 Hz, C16), 2.02–1.97 (m, 1H, C17), 1.88–1.81 (m, 1H, C12), 1.69–1.63 (m, 2H, C11), 1.62–1.43 (m, 3H, C15 and C17), 1.30–1.21 (m, 1H, C12), 1.01 (s, 3H, CH3), 0.83 (s, 3H, CH3). 13C-NMR (125 MHz, CDCl3); 212.4 (C14), 199.3 (C9), 169.6 (C2), 160.7 (C4), 150.3 (C6), 95.1 (C3), 54.8 (C16), 49.1 (C13), 37.9 (C10), 36.9 (C17), 36.6 (C18), 31.3 (CH3), 29.4 (C12 or C15), 28.8 (C12 or C15), 27.9 (N-CH3), 27.7 (N-CH3), 25.5 (CH3), 23.3 (C11). MS (ES−); 349.19 (M−H); MS (ES+); 351.21 (M+H), 373.19 (M+Na); HRMS (M+Na); calcd for C18H26N2Na1O5; 373.1734; found; 373.1723.
3.4.14. Synthesis of Compound cis-11
Yield; 46 % (oil); 1H-NMR (400 MHz, CDCl3); 3.99–3.91 (m, 4H, C7 and C9), 3.20 (dd, 1H, J1 = 15.2 Hz, J2 = 6.4 Hz, C12), 3.05 (dd, 1H, J1 = 15.2 Hz, J2 = 8.0 Hz, C12), 2.84 (dd, 1H, J1 = 10.0 Hz, J2 = 7.6 Hz, C15), 2.43–2.34 (m, 1H, C13), 2.11–2.03 (m, 1H, C14), 2.01 (s, 3H, C20), 1.92–1.85 (m, 1H, C14), 1.31 (s, 3H, C17), 1.23–1.16 (m, 6H, C8 and C10), 0.90 (s, 3H, C17). 13C NMR (100 MHz, CDCl3); 207.3 (C20), 198.8 (C11), 169.5 (C2), 160.4 (C4), 149.4 (C6), 95.2 (C3), 54.2 (C15), 43.7 (C16), 38.5 (C13), 37.3 (C12), 36.6 (NCH2), 36.5 (NCH2), 30.0 (C18 and C20), 23.2 (C14), 17.5 (C17), 13.1 (C8 and C10). MS (ES−); 349.18 (M−H); MS (ES+); 373.18 (M+Na); HRMS (M+Na); calcd for C18H26Na1N2O5; 373.1734; found; 373.1728.
3.4.15. Synthesis of Compound (±)-12
Yield; 35% (oil); 1H-NMR (400 MHz, CDCl3); 4.73 (brs, 1H, C18), 4.21 (d, 1H, J = 15.6 Hz, C17), 4.12 (d, 1H, J = 15.6 Hz, C17), 3.79–3.74 (m, 1H, C22), 3.47–3.43 (m, 1H, C22), 3.30 (s, 3H, C7), 3.25 (s, 3H, C8), 3.07 (t, 2H, J = 7.2 Hz, C10), 2.28–2.16 (m, 2H, C14), 1.78–1.45 (m, 10H, CH2). 13C-NMR (100 MHz, CDCl3); 199.3 (C9), 169.6 (C2), 160.6 (C4), 150.1 (C6), 96.3 (C18), 95.0 (C3), 86.0 (C15), 75.9 (C16), 61.7 (C22), 54.4 (C17), 36.4 (C10), 30.1 (CH2), 28.4 (CH2), 28.0 (CH2), 27.8 (N-CH3), 27.6 (N-CH3), 25.2 (CH2), 25.1 (CH2), 18.9 (CH2), 18.5 (CH2). MS (ES−); 391.19 (M−H); HRMS (M+Na); calcd for C20H28N2Na1O6; 415.1840; found; 415.1821.
3.4.16. Synthesis of Compound 13
Yield; 87%; M.P.; 245 °C (decomposed); 1H-NMR (400 MHz, CDCl3); 3.35 (s, 3H, C7), 3.32 (s, 3H, C8), 3.20–3.05 (m, 2H), 2.94–2.81 (m, 3H), 2.37–1.81 (m, 15H), 1.70–1.57 (m, 2H), 1.39 (s, 3H, C32), 1.33–1.24 (m, 2H), 1.07 (s, 3H, C31), 0.92 (d, 3H, J = 6.4 Hz, C13). 13C-NMR (100 MHz, CDCl3); 211.8 (C=O), 209.0 (C=O), 208.7 (C=O), 200.1 (C=O), 169.7 (C2), 160.7 (C4), 150.3 (C6), 95.2 (C3), 56.9 (C18), 51.7 (CH), 48.9 (CH), 46.8 (CH), 45.7 (CH), 45.5 (CH), 44.9 (CH2), 42.7 (CH2), 38.6 (CH2), 36.4 (CH2), 36.3 (C12), 35.9 (C26), 35.2 (CH2), 34.3 (CH2), 33.9 (CH2), 31.5 (CH2), 28.0 (N-CH3), 27.8 (N-CH3), 27.6 (CH2), 25.6 (CH2), 25.1 (CH2), 24.9 (CH2), 21.8 (C13), 18.7 (C32), 11.8 (C31). MS (ES−); 539.30 (M−H), HRMS (M-H); calcd for C30H39N2O7; 539.2763; found; 539.2763.
3.4.17. Synthesis of Compound 14
Yield; 66%; M.P. 96 °C; 1H-NMR (500 MHz, CDCl3); 5.79 (d, 1H, J = 10.0 Hz, C13), 5.42 (s, 1H, C17), 3.62–3.56 (m, 1H, C11), 3.50–3.44 (m, 1H, C11), 3.36 (s, 3H, C7), 3.32 (s, 3H, C8), 2.81 (t, 2H, J = 6.5 Hz, C10), 2.59–2.52 (m, 1H, C14), 2.36 (td, 1H, J1 = 13.5 Hz, J2 = 3.5 Hz, CH2), 2.05–2.00 (m, 1H, CH2), 1.91–1.85 (m, 1H, CH2), 1.79–1.69 (m, 2H, C24 and CH2), 1.63–1.59 (m, 1H, C15), 1.51–1.40 (m, 1H, CH2), 1.43 (s, 3H, C26), 1.38–1.24 (m, 3H, C21, C22 and C24), 1.04–0.99 (m, 1H, CH2), 0.95 (d, 3H, J = 6.0 Hz, C27), 0.85 (d, 3H, J = 7.0 Hz, C25). 13C-NMR (125 MHz, CDCl3); 197.4 (C9), 171.0 (C12), 169.7 (C2), 160.7 (C4), 150.2 (C6), 104.4 (C18), 95.2 (C3), 92.2 (C13), 91.4 (C17), 80.0 (C16), 51.5 (C21), 45.2 (C15), 37.2 (C22), 36.1 (CH2), 34.0 (CH2), 31.9 (C11), 31.8 (C14), 28.6 (C10), 27.9 (N-CH3), 27.8 (N-CH3), 25.9 (C26), 24.5 (CH2), 21.9 (C24), 20.2 (C27), 12.0 (C25). MS (ES−); 521.22 (M−H); HRMS (M−H); calcd for C25H33N2O10; 521.2141; found; 521.2148.
3.4.18. Synthesis of Compound 15a
Yield; 27%; M.P.; 118 °C; 1H-NMR (400 MHz, CDCl3); 6.83 (brs, 1H, NH), 4.98 (s, 2H, CH2), 4.07 (s, 2H, CH2), 3.38 (s, 3H, C7), 3.34–3.28 (m, 5H, C8 and C13), 2.38–2.32 (m, 1H, C18), 2.16–2.18 (m, 1H, C14), 1.98–1.81 (m, 5H, C15, C16, C19 and C20), 1.54–1.44 (m, 1H, C19), 1.18 (s, 3H, C21), 1.03 (s, 3H, C22), 0.89 (d, 1H, J = 9.6 Hz, C18). 13C-NMR (100 MHz, CDCl3); 195.2 (C9), 169.5 (C12), 168.6 (C2), 160.4 (C4), 150.0 (C6), 93.9 (C3), 72.1 (CH2), 71.3 (CH2), 44.4 (C13), 43.7 (CH), 41.2 (CH), 41.2 (CH), 38.6 (C17), 33.1 (CH2), 28.0 (CH3), 27.9 (CH3), 27.8 (CH3), 25.9 (CH2), 23.1 (CH3), 19.7 (CH2). MS (ES−); 406.20 (M−H); HRMS (M−H); calcd for C20H28N3O6; 406.1984; found; 406.1986.
