
X-ray Structures of Precursors of Styrylpyridine-Derivatives Used to Obtain 4-((E)-2-(Pyridin-2-yl)vinyl)benzamido-TEMPO: Synthesis and Characterization

Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I | II | III | |
|---|---|---|---|
| Empirical formula | C14H11NO | C14H11NO | C14H11NO2 |
| Crystal system | monoclinic | monoclinic | monoclinic |
| Color, Habit | colorless irregular block | yellow, block | Needle, colorless |
| Formula weight | 209.24 | 209.24 | 225.24 |
| Space group | P 21/c | P21 | P 21 |
| T (K) | 110(2) | 100(2) | 110(2) |
| A (Å) | 12.6674(19) | 3.85728(9) | 3.89359(7) |
| b (Å) | 7.2173(11) | 10.62375(19) | 17.7014(3) |
| c (Å) | 11.5877(14) | 12.8625(2) | 8.04530(12) |
| α (°) | 90.00 | 90.00 | 90.00 |
| β (°) | 97.203(13) | 91.722(2) | 94.4030(16) |
| γ (°) | 90.00 | 90.00 | 90.00 |
| V (Å3) | 1051.0(3) | 526.852(18) | 552.862(16) |
| Z | 4 | 2 | 2 |
| Dc(g cm−3) | 1.322 | 1.319 | 1.353 |
| F (000) | 592 | 220 | 440 |
| μ (mm−1) | 0.663 | 0.662 | 0.740 |
| λ (Å) | 1.5418 | 1.5418 | 1.5418 |
| Crystal size (mm3) | 0.25 × 0.10 × 0.03 | 0.39 × 0.28 × 0.22 | 0.24 × 0.09 × 0.06 |
| 2θmax (°) | 143.8 | 143.8 | 143.8 |
| No of reflections | 6697 | 6136 | 7517 |
| N° of unique reflections, I > 2 σ(I) | 1933 | 2037 | 2165 |
| R1 (I > 2 σ(I)), R1 (all) | 6.08, 8.29 | 3.03, 3.05 | 2.75, 2.79 |
| wR2 (I > 2 σ(I)), wR2 (all) | 17.16, 18.39 | 8.63, 8.65 | 7.43, 7.48 |
| goodness-of-fit | 1.169 | 1.085 | 1.054 |
| Largest diff peak and hole (e Å−3) | 0.22 and −0.27 | 0.21 and −0.21 | 0.21 and −0.16 |
| I | II | III | |
|---|---|---|---|
| C(1)-O(1) | 1.212(4) | 1.212(2) | 1.219(2) |
| C(1)-C(2) | 1.470(4) | 1.477(2) | 1.493(2) |
| C(2)-C(3) | 1.391(4) | 1.392(2) | 1.392(2) |
| C(2)-C(7) | 1.398(4) | 1.394(2) | 1.398(3) |
| C(4)-C(5) | 1.400(4) | 1.402(2) | 1.399(3) |
| C(5)-C(6) | 1.406(4) | 1.403(2) | 1.401(2) |
| C(5)-C(8) | 1.462(4) | 1.470(2) | 1.466(2) |
| C(6)-C(7) | 1.376(4) | 1.386(2) | 1.383(3) |
| C(8)-C(9) | 1.331(4) | 1.331(2) | 1.335(3) |
| C(9)-C(10) | 1.469(4) | 1.469(2) | 1.470(2) |
| C(10)-C(11) | 1.399(4) | 1.393(2) | 1.397(3) |
| C(11)-C(12) | 1.382(5) | 1.391(2) | 1.381(3) |
| C(13)-C(14) | 1.395(5) | 1.388(2) | 1.386(3) |
