
(−)-Tarchonanthuslactone Exerts a Blood Glucose-Increasing Effect in Experimental Type 2 Diabetes Mellitus

Abstract
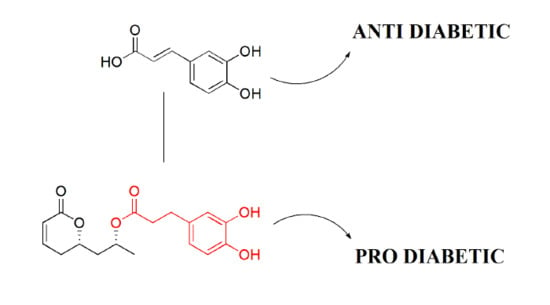
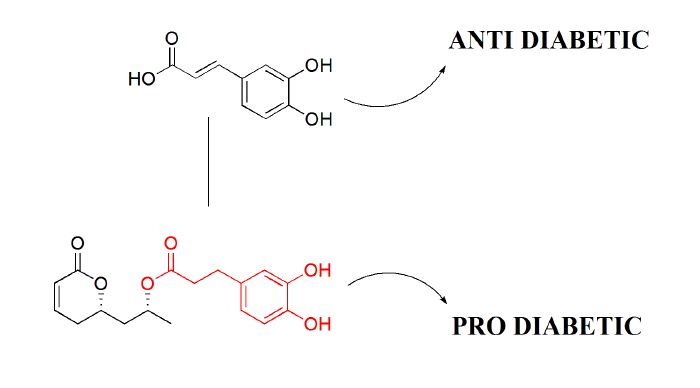
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

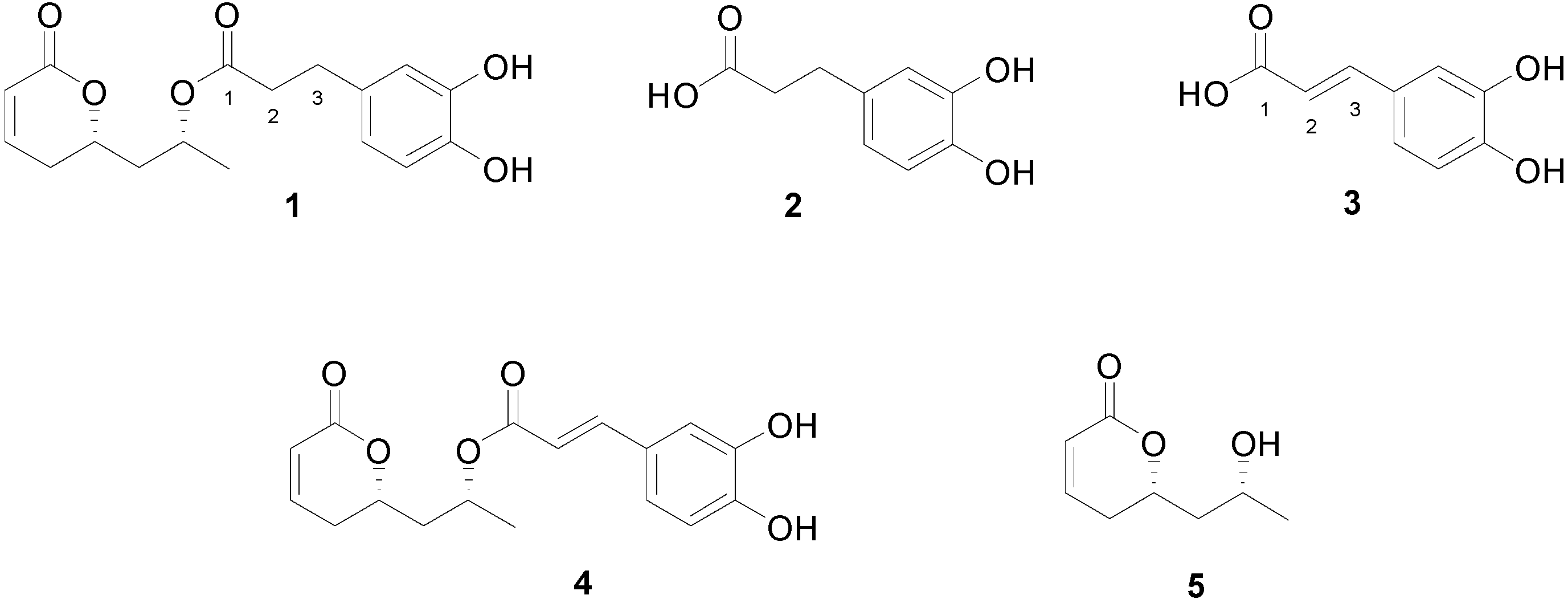
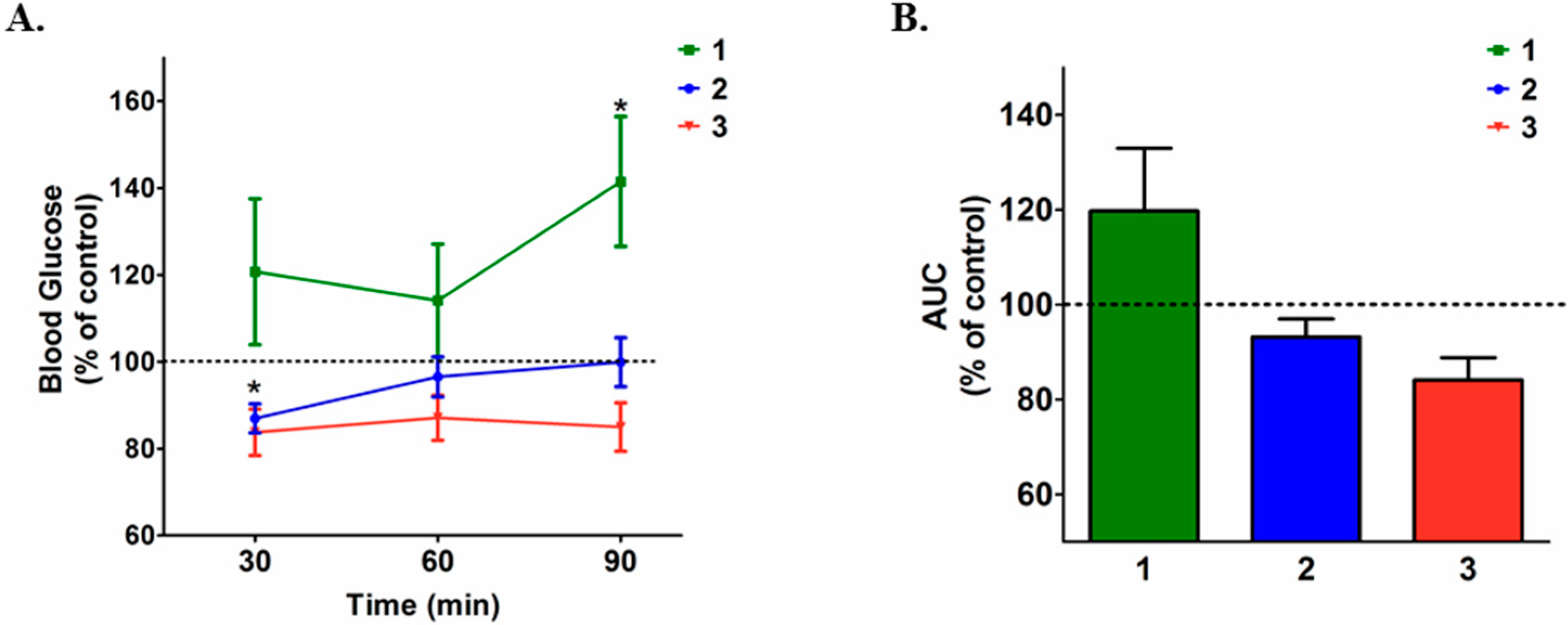
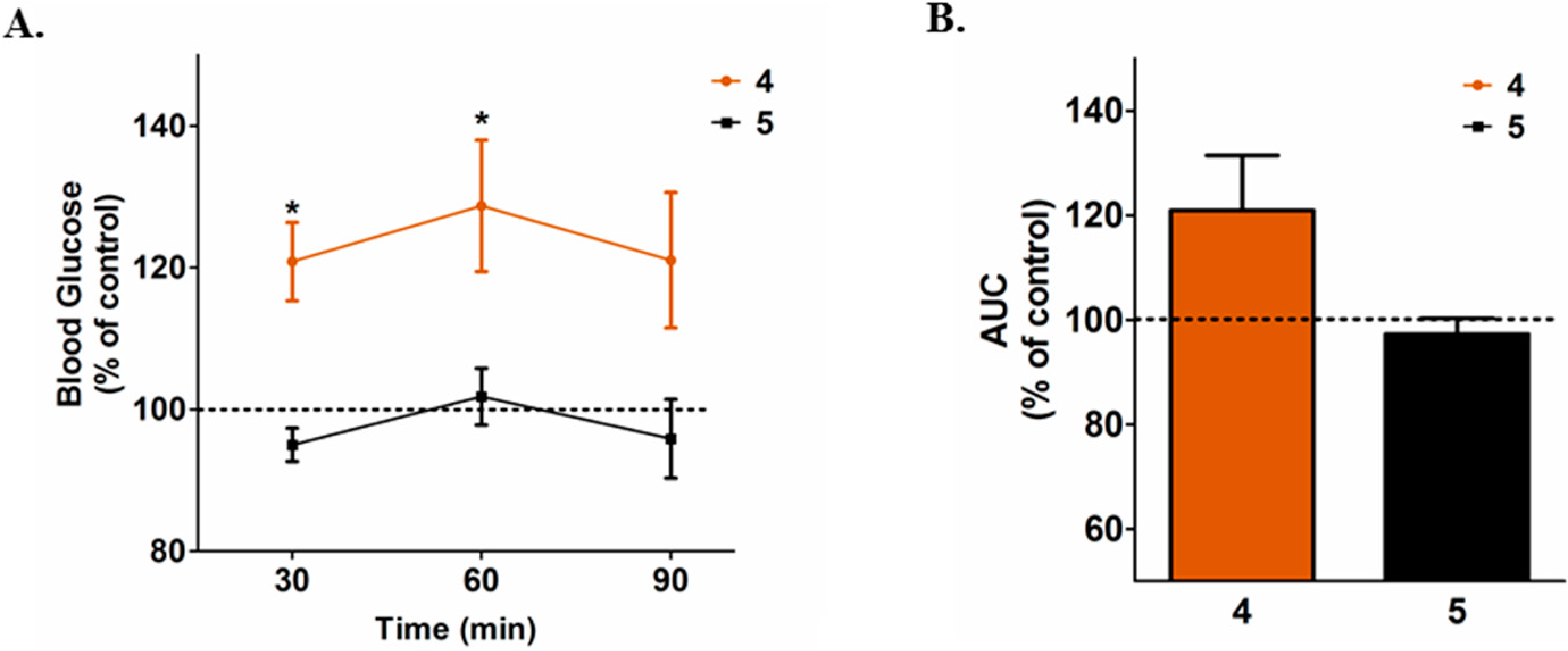
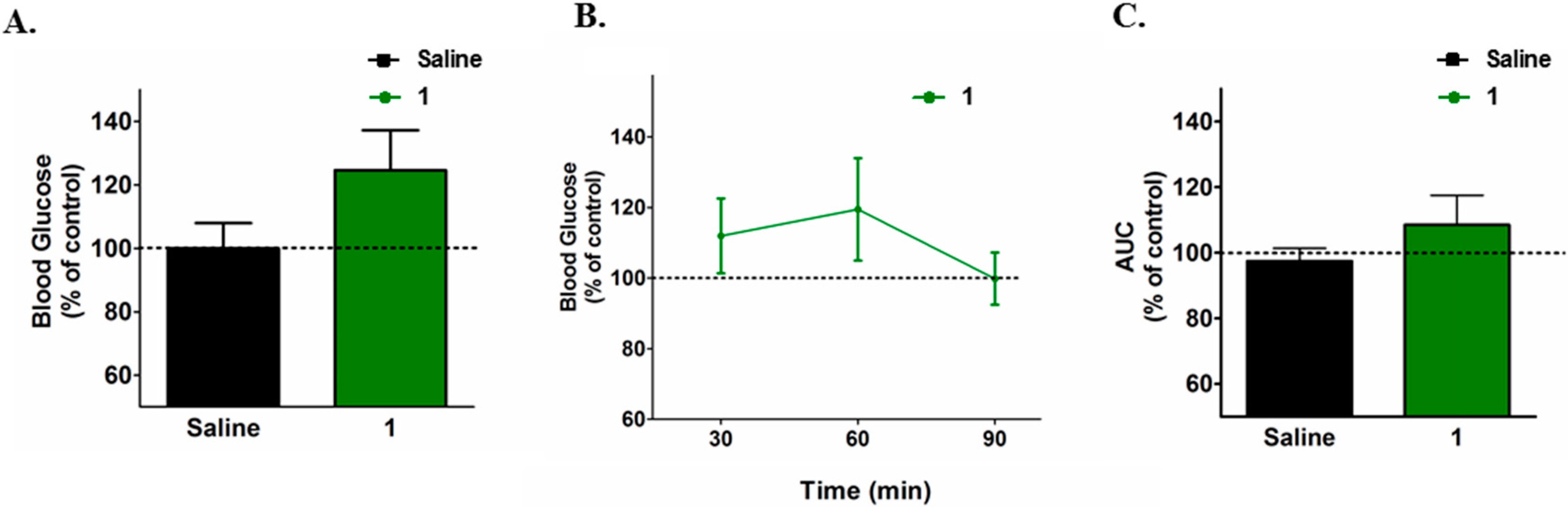
2. Results and Discussion



3. Experimental Section
3.1. Chemistry
3.1.1. General Procedures
3.1.2. Experimental Procedures
3.2. Biological Experiments
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Akash, M.S.H.; Rehman, K.; Chen, S. Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J. Cell. Biochem. 2013, 114, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, A.B.; Fonseca, V.; Jablonski, K.A.; Pyle, L.; Staten, M.A.; Shoelson, S.E. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann. Intern. Med. 2010, 152, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Rehman, K.; Chen, S. An overview of valuable scientific models for diabetes mellitus. Curr. Diabetes Rev. 2013, 9, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Agunloye, O.M.; Adefegha, S.A.; Akinyemi, A.J.; Ademiluyi, A.O. Caffeic and chlorogenic acids inhibit key enzymes linked to type 2 diabetes (in vitro): A comparative study. J. Basic Clin. Physiol. Pharmacol. 2014, 26, 165–170. [Google Scholar]