3.4.19. Synthesis of Compound 15b
Yield; 43% (oil); 1H-NMR (500 MHz, CDCl3); 5.95 (brs, 1H, NH), 3.34–3.16 (m, 10H, C7, C8, C10 and C14), 2.30–2.27 (m, 1H, C21), 2.25 (s, 2H, C12), 2.17–2.10 (m, 1H, C17), 1.92–1.77 (m, 5H, C18, C20, C22 and C23), 1.47–1.41 (m, 1H, C22), 1.12 (s, 9H, CH3), 0.98 (s, 3H, CH3), 0.84 (d, 1H, J = 9.5 Hz, C21). 13C-NMR (125 MHz, CDCl3); 198.4 (C9), 170.7 (C13), 169.9 (C2), 160.8 (C4), 149.9 (C6), 96.8 (C3), 48.3 (CH2), 45.0 (CH2), 44.5 (CH2), 43.8 (CH), 41.3 (CH), 41.2 (CH), 38.5 (C19), 35.4 (C11), 33.0 (CH2), 28.1 (CH3), 28.1 (CH3), 27.8 (CH3), 25.8 (CH2), 23.1 (CH3), 19.8 (CH2). MS (ES−); 432.24 (M−H); MS (ES+); 434.29 (M+H), 456.27 (M+Na); HRMS (M+H); calcd for C23H36N3O5; 434.2649; found; 434.2634.
3.4.20. Synthesis of Compound 16
Yield; 49%; M.P.; 102 °C; 1H-NMR (500 MHz, CDCl3); 8.39 (brs, 1H, ArH), 7.39 (brs, 1H, ArH), 7.14 (d, 1H, J = 7.5 Hz, C20), 3.98 (brs, 4H, C24 and C25), 3.43 (s, 2H, C10), 3.37 (s, 3H, C7), 3.31 (s, 3H, C8), 3.21 (brs, 4H, C22 and C23), 2.43 (s, 2H, C12), 1.22 (s, 6H, C14 and C15). 13C-NMR (125 MHz, CDCl3); 198.2 (C9), 170.0 (C2), 162.7 (C13), 162.0 (C4), 155.5 (d, JC-F = 245 Hz, C19), 149.9 (C6), 135.6 (C21), 120.3 (C16), 115.5 (Ar-tert C), 115.5 (Ar-tert C), 109.0 (d, JC-F = 25.5 Hz, C17), 97.0 (C3), 66.0 (C24 and C25), 51.8 (C22 and C23), 48.2 (CH2), 44.4 (CH2), 36.0 (C11), 28.3 (C14 and C15), 28.2 (N-CH3), 28.0 (N-CH3). MS (ES−); 475.20 (M−H), MS (ES+); 499.23 (M+Na), HRMS (M+Na); calcd for C23H29F1N4Na1O6; 499.1963; found; 499.1964.
3.4.21. Synthesis of Compound 17a
Yield; 84%; M.P.; 95 °C; 1H-NMR (500 MHz, CDCl3); 7.27–7.23 (m, 2H, C16 and C17), 6.91 (t, 1H, J = 7.0 Hz, C18), 6.85 (d, 2H, J = 8.0 Hz, C14 and C15), 4.05 (t, 2H, J = 6.0 Hz, C12), 3.37–3.34 (m, 5H, C10 and C7), 3.29 (s, 3H, C8), 2.23–2.17 (m, 2H, C11). 13C-NMR (125 MHz, CDCl3); 198.8 (C9), 169.6 (C2), 160.7 (C4), 158.6 (C13), 150.1 (C6), 129.2 (C16 and C17), 120.6 (C18), 114.3 (C14 and C15), 95.3 (C3), 66.7 (C12), 33.6 (C10), 27.8 (N-CH3), 27.7 (N-CH3), 25.1 (C11). MS (ES−); 317.13 (M−H), HRMS (M−H); calcd for C16H17N2O5; 317.1143; found; 317.1141.
3.4.22. Synthesis of Compound 17b
Yield; 63%; M.P.; 84 °C; 1H-NMR (400 MHz, CDCl3); 7.34–7.23 (m, 5H, C15-C19), 4.49 (s, 2H, C13), 3.58 (t, 2H, J = 6.0 Hz, C12), 3.33 (s, 3H, C7), 3.31 (s, 3H, C8), 3.27 (t, 2H, J = 6.0 Hz, C10), 2.07–2.00 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 199.4 (C9), 169.6 (C2), 160.8 (C4), 150.2 (C6), 138.3 (C14), 128.2 (C17 and C18), 127.5 (C15 and C16), 127.4 (C19), 95.2 (C3), 72.8 (C13), 69.4 (C12), 33.8 (C10), 27.9 (N-CH3), 27.7 (N-CH3), 25.7 (C11). MS (ES−); 331.13 (M−H); MS (ES+); 355.14 (M+Na); HRMS (M+Na); calcd for C17H20N2Na1O5; 355.1264; found; 355.1262.
3.4.23. Synthesis of Compound 17c
Yield; 70%; M.P.; 142 °C; 1H-NMR (400 MHz, CDCl3); 7.77–7.70 (m, 3H, C16, C19 and C20), 7.44 (dd, 1H, J1=J2 = 6.8 Hz, C21), 7.33 (dd, 1H, J1=J2 = 6.8 Hz, C22), 7.12–7.11 (m, 2H, C14 and C15), 4.20 (t, 2H, J = 5.6 Hz, C12), 3.41 (t, 2H, J = 5.6 Hz, C10), 3.36 (s, 3H, C7), 3.29 (s, 3H, C8), 2.31–2.25 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 198.9 (C9), 169.7 (C2), 160.8 (C4), 156.6 (C13), 150.2 (C6), 134.5 (quart C), 129.3 (tert C), 128.9 (quart C), 127.6 (tert C), 126.6 (tert C), 126.3 (tert C), 123.6 (tert C), 118.8 (tert C), 95.4 (C3), 66.9 (C12), 33.7 (C10), 27.9 (N-CH3), 27.8 (N-CH3), 25.2 (C11). MS (ES−); 367.14 (M−H); MS (ES+); 369.19 (M+H), 391.14 (M+Na); HRMS (M+Na); calcd for C20H20N2Na1O5; 391.1264; found; 391.1261.
3.4.24. Synthesis of Compound 18a
Yield; 75%; M.P.; 87 °C; 1H-NMR (400 MHz, CDCl3); 7.09 (d, 2H, J = 8.0 Hz, C16 and C17), 6.80 (d, 2H, J = 8.0 Hz, C14 and C15), 4.05 (t, 2H, J = 6.0 Hz, C12), 3.38–3.34 (m, 5H, C7 and C10), 3.31 (s, 3H, C8), 2.58 (q, 2H, J = 7.6 Hz, C19), 2.23–2.17 (m, 2H, C11), 1.21 (t, 3H, J = 7.6 Hz, C20). 13C-NMR (100 MHz, CDCl3); 199.0 (C9), 169.7 (C2), 160.8 (C4), 156.7 (C13), 150.3 (C6), 130.5 (C18), 128.6 (C16 and C17), 114.3 (C14 and C15), 95.4 (C3), 66.9 (C12), 33.7 (C10), 28.0 (N-CH3), 27.9 (C19), 27.8 (N-CH3), 25.3 (C11), 15.8 (C20). MS (ES−); 345.15 (M−H); MS (ES+); 369.16 (M+Na); HRMS (M+Na); calcd for C18H22N2Na1O5; 369.1421; found; 369.1412.