| C(12)-C(13) | 1.370(5) | 1.386(3) | |
| C(10)-N(1) | 1.351(4) | 1.352(2) | |
| C(14)-N(1) | 1.330(4) | 1.335(2) | |
| C(1)-O(2) | 1.317(2) | ||
| C(10)-C(14) | 1.397(2) | ||
| C(12)-N(1) | 1.338(2) | ||
| C(13)-N(1) | 1.343(2) |
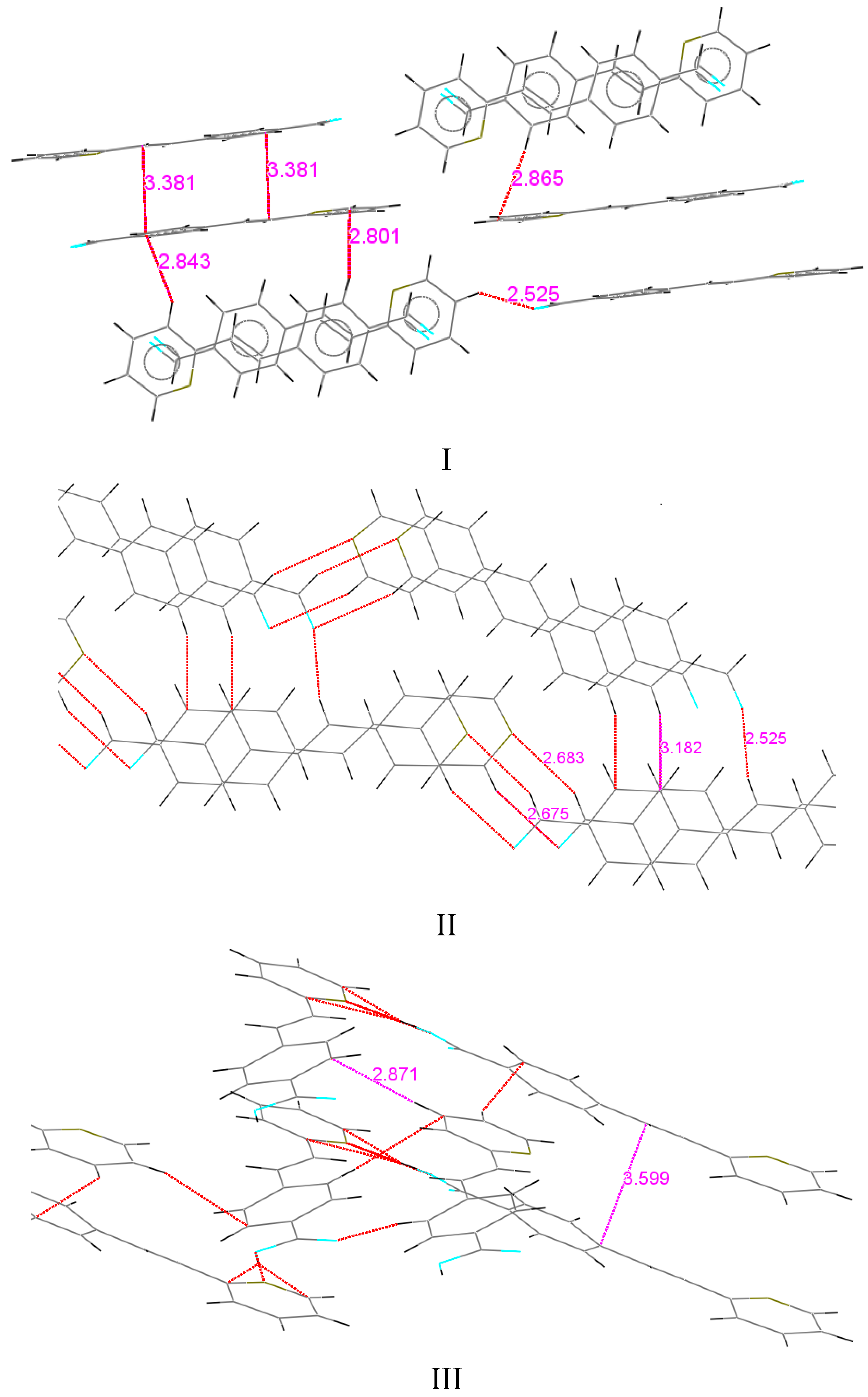
 Car in the phenyl ring of 1.387 Å, in the two six-membered rings through the C(8)-C(9) bond. Similar distances were observed for the 2-styrylpyridine derivatives and were slightly different than the reported values [55]. Bond lengths C(1)-O(1) for I and II were of the equal value, 1.212(4) Å for aldehyde (Csp2=O in Car-C=O of 1.221 Å) [55]. Also, the results showed that structures I and II were almost planar molecules, with the pyridyl ring and 4-phenylcarbaldehyde group being coplanar with the double bond (Table 3 and Table 4). For I, the torsion angles between the atoms C(8)-C(9)-C(10)-N(1) were 0.4(5)°, C(4)-C(5)-C(8)-C(9) of 1.0(5)° and between O(1)-C(1)-C(2)-C(3) were −172.1(3)°. Compound II showed torsion angles between C(8)-C(9)-C(10)-C(14) of 1.2(3)°, C(4)-C(5)-C(8)-C(9) of −4.6(2)°, and between O(1)-C(1)-C(2)-C(3) of 179.57(16)°. The partial π character of the C(9)-C(8), C(10)-C(9) and C(8)-C(5) bonds (Table 2) helps to explain the aromatic planar nature of the molecules. The difference between both compounds is in the molecular crystal packing (Figure 2). For I, the molecular packing does not present regular hydrogen bonding between molecules. However, the packing motif showed a non-classical herringbone packing with a weak π-π overlap between neighboring molecules. For compound I, the distances between the co-facial π-π overlap interaction C/π…C/π was 3.381 Å, between CH(py)…C(Ar) (2.843 Å) and C(Ar)…CH(py) (2.865 Å). Within each stack, however, the molecules are translated (slipped) along the short axis, thus minimizing the π-overlapping between them. A similar interaction arises for compound II between CH(COH)…N(Py) of 2.683 Å, and C(CO)…CH(Py) 2.615 Å, but II did not present short contact interactions such as CH/π∙∙∙∙–CH/π.