- Akash, M.S.H.; Rehman, K.; Chen, S. Effects of coffee on type 2 diabetes mellitus. Nutrition 2014, 30, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Rehman, K.; Chen, S. Spice plant Allium cepa: Dietary supplement for treatment of type 2 diabetes mellitus. Nutrition 2014, 30, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, F.; Suwita, A. Ein neues bisabolen-derivat und ein neues dihydro kaffesaure-derivat aus tarchonanthus tribolus. Phytochemistry 1979, 18, 677–678. [Google Scholar]
- Van Huyssteen, M.; Milne, P.J.; Campbell, E.E.; van de Venter, M. Antidiabetic and cytotoxicity screening of five medicinal plants used by traditional African health practitioners in the Nelson Mandela Metropole, South Africa. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 150–158. [Google Scholar] [PubMed]
- Kalesse, M.; Christmann, M.; Bhatt, U.; Quitschalle, M.; Claus, E.; Saeed, A.; Burzlaff, A.; Kasper, C.; Haustedt, L.O.; Hofer, E.; et al. The chemistry and biology of ratjadone. ChemBioChem 2001, 2, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Kalesse, M.; Christmann, M. The chemistry and biology of the leptomycin family. Synthesis 2002, 8, 981–1003. [Google Scholar] [CrossRef]
- Bialy, L.; Waldmann, H. Synthesis and biological evaluation of cytostatin analogues. Chem. Commun. 2003, 1872–1873. [Google Scholar] [CrossRef]