3.4.25. Synthesis of Compound 18b
Yield; 58%; M.P.; 92 °C; 1H-NMR (400 MHz, CDCl3); 7.29 (d, 2H, J = 8.8 Hz, C16 and C17), 6.81 (d, 2H, J = 8.8 Hz, C14 and C15), 4.06 (t, 2H, J = 6.4 Hz, C12), 3.39–3.35 (m, 5H, C7 and C10), 3.31 (s, 3H, C8), 2.24–2.17 (m, 2H, C11), 1.30 (s, 9H, C20–C22). 13C-NMR (100 MHz, CDCl3); 199.0 (C9), 169.7 (C2), 160.8 (C4), 156.4 (C13), 150.2 (C6), 143.4 (C18), 126.1 (C16 and C17), 113.4 (C14 and C15), 95.4 (C3), 66.9 (C12), 34.0 (C19), 33.7 (C10), 31.5 (C20–C22), 27.9 (N-CH3), 27.8 (N-CH3), 25.3 (C11). MS (ES−); 373.19 (M−H); MS (ES+); 397.20 (M+Na); HRMS (M+Na); calcd for C20H26N2Na1O5; 397.1734; found; 397.1740.
3.4.26. Synthesis of Compound 18c
Yield; 67%; M.P.; 150 °C; 1H-NMR (400 MHz, CDCl3); 7.00 (d, 2H, J = 7.6 Hz, C16 and C17), 6.91 (dd, 1H, J1 = 7.6 Hz, J2= 7.6 Hz, C18), 3.87 (t, 2H, J = 6.4 Hz, C12), 3.44 (t, 2H, J = 6.4 Hz, C10), 3.39 (s, 3H, C7), 3.35 (s, 3H, C8), 2.29 (s, 6H, C19 and C20), 2.25–2.18 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 199.1 (C9), 169.7 (C2), 160.8 (C4), 155.7 (C13), 150.3 (C6), 130.8 (C14 or C15), 128.8 (C16 and C17), 123.7 (C18), 95.3 (C3), 70.9 (C12), 33.7 (C10), 28.0 (N-CH3), 27.8 (N-CH3), 26.1 (C11), 16.3 (C19 and C20). MS (ES−); 345.16 (M−H); MS (ES+); 347.20 (M+H), 369.17 (M+Na); HRMS (M+Na); calcd for C18H22N2Na1O5; 369.1421; found; 369.1415.
3.4.27. Synthesis of Compound 18d
Yield; 72%; M.P.; 139 °C; 1H-NMR (400 MHz, CDCl3); 7.00 (d, 1H, J = 7.2 Hz, C16), 6.67 (d, 1H, J = 7.2 Hz, C18), 6.63 (s, 1H, C15), 4.07 (t, 2H, J = 6.0 Hz, C12), 3.42–3.38 (m, 5H, C7 and C10), 3.33 (s, 3H, C8), 2.32 (s, 6H, C20), 2.27–2.20 (m, 2H, C11), 2.17 (s, 6H, C19). 13C-NMR (100 MHz, CDCl3); 199.0 (C9), 169.7 (C2), 160.8 (C4), 156.6 (C13), 150.3 (C6), 136.4 (C17), 130.3 (C16), 123.5 (C14), 120.8 (C18), 111.8 (C15), 95.3 (C3), 66.7 (C12), 33.7 (C10), 27.9 (N-CH3), 27.8 (N-CH3), 25.3 (C11), 21.3 (C20), 15.7 (C19). MS (ES−); 345.15 (M−H); MS (ES+); 347.18 (M+H), 369.16 (M+Na); HRMS (M+Na); calcd for C18H22N2Na1O5; 369.1421; found; 369.1407.
3.4.28. Synthesis of Compound 18e
Yield; 43%; M.P.; 127 °C; 1H-NMR (400 MHz, CDCl3); 7.17 (d, 1H, J = 8.8 Hz, C16), 6.72 (d, 1H, J = 2.8 Hz, C15), 6.62 (dd, 1H, J1 = 8.8 Hz, J2= 2.8 Hz, C14), 4.01 (t, 2H, J = 6.0 Hz, C12), 3.36–3.29 (m, 8H, C7, C8 and C10), 2.30 (s, 3H, C19), 2.21–2.14 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 198.7 (C9), 169.7 (C2), 160.7 (C4), 157.2 (C13), 150.2 (C6), 136.8 (C17 or C18), 129.4 (C16), 125.7 (C17 or C18), 116.9 (C14 or C15), 112.9 (C14 or C15), 95.3 (C3), 67.1 (C12), 35.6 (C10), 27.9 (N-CH3), 27.8 (N-CH3), 25.1 (C11), 20.2 (C19). MS (ES−); 365.11 (M−H), HRMS (M+Na); calcd for C17H19Cl1N2Na1O5; 389.0875; found; 389.0863.
3.4.29. Synthesis of Compound 18f
Yield; 70%; M.P.; 123 °C; 1H-NMR (400 MHz, CDCl3); 7.31 (s, 1H, C17), 7.14 (d, 1H, J = 8.8 Hz, C16), 6.81 (d, 1H, J = 8.8 Hz, C14), 4.09 (t, 2H, J = 6.0 Hz, C12), 3.39–3.35 (m, 5H, C10 and C7), 3.29 (s, 3H, C8), 2.28–2.21 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 198.5 (C9), 169.7 (C2), 160.8 (C4), 153.0 (C13), 150.2 (C6), 129.8 (C17), 127.4 (C16), 125.6 (C18), 123.6 (C15), 113.8 (C14), 95.4 (C3), 68.4 (C12), 33.5 (C10), 27.9 (N-CH3), 27.8 (N-CH3), 24.9 (C11). MS (ES−); 385.06 (M−H), HRMS (M−H); calcd for C16H15Cl2N2O5; 385.0364; found; 385.0368.
3.4.30. Synthesis of Compound 18g
Yield; 56%; M.P.; 148 °C; 1H-NMR (400 MHz, CDCl3); 7.09–7.07 (m, 2H, C16 and C17), 6.70 (d, 1H, J = 8.0 Hz, C14), 4.04 (t, 2H, J = 6.0 Hz, C12), 3.40–3.36 (m, 5H, C10 and C7), 3.33 (s, 3H, C8), 2.26–2.20 (m, 2H, C11), 2.18 (s, 3H, C19). 13C-NMR (100 MHz, CDCl3); 198.8 (C9), 169.8 (C2), 160.8 (C4), 155.4 (C13), 150.3 (C6), 130.3 (C17), 128.7 (C15), 126.2 (C16), 125.0 (C18), 111.8 (C14), 95.4 (C3), 67.2 (C12), 33.7 (C10), 28.0 (N-CH3), 27.8 (N-CH3), 25.2 (C11), 16.0 (C19). MS (ES−); 365.10 (M−H), HRMS (M−H); calcd for C17H18Cl1N2O5; 365.0910; found; 365.0901.
3.4.31. Synthesis of Compound 19a
Yield; 37%; M.P. 77 °C; 1H-NMR (400 MHz, CDCl3); 7.07–7.05 (m, 2H, C18 and C19), 6.69 (d, 1H, J = 8.4 Hz, C17), 4.04–3.94 (m, 6H, C7, C9 and C14), 3.37 (t, 2H, J = 7.2 Hz, C12), 2.25–2.18 (m, 2H, C13), 2.17 (s, 3H, C21), 1.25 (t, 3H, J = 7.6 Hz, CH3), 1.20 (t, 3H, J = 7.6 Hz, CH3). 13C-NMR (100 MHz, CDCl3); 198.9 (C11), 169.5 (C2), 160.4 (C4), 155.4 (C15), 149.3 (C6), 130.3 (C18), 128.6 (quart C), 126.2 (C19), 124.9 (quart C), 111.7 (C17), 95.4 (C3), 67.1 (C14), 36.7 (N-CH2), 36.5 (N-CH2), 33.7 (C12), 25.0 (C13), 16.0 (C21), 13.2 (CH3), 13.1 (CH3). MS (ES−); 393.12 (M−H); HRMS (M−H); calcd for C19H22Cl1N2O5; 393.1223; found; 393.1215.