Car in the phenyl ring of 1.387 Å, in the two six-membered rings through the C(8)-C(9) bond. Similar distances were observed for the 2-styrylpyridine derivatives and were slightly different than the reported values [55]. Bond lengths C(1)-O(1) for I and II were of the equal value, 1.212(4) Å for aldehyde (Csp2=O in Car-C=O of 1.221 Å) [55]. Also, the results showed that structures I and II were almost planar molecules, with the pyridyl ring and 4-phenylcarbaldehyde group being coplanar with the double bond (Table 3 and Table 4). For I, the torsion angles between the atoms C(8)-C(9)-C(10)-N(1) were 0.4(5)°, C(4)-C(5)-C(8)-C(9) of 1.0(5)° and between O(1)-C(1)-C(2)-C(3) were −172.1(3)°. Compound II showed torsion angles between C(8)-C(9)-C(10)-C(14) of 1.2(3)°, C(4)-C(5)-C(8)-C(9) of −4.6(2)°, and between O(1)-C(1)-C(2)-C(3) of 179.57(16)°. The partial π character of the C(9)-C(8), C(10)-C(9) and C(8)-C(5) bonds (Table 2) helps to explain the aromatic planar nature of the molecules. The difference between both compounds is in the molecular crystal packing (Figure 2). For I, the molecular packing does not present regular hydrogen bonding between molecules. However, the packing motif showed a non-classical herringbone packing with a weak π-π overlap between neighboring molecules. For compound I, the distances between the co-facial π-π overlap interaction C/π…C/π was 3.381 Å, between CH(py)…C(Ar) (2.843 Å) and C(Ar)…CH(py) (2.865 Å). Within each stack, however, the molecules are translated (slipped) along the short axis, thus minimizing the π-overlapping between them. A similar interaction arises for compound II between CH(COH)…N(Py) of 2.683 Å, and C(CO)…CH(Py) 2.615 Å, but II did not present short contact interactions such as CH/π∙∙∙∙–CH/π.| I | II | III | |
|---|---|---|---|
| O(1)-C(1)-C(2) | 124.5(3) | 124.68(16) | 122.50(17) |
| C(3)-C(2)-C(1) | 119.0(3) | 118.97(15) | 121.85(17) |
| C(7)-C(2)-C(1) | 122.1(3) | 121.11(15) | 119.15(16) |
| C(4)-C(5)-C(8) | 122.8(3) | 122.25(14) | 119.27(16) |
| C(6)-C(5)-C(8) | 119.4(3) | 118.76(13) | 122.46(16) |
| C(9)-C(8)-C(5) | 126.4(3) | 126.24(14) | 125.65(17) |
| C(8)-C(9)-C(10) | 124.7(3) | 125.54(14) | 124.62(17) |
| C(11)-C(10)-C(9) | 119.9(3) | 119.68(14) | 123.49(16) |
| N(1)-C(14)-C(13) | 124.0(3) | 124.18(15) | 122.86(19) |
| N(1)-C(10)-C(11) | 122.0(3) | 120.93(17) | |
| N(1)-C(10)-C(9) | 118.1(3) | 115.58(16) | |