- Buck, S.B.; Hardouin, C.; Ichikawa, S.; Soenen, D.R.; Gauss, C.M.; Hwang, I.; Swingle, M.R.; Bonness, K.M.; Honkanen, R.E.; Boger, D.L. Fundamental role of the fostriecin unsaturated lactone and implications for selective protein phosphatase inhibition. J. Am. Chem. Soc. 2003, 125, 15694–15695. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Steinbusch, D. An efficient asymmetric synthesis of tarchonanthuslactone. Eur. J. Org. Chem. 2003, 22, 4450–4454. [Google Scholar] [CrossRef]
- Baktharama, S.; Selvakumar, S.; Singh, V.K. Asymmetric synthesis of all the stereoisomers of tarchonanthuslactone. Tetrahed. Lett. 2005, 46, 7527–7529. [Google Scholar] [CrossRef]
- George, S.; Sudalai, A. Enantioselective synthesis of tarchonanthuslactone using proline-catalyzed asymmetrica-aminooxylation. Tetrahedron Asymmetry 2007, 18, 975–981. [Google Scholar] [CrossRef]
- Yadav, J.S.; Kumar, N.N.; Reddy, M.S.; Prasad, A.R. Stereoselective synthesis of tarchonanthuslactone via the Prins cyclisation. Tetrahedron 2007, 63, 2689–2694. [Google Scholar] [CrossRef]
- Lee, H.; Sampath, V.; Yoon, Y. A facile total synthesis of all stereoisomers of tarchonanthuslactone and euscapholide from chiral epichlorohydrin. Synlett 2009, 2, 249–252. [Google Scholar] [CrossRef]
- Hsu, F.; Chen, Y.; Cheng, J. Caffeic acid as active principle from the fruit of Xanthium strumarium to lower plasma glucose in diabetic rats. Planta Med. 2000, 66, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.S.; Reddy, P.M.K.; Gupta, M.K.; Chary, J. Stereoselective total synthesis of tarchonanthuslactone and formal synthesis of (–)-colletol. Synthesis 2007, 23, 3639–3646. [Google Scholar] [CrossRef]