3.4.32. Synthesis of Compound 19b
Yield; 40% (oil); 1H-NMR (400 MHz, CDCl3); 7.04 (d, 2H, J = 8.4 Hz, C18 and C19), 6.76 (d, 2H, J = 8.4 Hz, C17 and C16), 4.05–3.93 (m, 6H, C7, C9 and C14), 3.35 (t, 2H, J = 7.2 Hz, C12), 2.27 (s, 3H, C21), 2.23–2.16 (m, 2H, C13), 1.28–1.17 (m, 6H, C8 and C10). 13C-NMR (100 MHz, CDCl3); 199.1 (C11), 169.5 (C2), 160.4 (C4), 156.5 (C15), 149.3 (C6), 129.8 (C20), 129.7 (C18 and C19), 114.1 (C16 and C17), 95.4 (C3), 66.9 (C14), 36.6 (N-CH2), 36.5 (N-CH2), 33.7 (C12), 25.2 (C13), 20.3 (C21), 13.1 (CH3), 13.0 (CH3). MS (ES−); 359.17 (M−H); HRMS (M−H); calcd for C19H23N2O5; 359.1612; found; 359.1611.
3.4.33. Synthesis of Compound 20
Yield; 15% (oil); 1H-NMR (400 MHz, CDCl3); 7.93 (d, 2H, J = 8.4 Hz, C16 and C17), 7.49 (d, 2H, J = 8.4 Hz, C14 and C15), 3.63 (t, 2H, J = 6.0 Hz, C11), 3.41 (t, 2H, J = 6.0 Hz, C10), 3.37 (s, 3H, C7), 3.34 (s, 3H, C8), 1.34 (s, 9H, C20–C22). 13C-NMR (100 MHz, CDCl3); 198.6 (C9), 197.7 (C12), 169.7 (C2), 160.9 (C4), 157.0 (C18), 150.3 (C6), 133.7 (C13), 128.0 (C14 and C15), 125.5 (C16 and C17), 95.2 (C3), 35.1 (C19), 32.9 (CH2), 31.3 (CH2), 31.0 (C20-C22), 28.0 (N-CH3), 27.8 (N-CH3). MS (ES−); 371.17 (M−H), HRMS (M+Na); calcd for C20H24N2Na1O5; 395.1577; found; 395.1567.
3.4.34. Synthesis of Compound 21
Yield; 70%; M.P.; 173 °C; 1H-NMR (400 MHz, CDCl3); 7.52 (d, 2H, J = 8.0 Hz, C16 and C17), 7.36 (d, 2H, J = 8.0 Hz, C18 and C19), 7.20 (s, 1H, C14), 3.73 (t, 2H, J = 7.2 Hz, C11), 3.37 (s, 3H, C7), 3.32 (s, 3H, C8), 3.24 (t, 2H, J = 7.2 Hz, C10). 13C-NMR (100 MHz, CDCl3); 196.9 (C9), 169.7 (C2), 162.6 (quart C), 160.7 (C4), 150.3 (C6), 150.2 (quart C), 133.9 (C20), 129.1 (C18 and C19), 126.5 (C15), 125.2 (C16 and C17), 122.3 (C14), 95.4 (C3), 33.9 (C10), 28.0 (N-CH3), 27.9 (N-CH3), 23.2 (C11). MS (ES−); 388.08 (M−H), HRMS (M−H); calcd for C18H15Cl1N3O5; 388.0706; found; 388.0689.
3.4.35. Synthesis of Compound 22
Yield; 69% (oil); 1H-NMR (400 MHz, CDCl3); 7.31–7.27 (m, 2H, C16 and C17), 7.22–7.18 (m, 3H, C14, C15 and C18), 3.37 (s, 3H, C7), 3.34 (s, 3H, C8), 3.20 (t, 2H, J = 8.0 Hz, C10), 2.75 (t, 2H, J = 8.0 Hz, C10), 2.08–2.01 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 199.3 (C9), 169.7 (C2), 160.8 (C4), 150.3 (C6), 141.3 (C13), 128.4 (C16 and C17), 128.3 (C14 and C15), 126.0 (C18), 95.3 (C3), 36.3 (C10), 35.5 (C12), 28.0 (N-CH3), 27.8 (N-CH3), 27.2 (C11). MS (ES−); 301.13 (M−H), HRMS (M−H); calcd for C16H17N2O4; 301.1194; found; 301.1193.
3.4.36. Synthesis of Compound 23a
Yield; 20%; M.P.; 156 °C; 1H-NMR (400 MHz, CDCl3); 7.77 (d, 1H, J = 8.0 Hz, C15), 7.31–7.15 (m, 4H, C12 and C16-C18), 4.62 (s, 2H, C10), 3.77 (s, 3H, C19), 3.39 (s, 3H, C7), 3.33 (s, 3H, C8). 13C-NMR (100 MHz, CDCl3); 196.7 (C9), 169.8 (C2), 160.7 (C4), 150.2 (C6), 136.7 (C14), 128.6 (C12), 127.8 (C13), 121.7 (C18), 119.3 (C15 or C17), 119.2 (C15 or C17), 109.2 (C16), 106.5 (C11), 94.6 (C3), 32.6 (C19), 32.2 (C10), 28.0 (N-CH3), 27.7 (N-CH3). MS (ES−); 326.13 (M−H), HRMS (M+Na); calcd for C17H17N3Na1O4; 350.1111; found; 350.1099.
3.4.37. Synthesis of Compound 23b
Yield; 57%; M.P.; 182 °C; 1H-NMR (400 MHz, CDCl3); 7.97 (brs, 1H, NH), 7.62 (d, 1H, J = 8.0 Hz, C18), 7.35 (d, 1H, J = 8.4 Hz, C17), 7.19 (dd, 1H, J1 = J2 = 8.0 Hz, C19 or C20), 7.12 (dd, 1H, J1 = J2 = 8.0 Hz, C19 or C20), 7.04 (s, 1H, C14), 3.36 (s, 3H, C7), 3.33 (s, 3H, C8), 3.26 (t, 2H, J = 7.6 Hz, C10), 2.91 (t, 2H, J = 7.6 Hz, C12), 2.19–2.12 (m, 2H, C11). 13C-NMR (100 MHz, CDCl3); 199.7 (C9), 169.7 (C2), 160.8 (C4), 150.4 (C6), 136.3 (C15), 127.4 (C16), 121.9 (C19 or C20), 121.5 (C19 or C20), 119.2 (C17 or C18), 118.9 (C17 or C18), 115.6 (C13), 111.0 (C14), 95.3 (C3), 36.6 (C10), 28.0 (N-CH3), 27.8 (N-CH3), 26.0 (C12), 24.9 (C11). MS (ES−); 340.14 (M−H), HRMS (M−H); calcd for C18H18N3O4; 340.1303; found; 340.1295.
3.4.38. Synthesis of Compound 24
Yield; 77%; M.P.; 184 °C; 1H-NMR (400 MHz, CDCl3); 6.91 (d, 1H, J = 7.6 Hz, C14), 6.86 (d, 1H, J = 7.6 Hz, C16), 5.24 (s, 2H, C10), 3.42 (s, 3H, C7), 3.30 (s, 3H, C8), 2.26 (s, 3H, CH3), 2.23 (s, 3H, CH3), 2.20 (s, 3H, CH3). 13C-NMR (100 MHz, CDCl3); 194.7 (C9), 169.6 (C2), 160.5 (C4), 155.0 (C11), 150.1 (C6), 136.0 (quart C), 129.2 (quart C), 127.9 (C14), 127.8 (quart C), 125.8 (C16), 93.6 (C3), 72.5 (C10), 27.9 (N-CH3), 27.8 (N-CH3), 19.8 (CH3), 16.1 (CH3), 12.3 (CH3). MS (ES−); 331.15 (M−H), HRMS (M+Na); calcd for C17H20N2Na1O5; 355.1264; found; 355.1271.