| C(13)-C(12)-C(11) | 119.4(3) | 119.76(18) | |
| C(12)-C(13)-C(14) | 118.1(3) | 118.04(18) | |
| C(14)-N(1)-C(10) | 117.5(3) | 119.27(16) | |
| C(11)-C(10)-C(14) | 116.77(14) | ||
| C(14)-C(10)-C(9) | 123.54(15) | ||
| N(1)-C(12)-C(11) | 124.19(16) | ||
| C(13)-C(14)-C(10) | 119.44(15) | ||
| C(12)-N(1)-C(13) | 115.91(14) | ||
| C(1)-O(2)-H(2) | 114.4(18) | ||
| O(2)-C(1)-C(2) | 113.39(16) | ||
| O(1)-C(1)-O(2) | 124.11(17) |
| I | II | III | |
|---|---|---|---|
| O(1)-C(1)-C(2)-C(3) | −172.1(3) | 179.57(16) | 170.38(19) |
| O(1)-C(1)-C(2)-C(7) | 7.5(5) | −1.9(3) | −8.9(3) |
| C(4)-C(5)-C(8)-C(9) | 1.0(5) | −4.6(2) | −165.4(2) |
| C(6)-C(5)-C(8)-C(9) | 179.5(3) | 175.87(15) | 15.8(3) |
| C(5)-C(8)-C(9)-C(10) | 178.9(3) | 178.74(15) | −179.94(17) |
| C(8)-C(9)-C(10)-C(11) | −178.5(3) | −178.21(15) | 19.0(3) |
| C(8)-C(9)-C(10)-N(1) | 0.4(5) | −161.1(2) | |
| N(1)-C(10)-C(11)-C(12) | −0.8(5) | 0.0(3) | 1.9(3) |
| C(9)-C(10)-C(11)-C(12) | 178.1(3) | 178.96(14) | −178.31(19) |
| C(9)-C(10)-N(1)-C(14) | −178.8(3) | 178.04(16) | |
| C(10)-C(11)-C(12)-C(13) | 0.7(5) | 0.0(3) | |
| C(11)-C(12)-C(13)-C(14) | −0.1(5) | −1.6(3) | |
| C(12)-C(13)-C(14)-N(1) | −0.6(5) | 1.4(3) | |
| C(13)-C(14)-N(1)-C(10) | 0.6(5) | 0.1(2) | 0.5(3) |
| C(11)-C(10)-N(1)-C(14) | 0.1(4) | −2.1(3) | |
| C(8)-C(9)-C(10)-C(14) | 1.2(3) | ||
| C(14)-C(10)-C(11)-C(12) | −0.5(2) | ||
| C(11)-C(10)-C(14)-C(13) | 0.4(2) | ||
| C(9)-C(10)-C(14)-C(13) | −178.99(15) | ||
| C(11)-C(12)-N(1)-C(13) | 0.5(2) | ||
| C(14)-C(13)-N(1)-C(12) | −0.6(2) | ||
| O(2)-C(1)-C(2)-C(3) | −9.3(3) | ||
| O(2)-C(1)-C(2)-C(7) | 171.38(17) |


2.2. Characterization of trans-4-((E)-2-(Pyridin-2-yl)vinyl)benzamido-TEMPO (V)

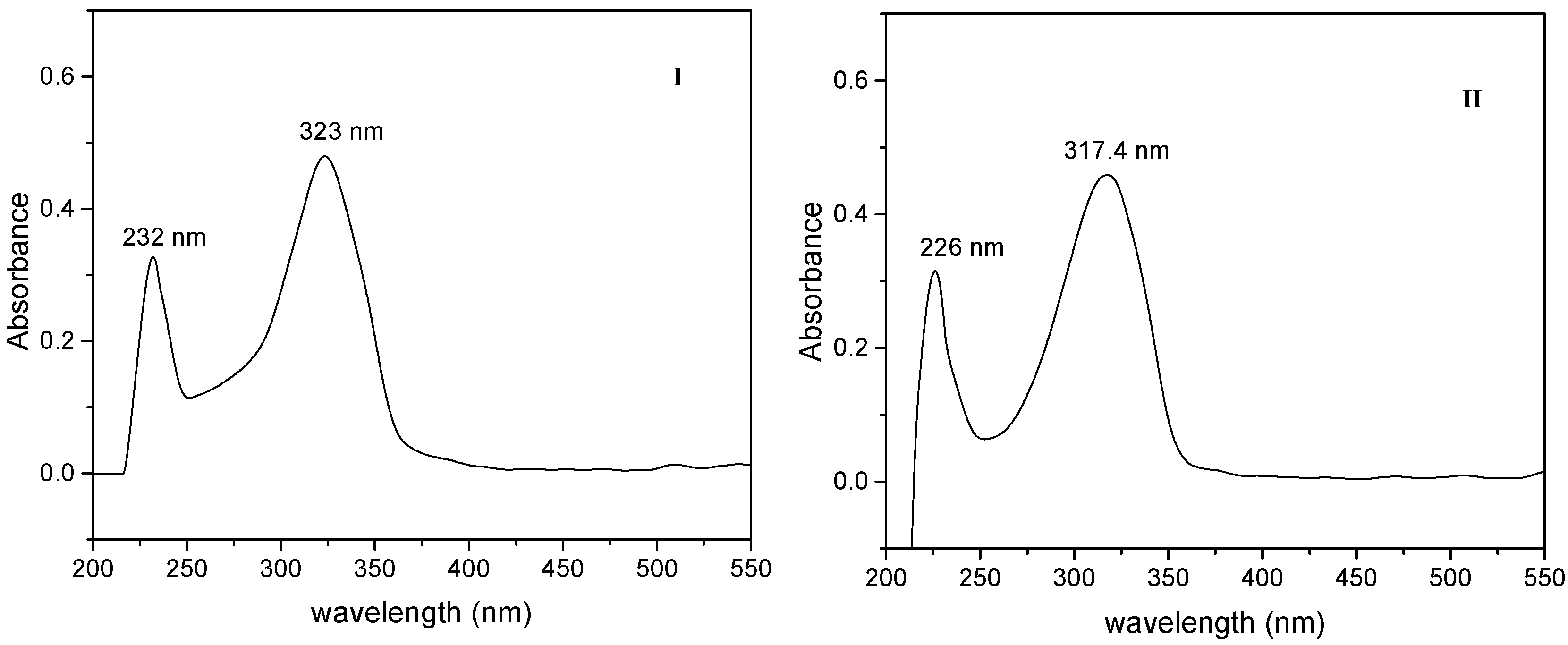
2.3. One Photon Absorption Characterization




3. Experimental Section
3.1. Materials and Instrumentation
3.2. Synthesis and Characterization of Precursors I–IV


3.2.1. Synthesis and Characterization of trans-(E)-4-(2-(Pydridin-2-yl)vinylbenzoic acid and trans-(E)-4-(4-(pydridin-4-yl)vinylbenzoic acid (III and IV)
3.2.2. Condensation Reaction to Obtain the trans-4-((E)-2-(Pyridin-2-yl)vinyl)benzamido-TEMPO (V)
3.3. Crystallization of Compounds I, II and III
3.4. X-ray Crystallography