- Scott, M.S.; Luckhurst, C.A.; Dixon, D.J. A total synthesis of tarchonanthuslactone exploiting N-pyrrole carbinols as efficient stereocontrolling elements. Org. Lett. 2005, 7, 5813–5816. [Google Scholar] [CrossRef] [PubMed]
- Garaas, S.D.; Hunter, T.J.; O’Doherty, G.A. An enantioselective synthesis of tarchonanthuslactone. J. Org. Chem. 2002, 67, 2682–2685. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, M.; Rim, J.S.; Koza, R.A.; Kozak, L.P. Variation in type 2 diabetes-related traits in mouse strains susceptible to diet-induced obesity. Diabetes 2003, 52, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araújo, E.P.; Stoppiglia, L.F.; Pauli, J.R.; Ropelle, E.; Rocco, S.A.; Marin, R.M.; Franchini, K.G.; Carvalheira, J.B.; Saad, M.J.; et al. Inhibition of UCP2 expression reverses diet-induced diabetes mellitus by effects on both insulin secretion and action. FASEB J. 2007, 21, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, P.H.G.; de Oliveira, A.R.M.; Delay, C.E. Diastereoselective route to (2R,5S)- and (2S,5S)-2-methyl-1,6-dioxaspiro[4.5]decane, a pheromone component of the wasp Paravespula vulgaris. Tetrahedron Lett. 2003, 44, 6849–6851. [Google Scholar] [CrossRef]
- Moore, C.G.; Murphy, P.J.; Williams, H.L.; McGown, A.T.; Smith, N.K. Synthetic studies towards ptilomycalin A: Total synthesis of crambescidin 359. Tetrahedron 2007, 63, 11771–11780. [Google Scholar] [CrossRef]