3.4.39. Synthesis of Compound 25a
Yield; 58%; M.P.; 181 °C; Mixture of two exo-enol tautomers (E1: E2 = 40: 60); 1H-NMR (500 MHz, CDCl3); 8.84 (brs, 1H, NH E1), 8.54 (brs, 1H, NH E2), 5.92–5.83 (m, 1H, C8), 5.32–5.20 (m, 2H, C9), 4.53–4.50 (m, 2H, C7), 2.73 (s, 3H, C11). 13C-NMR (125 MHz, CDCl3); 196.9 (C10 E1), 196.4 (C10 E2), 170.1 (C2 E2), 169.0 (C4 E1), 161.2 (C2 E1), 161.0 (C4 E2), 149.0 (C6 E1), 148.8 (C6 E2), 131.5 (C8 E1), 130.8 (C8 E2), 118.8 (C9 E2), 118.1 (C9 E1), 95.7 (C3 E2), 95.6 (C3 E1), 42.8 (C7 E1), 42.7 (C7 E2), 24.5 (C11 E1), 24.4 (C11 E2). MS (ES−); 209.07 (M−H); HRMS (M−H); calcd for C9H9N2O4; 209.0568; found; 209.0570.
3.4.40. Synthesis of Compound (±)-25b
Yield; 40%; M.P.; 76 °C; Mixture of two exo-enol tautomers (E1:E2 = 40:60); 1H-NMR (400 MHz, CDCl3); 9.94 (brs, 1H, NH E1), 9.39 (brs, 1H, NH E2), 5.92–5.81 (m, 1H, C8), 5.30–5.16 (m, 2H, C9), 4.53–4.50 (m, 2H, C7), 3.09–3.07 (m, 2H, C11), 2.11–2.13 (m, 1H, C12), 1.36 (dd, 1H, J1 = 6.4 Hz, J2 = 6.0 Hz, C13 E1), 1.33 (dd, 1H, J1 = 6.0 Hz, J2 = 4.0 Hz, C13 E2), 1.19 (dd, 1H, J1 = 6.0 Hz, J2 = 4.0 Hz, C13 E2), 1.16 (dd, 1H, J1 = 6.4 Hz, J2 = 6.0 Hz, C13 E1), 1.02–1.00 (m, 3H, C18), 0.89 (s, 9H, C15-C17). 13C-NMR (125 MHz, CDCl3); 200.1 (C10 E1), 199.5 (C10 E2), 170.4 (C2 E2), 169.7 (C4 E1), 161.2 (C2 E1), 160.9 (C4 E2), 149.6 (C6 E1), 149.0 (C6 E2), 131.6 (C8 E1), 130.9 (C8 E2), 118.6 (C9 E2), 117.8 (C9 E1), 95.7 (C3 E2), 95.6 (C3 E1), 50.6 (CH2 E2), 50.5 (CH2 E1), 45.2 (CH2 E2), 45.1 (CH2 E1), 42.6 (C7 E1), 42.5 (C7 E2), 31.0 (C14), 29.9 (C15-C17), 28.1 (CH3 E2), 27.8 (CH3 E1), 22.7 (CH3 E1), 22.6 (CH3 E2). MS (ES−); 307.19 (M−H); HRMS (M−H); calcd for C16H23N2O4; 307.1663; found; 307.1657.
3.4.41. Synthesis of Compound 26a
Yield; 56%; M.P.; 131 °C; Mixture of two exo-enol tautomers (E1: E2 = 40: 60); 1H-NMR (500 MHz, CDCl3); 9.36 (brs, 1H, NH E1), 8.90 (brs, 1H, NH E2), 7.29–7.25 (m, 2H, C17 and C18), 6.96–6.92 (m, 1H, C19), 6.87 (d, 2H, J = 8.0 Hz, C15 and C16), 5.93–5.80 (m, 1H, C8), 5.33–5.18 (m, 2H, C9), 4.53 (d, 2H, J = 6.0 Hz, C7 E2), 4.48 (d, 2H, J = 6.0 Hz, C7 E1), 4.09–4.06 (m, 2H, C13), 3.38 (t, 2H, J = 7.5 Hz, C11), 2.25–2.20 (m, 2H, C12). 13C-NMR (125 MHz, CDCl3); 199.9 (C10 E1), 199.3 (C10 E2), 170.4 (C2 E2), 169.5 (C4 E1), 160.9 (C4 E2 and C2 E1), 158.6 (C14), 149.2 (C6 E1), 148.8 (C6 E2), 131.5 (C8 E1), 130.8 (C8 E2), 129.4 (C17 and C18 E1), 129.4 (C17 and C18 E2), 120.7 (C19), 118.8 (C9 E2), 118.0 (C9 E1), 114.4 (C15 and C16), 95.2 (C3 E2), 95.2 (C3 E1), 66.7 (C13), 42.7 (C7 E2), 42.7 (C7 E1), 33.6 (C11 E1), 33.5 (C11 E2), 25.3 (C12 E2), 25.1 (C12 E1). MS (ES−); 329.11 (M−H); HRMS (M−H); calcd for C17H17N2O5; 329.1143; found; 329.1143.
3.4.42. Synthesis of Compound 26b
Yield; 59%; M.P.; 128 °C; Mixture of two exo-enol tautomers (E1: E2 = 40: 60); 1H-NMR (400 MHz, CDCl3); 9.56 (brs, 1H, NH E1), 9.10 (brs, 1H, NH E2), 7.09–7.07 (m, 2H, C17 and C18), 6.71 (d, 1H, J = 8.4 Hz, C16), 5.93–5.80 (m, 1H, C8), 5.32–5.18 (m, 2H, C9), 4.53 (d, 2H, J = 5.6 Hz, C7 E2), 4.50 (d, 2H, J = 5.6 Hz, C7 E1), 4.04 (t, 2H, J = 6.0 Hz, C11), 3.38 (t, 2H, J = 7.6 Hz, C13), 2.26–2.19 (m, 2H, C12), 2.18 (s, 3H, C20 E1), 2.17 (s, 3H, C20 E2). 13C-NMR (100 MHz, CDCl3); 199.7 (C10 E1), 199.1 (C10 E2), 170.4 (C2 E2), 169.5 (C4 E1), 161.1 (C4 E2), 160.9 (C2 E1), 155.4 (C14), 149.3 (C6 E1), 148.8 (C6 E2), 131.5 (C8 E1), 130.8 (C8 E2), 130.3 (C17), 128.6 (quart C), 126.2 (C18), 125.0 (quart C), 118.9 (C9 E2), 118.1 (C9 E1), 111.8 (C16), 95.2 (C3), 66.1 (C13 E2), 67.1 (C13 E1), 42.7 (C7), 33.6 (C11 E1), 33.5 (C11 E2), 25.2 (C12 E2), 24.9 (C12 E1), 16.0 (C20). MS (ES−); 377.11 (M−H); HRMS (M−H); calcd for C18H18Cl1N2O5; 377.0910; found; 377.0908.
3.4.43. Synthesis of Compound 26c
Yield; 37%; M.P.; 201 °C; 1H-NMR (500 MHz, DMSO); 11.86 (brs, 1H, NH), 11.05 (brs, 1H, NH), 7.30 (d, 2H, J = 9.0 Hz, C14 and C15), 6.93 (d, 2H, J = 9.0 Hz, C12 and C13), 4.02 (t, 2H, J = 6.5 Hz, C10), 3.20 (t, 2H, J = 6.5 Hz, C8), 2.07–2.01 (m, 2H, C9). 13C-NMR (125 MHz, DMSO); 197.9 (C7), 157.3 (C11), 149.0 (C2, C4 and C6), 129.2 (C14 and C15), 124.2 (C16), 116.2 (C12 and C13), 94.9 (C3), 67.2 (C10), 32.8 (C8), 24.4 (C9). MS (ES−); 323.07 (M−H); HRMS (M−H); calcd for C14H12N2O5; 323.0440; found; 323.0436.