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffman, A.K.; Henderson, A.T. A new stable free radical: Di-t-butylnitroxide. J. Am. Chem. Soc. 1961, 83, 4671–4672. [Google Scholar] [CrossRef]
- Korshak, Y.V.; Medvedeva, T.V.; Ovchinnikov, A.A.; Spector, V.N. Organic polymer ferromagnet. Nature 1987, 326, 370–372. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Samuni, A.; Krishna, M.C.; DeGraff, W.G.; Ahn, M.S.; Samuni, U.; Russo, A. Biologically active metal-independent superoxide dismutase mimics. Biochemistry 1990, 29, 2802–2807. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; McDonald, M.C.; Mazzon, E.; Siriwardena, D.; Costantino, G.; Fulia, F.; Cucinotta, G.; Gitto, E.; Cordaro, S.; Barberi, I.; et al. Effects of tempol, a membrane-permeable radical scavenger, in a gerbil model of brain injury. Brain Res. 2000, 875, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Utsumi, H.; Hamada, A. Antioxidant activity of nitroxide radicals in lipid peroxidation of rat liver microsomes. Arch. Biochem. Biophys. 1993, 300, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, K.Y.; Zhou, L.; Kálai, T.; Zweier, J.L.; Hideg, K.; Kuppusamy, P. A pyrroline derivative of mexiletine offers marked protection against ischemia/reperfusion-induced myocardial contractile dysfunction. J. Pharmacol. Exp. Ther. 2000, 295, 563–571. [Google Scholar] [PubMed]
- Mitchell, J.B.; DeGraff, W.; Kaufman, D.; Krishna, M.C.; Samuni, A.; Finkelstein, E.; Ahn, M.S.; Hahn, S.M.; Gamson, J.; Russo, A. Inhibition of oxygen-dependent radiation-induced damage by the nitroxide superoxide dismutase mimic, Tempol. Arch. Biochem. Biophys. 1991, 289, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Fritscher, J.; Beyer, M.; Schiemann, O. Synthesis, crystal structure and magnetic properties of a novel nitroxide biradical. Theoretical investigation of the exchange mechanism. Chem. Phys. Lett. 2002, 364, 393–401. [Google Scholar] [CrossRef]
- Fujiwara, H.; Fujiwara, E.; Kobayashi, H. Synthesis, structures and properties of new organic donors connecting to a TEMPO radical through a pyrrolidine ring. Synth. Met. 2003, 133–134, 359–360. [Google Scholar] [CrossRef]