- Wattanasereekul, S.; Maier, M.E. Synthesis of the 8-hydroxy acid of jasplakinolide. Adv. Synth. Catal. 2004, 346, 855–861. [Google Scholar] [CrossRef]
- Kumar, P.; Gupta, P.; Naidu, S.V. A simple and efficient approach to 1,3-polyols: Application to the synthesis of cryptocarya diacetate. Chem. Eur. J. 2006, 12, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Naidu, S.V.; Kumar, P. Enantioselective synthesis of tarchonanthuslactone via iterative hydrolytic kinetic resolution. Tetrahedron Lett. 2005, 46, 6571–6573. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2 and 3 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Souza, G.F.P.; Novaes, L.F.T.; Avila, C.M.; Nascimento, L.F.R.; Velloso, L.A.; Pilli, R.A. (−)-Tarchonanthuslactone Exerts a Blood Glucose-Increasing Effect in Experimental Type 2 Diabetes Mellitus. Molecules 2015, 20, 5038-5049. https://doi.org/10.3390/molecules20035038
De Souza GFP, Novaes LFT, Avila CM, Nascimento LFR, Velloso LA, Pilli RA. (−)-Tarchonanthuslactone Exerts a Blood Glucose-Increasing Effect in Experimental Type 2 Diabetes Mellitus. Molecules. 2015; 20(3):5038-5049. https://doi.org/10.3390/molecules20035038
Chicago/Turabian StyleDe Souza, Gabriela F. P., Luiz F. T. Novaes, Carolina M. Avila, Lucas F. R. Nascimento, Licio A. Velloso, and Ronaldo A. Pilli. 2015. "(−)-Tarchonanthuslactone Exerts a Blood Glucose-Increasing Effect in Experimental Type 2 Diabetes Mellitus" Molecules 20, no. 3: 5038-5049. https://doi.org/10.3390/molecules20035038