3.4.44. Synthesis of Compound 26d
Yield; 44%; M.P.; 207 °C; 1H-NMR (400 MHz, DMSO); 11.86 (brs, 1H, NH), 11.06 (brs, 1H, NH), 7.17–7.14 (m, 2H, C14 and C15), 6.91 (d, 1H, J = 8.8 Hz, C12), 4.01 (t, 2H, J = 6.0 Hz, C10), 3.24 (t, 2H, J = 6.0 Hz, C8), 2.12–2.03 (m, 5H, C9 and C17). 13C-NMR (125 MHz, DMSO); 198.0 (C7), 155.4 (C11), 149.1 (C2, C4 and C6), 129.9 (C15), 128.3 (tert-C), 126.5 (C14), 123.7 (tert-C), 112.7 (C12), 94.9 (C3), 67.2 (C10), 32.9 (C8), 24.5 (C9), 15.6 (C17). MS (ES−); 337.08 (M−H); HRMS (M−H); calcd for C15H14N2O5; 337.0597; found; 337.0587.
3.4.45. Synthesis of Compound 27
Yield; 50%; M.P.; over 260 °C; 1H-NMR (500 MHz, mixture of CDCl3 and CD3OD); 2.90 (dd, 1H, J1 = 14.0 Hz, J2 = 8.0 Hz, C8), 2.84 (dd, 1H, J1 = 14.0 Hz, J2 = 8.0 Hz, C8), 2.07 (brs, 1H, C14), 1.87 (brs, 1H, C15), 1.83–1.78 (m, 1H, C9), 1.39–1.25 (m, 4H, CH2), 1.06–0.95 (m, 4H, CH2). 13C-NMR (125 MHz, mixture of CDCl3 and CD3OD); 199.1 (C7), 171.8 (C2), 163.1 (C4), 150.0 (C6), 95.8 (C3), 42.8 (C8), 41.5 (C15), 39.6 (C9), 38.1 (CH2), 37.2 (C14), 35.7 (CH2), 30.1 (CH2), 29.0 (CH2). MS (ES−); 263.12 (M−H), HRMS (M−H); calcd for C13H15N2O4; 263.1037; found; 263.1040.
3.4.46. Synthesis of Compound cis-28
Yield; 61%; M.P.; 192 °C; 1H-NMR (500 MHz, CD3OD); 3.20 (dd, 1H, J1 = 14.0 Hz, J2 = 6.5 Hz, C11), 3.06–2.97 (m, 2H, C8), 2.45–2.40 (m, 1H, C9), 2.09–2.03 (m, 4H, C10 and C16), 1.91–1.86 (m, 1H, C10), 1.34 (s, 3H, C13), 0.92 (s, 3H, C14). 13C-NMR (125 MHz, CD3OD); 210.5 (C15), 199.3 (C7), 173.2 (C2), 164.3 (C4), 151.1 (C6), 96.2 (C3), 55.4 (C11), 45.1 (C12), 40.4 (C9), 37.7 (C8), 30.4 (C16), 30.3 (C13), 24.4 (C10), 18.0 (C14). MS (ES−); 293.12 (M−H); MS (ES+); 317.13 (M+Na); HRMS (M+Na); calcd for C14H18N2Na1O5; 317.1108; found; 317.1099.
3.6. Synthesis of 3-Carboxamide an 3-Enamine Barbituric Acids
General procedure: To the solution of 3-alkoxycarbonyl or 3-acylbarbituric acid (1.0 equivalent) in toluene (or methanol for compound 36) was added primary amine (1.0 equivalent) and the mixture was refluxed for 4–24 h checking TLC. After completion of the reaction, the solution was evaporated and column chromatography (or precipitation in methanol for compound 36) gave metal-chelated 3-carboxamide tetramic acid or pure 3-enamine tetramic acid. In case of 3-carboxamide tetramic acid, the compound was dissolved in dichloromethane (50 mL) and washed with 1 N HCl (20 mL) to make metal free form. The organic layer was dried with MgSO4 and concentrated in vacuo to give metal free 3-carboxamide tetramic acid.
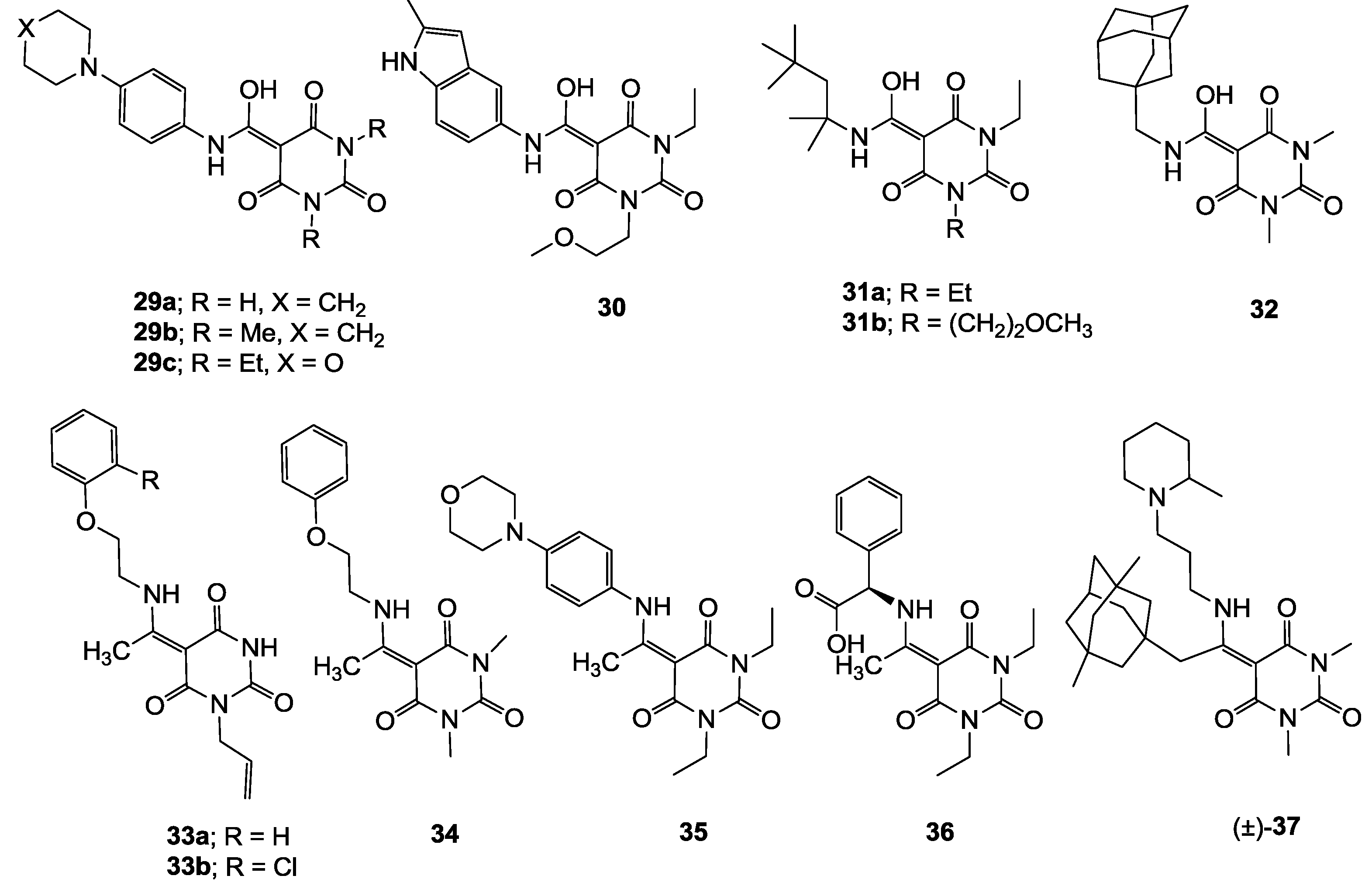
3.6.1. Synthesis of Compound 29a
Yield; 63%; M.P.: >250 °C; 1H-NMR (400 MHz, DMSO); 11.54 (brs, 2H, NH), 11.37 (s, 1H, NH), 7.32 (d, 2H, J = 8.4 Hz, C9 and C10), 6.93 (d, 2H, J = 8.4 Hz, C11 and C12), 3.11 (brs, 4H, C14 and C15), 1.60 (brs, 4H, C16 and C17), 1.53 (brs, 2H, C18). 13C-NMR; not determined because of peak broading and solubility problems. MS (ES−); 329.13 (M−H); MS (ES+); 331.15 (M+H), 353.13 (M+Na); HRMS (M+H); calcd for C16H19N4O4; 331.1401; found; 331.1392.