- Tudose, M.; Ionita, P.; Dunitrascu, F.; Draghici, C.; Caproiu, M.T.; Covaci, I.C.; Constantinescu, T.; Banciu, M.; Balaban, A.T. Synthesis and properties of dinitrobenzamido-TEMPO derivatives. ARKIVOC 2005, 225–237. [Google Scholar] [CrossRef]
- Kasumov, V.T.; Ucar, I.; Bulut, A.; Yerli, Y. Synthesis, crystal structure and intermolecular magnetic interactions of a new N-TEMPO-3,5-di-tert-butylsalicylaldimine radical. Solid State Sci. 2011, 13, 1852–1857. [Google Scholar] [CrossRef]
- Cicogna, F.; Coiai, S.; Pinzino, C.; Ciardelli, F.; Passaglia, E. Fluorescence polyolefins by free radical post-reactor modification with functional nitroxides. React. Funct. Polym. 2012, 72, 695–702. [Google Scholar] [CrossRef]
- Lozinsky, E.; Martin, V.V.; Berezina, T.A.; Shames, A.I.; Weis, A.L.; Likhtenshtein, G.I. Dual fluorophore–nitroxide probes for analysis of vitamin C in biological liquids. J. Biochem. Biophys. Methods 1999, 38, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-L.; Zhuo, S.-J.; Wu, Y.-Q.; Fang, F.; Li, L.; Zhu, C.-Q. High selective determination iron(II) by its enhancement effect on the fluorescence of pyrene-tetramethylpiperidinyl (TEMPO) as a spin fluorescence probe. Spectrochim. Acta Part A 2006, 63, 438–443. [Google Scholar] [CrossRef]
- Sato, S.; Suzuki, M.; Soma, T.; Tsunoda, M. Synthesis and properties of umbelliferone-nitroxide radical hybrid compounds as fluorescence and spin-label probes. Spectrochim. Acta Part A 2008, 70, 799–804. [Google Scholar] [CrossRef]
- Sato, S.; Tsunoda, M.; Suzuki, M.; Kutsuna, M.; Takido-uchi, K.; Shindo, M.; Mizuguchi, H.; Obara, H.; Ohya, H. Synthesis and spectral properties of polymethine-cyanine dye-nitroxide radical hybrid compounds for use as fluorescence probes to monitor reducing species and radicals. Spectrochim. Acta Part A 2009, 71, 2030–2039. [Google Scholar] [CrossRef]
- Ikeda, M.; Nagawa, H.; Suzuki, T.; Miyata, N. Novel bisbenzimide-nitroxides for nuclear redox imaging in living cells. Bioorg. Med. Chem. Lett. 2012, 22, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- π–Electron Magnetism: From Molecule to Magnetic Materials. Structure and Bonding; Rawson, J.M.; Palacio, F.; Veciana, J. (Eds.) Springer Verlag: Berlin, Germany, 2001; Volume 100, pp. 93–128.
- Hata, M.; Akutsu, H.; Yamada, J.-I.; Nakatsuji, S. N-Salicylideneamine derivatives with TEMPO substituents. Molecules 2004, 9, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Burroughes, J.H.; Bradley, D.D.C.; Brown, A.R.; Marks, R.N.; Mackay, K.; Friend, R.H.; Burn, P.L.; Holmes, A.B. Light-emitting diodes based on conjugated polymers. Nature 1990, 347, 539–541. [Google Scholar] [CrossRef]
- Goodson, T.; Li, W.; Gharavi, A.; Yu, L. Oligophenylenevinylenes for light-emitting diodes. Adv. Mater. 1997, 9, 639–643. [Google Scholar] [CrossRef]
- Müllen, K.; Wegner, G. Electronic Materials: The Oligomer Approach; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar]
- Gierschner, J.; Ehni, M.; Egelhaaf, H.-J.; Millian, B.; Beljonne, D.; Benmansour, H.; Bazan, G.C. Solid-state optical properties of linear polyconjugated molecules: π-Stack contra herringbone. J. Chem. Phys. 2005, 123, 144914. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.; Grimsdale, A.C.; Holmes, A.B. Elektrolumineszierende konjugierte polymere-polymere erstrahlen in neuem licht. Angew. Chem. 1998, 110, 416–443. [Google Scholar] [CrossRef]
- Kraft, A.; Grimsdale, A.C.; Holmes, A.B. Electroluminescent conjugated polymers—Seeing polymers in a new light. Angew. Chem. Int. Ed. Engl. 1998, 37, 402–428. [Google Scholar] [CrossRef]
- Mathy, A.; Ueberhofen, K.; Schenk, R.; Gregorius, H.; Garay, R.; Müllen, K.; Bubeck, C. Third-harmonic-generation spectroscopy of poly(p-phenylenevinylene): A comparison with oligomers and scaling laws for conjugated polymers. Phys. Rev. B 1996, 53, 4367–4375. [Google Scholar] [CrossRef]
- Gurge, R.M.; Hickl, M.; Krause, G.; Lahti, P.M.; Hu, B.; Yang, Z.; Karasz, F.E. Synthesis of a green-emitting alternating block copolymer. Polym. Adv. Technol. 1998, 9, 504–510. [Google Scholar] [CrossRef]
- Skotheim, T.A.; Reynolds, J.R. Handbook of Conducting Polymers, Part III: Properties and Characterization of Conducting Polymers; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Strehmel, B.; Sarker, A.M.; Detert, H. The Influence of σ and π Acceptors on Two-Photon Absorption and Solvatochromism of Dipolar and Quadrupolar Unsaturated Organic Compounds. ChemPhysChem 2003, 4, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Collette, J.C.; Harper, A.W. Properties and chemical environment effects of alkylamino styryl pyrazine two-photon fluorophores. Proc. SPIE 2003, 5212, 184–192. [Google Scholar]
- Woo, H.Y.; Liu, B.; Kohler, B.; Korystov, D.; Mikhailovsky, A.; Bazan, G.C. Solvent Effects on the Two-Photon Absorption of Distyrylbenzene Chromophores. J. Am. Chem. Soc. 2005, 127, 14721–14729. [Google Scholar] [CrossRef] [PubMed]
- Czerney, P.; Grummt, U.-W. New near-infrared-absorbing acidochromic dyes and their application in sensor techniques. Sensor. Actuat. B.-Chem. 1997, 39, 395–400. [Google Scholar] [CrossRef]
- Schmitt, V.; Glang, S.; Preis, J.; Detert, H. Proton-Induced Multiple Changes of the Absorption and Fluorescence Spectra of Amino-Aza-Oligo(phenylenevinylene)s. Sens. Lett. 2008, 6, 524–530. [Google Scholar] [CrossRef]
- Lippert, E.; Lüder, W.; Moll, F.; Nägele, W.; Boos, H.; Prigge, H.; Seybold-Blankenstein, I. Umwandlung von Elektronenanregungsenergie. Angew. Chem. 1961, 73, 695–706. [Google Scholar] [CrossRef]
- Rettig, W. Ladungstrennung in angeregten Zuständen entkoppelter Systeme—TICT-Verbindungen und Implikationen für die Entwicklung neuer Laserfarbstoffe sowie für den Primärprozeß von Sehvorgang und Photosynthese. Angew. Chem. 1986, 98, 969–986. [Google Scholar] [CrossRef]