3.6.2. Synthesis of Compound 29b
Yield; 77%; M.P.; 180 °C; 1H-NMR (500 MHz, CD3OD); 7.78 (d, 2H, J = 9.5 Hz, C11 and C12), 7.68 (d, 2H, J = 9.5 Hz, C13 and C14), 3.62 (t, 4H, J = 6.0 Hz, C16 and C17), 3.34 (s, 6H, C7 and C8), 2.07–2.03 (m, 4H, C18 and C19), 1.81 (brs, 2H, C20). 13C-NMR; not determined because of peak broading and solubility problems. MS (ES−); 357.16 (M−H); HRMS (M−H); calcd for C18H21N4O4; 357.1568; found; 357.1568.
3.6.3. Synthesis of Compound 29c
Yield; 48%; M.P.: 167 °C; 1H-NMR (400 MHz, CDCl3); 11.82 (brs, 1H, NH or OH), 7.41 (d, 2H, J = 8.8 Hz, C13 and C14), 6.91 (d, 2H, J = 8.8 Hz, C15 and C16), 4.02 (q, 4H, J = 6.8 Hz, C7 and C9), 3.86 (t, 4H, J = 4.8 Hz, C20 and C21), 3.16 (t, 4H, J = 4.8 Hz, C18 and C19), 1.29–1.24 (m, 6H, C8 and C9). 13C-NMR (100 MHz, CDCl3); 168.9 (C11), 168.4 (C4), 163.0 (C2), 149.4 (C6), 149.2 (C17), 127.8 (C12), 122.9 (C13 and C14), 115.9 (C15 and C16), 80.5 (C3), 66.7 (C20 and C21), 49.1 (C18 and C19), 36.6 (N-CH2), 36.3 (N-CH2), 13.3 (CH3), 13.2 (CH3). MS (ES−); 387.17 (M−H); HRMS (M+Na); calcd for C19H24N4Na1O5; 411.1639; found; 411.1627.
3.6.4. Synthesis of Compound 30
Yield; 53%; M.P. 160 °C; 1H-NMR (400 MHz, DMSO); 11.79 (s, 1H, NH or OH), 11.04 (s, 1H, indole NH), 7.55 (s, 1H, C15), 7.27 (d, 1H, J = 8.8 Hz, C14), 7.04 (dd, 1H, J1 = 8.8 Hz, J2 = 2.0 Hz, C16), 6.12 (s, 1H, C19), 3.99 (t, 2H, J = 6.0 Hz, C9), 3.85 (q, 2H, J = 6.8 Hz, C7), 3.50 (t, 2H, J = 6.0 Hz, C10), 3.25 (s, 3H, C11), 2.37 (s, 3H, C21), 1.13 (t, 3H, J = 6.8 Hz, C8). 13C-NMR (100 MHz, DMSO); 168.3 (C2, C4 and C12), 149.1 (C6), 137.3 (Ar quart-C), 134.2 (Ar quart-C), 128.7 (Ar quart-C), 126.7 (Ar quart-C), 115.1 (Ar tert-C), 112.5 (Ar tert-C), 110.8 (Ar tert-C), 99.5 (C19), 79.7 (C3), 68.4 (C10), 58.0 (C11), 39.5 (C9, overlapping with DMSO peak, confirmed in DEPT NMR), 35.9 (C7), 13.4 (CH3), 13.0 (CH3). MS (ES−); 385.16 (M−H); HRMS (M+Na); calcd for C19H22N4Na1O5; 409.1482; found; 409.1474.
3.6.5. Synthesis of Compound 31a
Yield; 67%; M.P.: 92 °C; 1H-NMR (400 MHz, CDCl3); 10.16 (brs, 1H, NH or OH), 3.97–3.90 (m, 4H, C7 and C9), 1.78 (s, 2H, C13), 1.37 (s, 6H, C18 and C19), 1.22–1.16 (m, 6H, C8 and C10), 0.98 (s, 9H, C15-C17). 13C-NMR (100 MHz, CDCl3); 170.4 (C11), 168.2 (C4), 163.1 (C2), 149.6 (C6), 80.1 (C3), 56.6 (C12), 51.7 (C13), 36.2 (N-CH2), 36.0 (N-CH2), 31.6 (C14), 31.2 (C15-C17), 29.5 (C18 and C19), 13.3 (CH3), 13.2 (CH3). MS (ES−); 338.22 (M−H); MS (ES+); 340.21 (M+H), 362.21 (M+Na); HRMS (M+Na); calcd for C17H29N3Na1O4; 362.2050; found; 362.2037.
3.6.6. Synthesis of Compound 31b
Yield; 68 % (oil); 1H-NMR (400 MHz, CDCl3); 10.21 and 10.14 (2 of s, 1H, NH or OH), 4.15–4.11 (m, 2H, C9), 3.98–3.92 (m, 2H, C7), 3.63–3.57 (m, 2H, C10), 3.35 and 3.34 (2 of s, 3H, C11), 1.79 (s, 2H, C14), 1.49 (s, 6H, C19 and C20), 1.24–1.18 (m, 3H, C8), 0.99 (s, 9H, C16-C18). 13C-NMR (100 MHz, CDCl3); 170.4 and 170.3 (C12), 168.4 and 168.2 (C4), 163.2 and 163.1 (C2), 149.9 and 149.8 (C6), 80.0 (C3), 69.5 and 69.4 (C10), 58.7 (C11), 56.7 and 56.6 (C13), 51.7 and 51.6 (C14), 39.9 and 39.8 (C9), 36.3 and 36.1 (C7), 31.6 (C15), 31.2 (C16-C18), 29.4 (C19 and C20), 13.3 and 13.2 (C8). MS (ES−); 368.23 (M−H); MS (ES+); 370.24 (M+H), 392.21 (M+Na); HRMS (M+Na); calcd for C18H31N3 Na1O5; 392.2156; found; 392.2141.
3.6.7. Synthesis of Compound 32
Yield; 74%; M.P.: 132 °C; 1H-NMR (500 MHz, CDCl3); 10.09 (brs, 1H, NH or OH), 3.33–3.32 (m, 6H, C7 and C8), 3.09 (d, 2H, J = 6.5 Hz, C10), 2.00 (brs, 3H, C18-C20), 1.73–1.62 (m, 6H, CH2), 1.53 (brs, 6H, CH2). 13C-NMR (125 MHz, CDCl3); 170.8 (C9), 168.2 (C4), 163.4 (C2), 150.6 (C6), 79.7 (C3), 51.6 (C10), 40.0 (C12, C13 and C17), 36.6 (C14–C16), 33.6 (C11), 28.0 (C18–C20), 27.5 (CH3), 27.4 (CH3). MS (ES−); 346.17 (M−H); HRMS (M−H); calcd for C18H24N3O4; 346.1772; found; 346.1772.
3.6.8. Synthesis of Compound 33a
Yield; 96%; M.P.; 142 °C; Mixture of two exo-enol tautomers (E1: E2 = 45: 55); 1H-NMR (400 MHz, CDCl3); 12.77 (brs, 1H, NH E2), 12.71 (brs, 1H, NH E1), 8.68 (brs, 1H, NH), 7.33–7.27 (m, 2H, C17 and C18), 7.02–6.97 (m, 1H, C19), 6.92 (d, 2H, J = 8.8 Hz, C15 and C16), 5.94–5.84 (m, 1H, C8), 5.22 (d, 1H, J = 17.2 Hz, C9), 5.16 (d, 1H, J = 10.4 Hz, C9), 4.50 (d, 2H, J = 5.2 Hz, C7), 4.20 (t, 2H, J = 5.2 Hz, C13), 3.89–3.85 (m, 2H, C12), 2.79 (s, 3H, C11 E2), 2.77 (s, 3H, C11 E1). 13C-NMR (100 MHz, CDCl3); 175.2 (C10 E1), 174.7 (C10 E2), 167.0 (C2 E2), 166.3 (C4 E1), 163.2 (C2 E1), 163.1 (C4 E2), 157.9 (C14), 149.8 (C6), 132.6 (C8 E1), 132.4 (C8 E2), 129.6 (C17 and C18), 121.7 (C19), 116.9 (C9 E2), 116.7 (C9 E1), 114.6 (C15 and C16), 90.3 (C3), 65.7 (C13), 43.2 (C12 E1), 43.1 (C12 E2), 42.4 (C7 E1), 42.2 (C7 E2), 17.8 (C11). MS (ES−); 328.15 (M−H); MS (ES+); 352.15 (M+Na); HRMS (M+Na); calcd for C17H19N3Na1O4; 352.1268; found; 352.1254.
3.6.9. Synthesis of Compound 33b
Yield; 84%; M.P.; 173 °C; Mixture of two exo-enol tautomers (E1: E2 = 45: 55); 1H-NMR (500 MHz, CDCl3); 12.82 (brs, 1H, NH E2), 12.73 (brs, 1H, NH E1), 8.34 (brs, 1H, NH E1), 8.25 (brs, 1H, NH E2), 7.39–7.37 (m, 1H, C18), 7.24–7.20 (m, 1H, C17), 6.98–6.92 (m, 2H, C15 and C19), 5.93–5.85 (m, 1H, C8), 5.24–5.14 (m, 2H, C9), 4.50 (d, 2H, J = 5.5 Hz, C7), 4.24 (t, 2H, J = 5.0 Hz, C13), 3.97–3.93 (m, 2H, C12), 2.85 (s, 3H, C11 E2), 2.83 (s, 3H, C11 E1). 13C-NMR (125 MHz, CDCl3); 175.7 (C10 E1), 175.1 (C10 E2), 167.1 (C2 E2), 166.2 (C4 E1), 163.2 (C2 E1), 162.8 (C4 E2), 153.6 (C14), 149.8 (C6 E1), 149.7 (C6 E2), 132.6 (C8 E1), 132.3 (C8 E2), 130.6 (C18), 127.8 (C17 E1), 127.6 (C17 E2), 123.4 (C16 E2), 123.3 (C16 E1), 122.6 (C19 E2), 122.6 (C19 E1), 117.0 (C9 E2), 116.8 (C9 E1), 113.9 (C15 E2), 113.9 (C15 E1), 90.4 (C3), 67.4 (C13 E1), 67.3 (C13 E2), 43.1 (C12 E1), 43.0 (C12 E2), 42.4 (C7 E1), 42.2 (C7 E2), 17.8 (C11 E1), 17.8 (C11 E2). MS (ES−); 362.11 (M−H); MS (ES+); 386.11 (M+Na); HRMS (M+Na); calcd for C17H18Cl1N3Na1O4; 386.0878; found; 386.0876.
3.6.10. Synthesis of Compound 34
Yield; 63%; M.P.; 129 °C; 1H-NMR (400 MHz, CDCl3); 12.90 (brs, 1H, NH), 7.29 (dd, 2H, J1 = J2 = 7.6 Hz, C16 and C17), 6.99 (t, 1H, J = 7.6 Hz, C18), 6.92 (d, 2H, J = 7.6 Hz, C14 and C15), 4.19 (t, 2H, J = 5.2 Hz, C12), 3.87–3.83 (m, 2H, C11), 3.30 (s, 6H, C7 and C8), 2.76 (s, 3H, C10). 13C-NMR (100 MHz, CDCl3); 174.5 (C9), 166.3 (C4), 162.9 (C2), 157.9 (C13), 151.3 (C6), 129.5 (C16 and C17), 121.6 (C18), 114.6 (C14 and C15), 90.7 (C3), 65.7 (C12), 43.1 (C11), 27.8 (NCH3), 27.5 (NCH3), 17.9 (C10). MS (ES−); 316.15 (M−H); MS (ES+); 318.17 (M+H), 340.15 (M+Na); HRMS (M+Na); calcd for C16H19N3Na1O4; 340.1268; found; 340.1256.
3.6.11. Synthesis of Compound 35
Yield; 66%; M.P.; 209 °C; 1H-NMR (400 MHz, CDCl3); 7.03 (d, 2H, J = 8.8 Hz, C16 and C17), 6.90 (d, 2H, J = 8.8 Hz, C14 and C15), 4.01–3.93 (m, 4H, C7 and C9), 3.84–3.82 (m, 4H, C21 and C22), 3.17–3.15 (m, 4H, C19 and C20), 2.59 (s, 3H, C12), 1.23–1.17 (m, 6H, C8 and C10). 13C-NMR (100 MHz, CDCl3); 173.3 (C11), 166.0 (C4), 162.5 (C2), 150.5 (C18), 150.4 (C6), 127.9 (C13), 126.5 (C14 and C15), 115.6 (C16 and C17), 91.3 (C3), 66.6 (C21 and C22), 48.6 (C19 and C20), 36.1 (N-CH2), 36.0 (N-CH2), 20.1 (C12), 13.35 (CH3), 13.33 (CH3). MS (ES-); 385.19 (M−H); HRMS (M−H); calcd for C20H25N4O4; 385.1881; found; 385.1888.
3.6.12. Synthesis of Compound 36
Yield; 60%; M.P.; 203 °C; 1H-NMR (400 MHz, CDCl3); 13.55 (d, 1H, J = 6.4 Hz, NH), 9.04 (brs, 1H, OH), 7.41–7.38 (m, 5H, C16-C20), 5.44 (d, 1H, J = 6.4 Hz, C13), 4.01 (q, 2H, J = 6.8 Hz, NCH2), 3.93 (q, 2H, J = 6.8 Hz, NCH2), 2.55 (s, 3H, C12), 1.21 (t, 3H, J = 6.8 Hz, CH3), 1.14 (t, 3H, J = 6.8 Hz, CH3). 13C-NMR (100 MHz, CDCl3); 173.8 (C11), 171.8 (C14), 165.9 (C4), 162.8 (C2), 150.5 (C6), 134.9 (C15), 129.5 (C16 and C17), 129.3 (C20), 127.0 (C18 and C19), 92.0 (C3), 60.5 (C13), 36.4 (N-CH2), 36.3 (N-CH2), 19.1 (C12), 13.4 (CH3), 13.3 (CH3).
3.6.13. Synthesis of Compound (±)-37
Yield; 82% (oil); 1H-NMR (400 MHz, CDCl3); 4.54–4.51 (m, 1H, NH), 3.65 (brs, 1H, C23), 3.43 (brs, 1H, C23), 3.29 (s, 3H, C7), 3.27 (s, 3H, C8), 2.75–2.68 (m, 2H, C10), 2.27 (brs, 3H, C26 and C27), 2.07–1.97 (m, 2H, C25), 1.77–1.75 (m, 2H, CH2), 1.61–0.99 (m, 21H, C12-C17, C20 and C24, 6H of CH2), 0.74 (s, 6H, C21 and C22). 13C-NMR (100 MHz, CDCl3); 175.0 (C9), 166.3 (C2), 163.2 (C4), 151.2 (C6), 91.2 (C3), 55.9 (C27), 51.6 (CH2), 50.6 (CH2), 50.4 (CH2), 50.1 (CH2), 48.9 (CH2), 43.1 (CH2), 42.7 (CH2), 40.6 (CH2), 39.4 (CH2), 38.7 (C18 and C19), 34.4 (CH2), 31.5 (C11), 30.5 (C21 and C22), 29.7 (C20), 28.0 (N-CH3), 27.6 (N-CH3), 26.6 (CH2), 26.1 (CH2), 23.4 (CH2), 18.4 (C31). MS (ES−); 497.38 (M−H); MS (ES+); 499.36 (M+H); HRMS (M+H); calcd for C29H47N4O3; 499.3643; found; 499.3629.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









































