- Rettig, W. Charge separation in excited states of decoupled systems—Tict compounds and implications regarding the development of new laser dyes and the primary process of vision and photosynthesis. Angew. Chem. Int. Ed. Engl. 1986, 25, 971–988. [Google Scholar] [CrossRef]
- Dobkowski, J.; Michl, J.; Waluk, J. Electronic spectroscopy and photophysics of 2-(N-methyl-N-isopropylamino)-5-cyanopyridine and related compounds. Phys. Chem. Chem. Phys. 2003, 5, 1027–1031. [Google Scholar] [CrossRef]
- Nemkovich, N.A.; Detert, H.; Schmitt, V. Localized excitation effect on dipole moments of oligophenylenevinylenes in their excited Franck–Condon state. Chem. Phys. 2010, 378, 37–41. [Google Scholar] [CrossRef]
- Detert, H.; Sadovski, O.; Sugiono, E. Acidochromism of C2-symmetrical aza-analogues of 1,4-distyrylbenzene. J. Phys. Org. Chem. 2004, 17, 1046–1050. [Google Scholar] [CrossRef]
- Detert, H.; Sugiono, E. Bis(pyridylvinyl)diaminobenzenes: Synthesis, acidochromism and solvatochromism of the fluorescence. J. Lumin. 2005, 112, 372–376. [Google Scholar] [CrossRef]
- Hancock, J.M.; Jenekhe, S.A. Unusual protonation-induced continuous tunability of optical properties and electroluminescence of a π-conjugated heterocyclic oligomer. Macromolecules 2008, 41, 6864–6867. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kobayashi, S.; Wakamiya, T.; Matsubara, Y.; Yoshida, Z.-I. Banana-shaped oligo(aryleneethynylene)s: Synthesis and light-emitting characteristics. Angew. Chem. Int. Ed. 2005, 44, 7040–7044. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Sánchez, A.; Maldonado-Rivera, J.L. Condensation reactions of methylpyridines and aromatic aldehydesunder catalyst and solvent free conditions. Chem. Indian J. 2006, 3, 262–267. [Google Scholar]
- Percino, M.J.; Chapela, V.M.; Salmón, M.; Toscano, R.A. Unexpected crystallization and X-ray crystal structure of racemic 1-phenyl-2-(4-pyridyl)ethanol intermediate. J. Chem. Crystallogr. 2000, 30, 385–388. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Salmón, M.; Espinoza-Pérez, G.; Herrera, A.M.; Flores, A. X-ray crystal structure of 2-styrylpyridine. J. Chem. Crystallogr. 1997, 27, 549–552. [Google Scholar] [CrossRef]
- Chapela, V.M.; Percino, M.J.; Rodríguez-Barbarín, C. Crystal structure of 2,6-distyrylpyridine. J. Chem. Crystallogr. 2003, 33, 77–83. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Montiel, L.-F.; Rodríguez-Barabrin, C. X-Ray crystal structures of a 1-(p-fluorophenyl)-2-(α-pyridyl)ethanol intermediate and the 1-(p-fluorophenyl)-2-(α-pyridyl)ethane dehydration compound obtained from the condensation reaction of 2-methylpyridine and p-fluorobenzaldehyde. Open Crystallogr. J. 2008, 1, 37–41. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Montiel, L.-F.; Pérez-Gutierrez, E.; Maldonado, J.L. Spectroscopic characterization of halogen- and cyano-substituted pyridinevinylenes synthesized without catalyst or solvent. Chem. Pap. 2010, 64, 360–367. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M. Unexpected intermediate 1-phenyl-2-(4-pyridyl)ethanol isolated from benzaldehyde and 4-picoline condensation reaction. Res. Chem. Intermed. 2000, 26, 303–307. [Google Scholar] [CrossRef]
- Pérez-Gutierrez, E.; Percino, M.J.; Chapela, V.M.; Maldonado, J.L. Optical and morphological characterization by atomic force microscopy of luminescent 2-styrylpyridine derivative compounds with poly(N-vinylcarbazole) films. Thin Solid Films 2011, 519, 6015–6020. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Urzua, O.; Montiel, L.-F.; Rodríguez-Barabrín, C. 1-(p-Fluorophenyl)-2-(2'-pyridyl)ethanol and 1-(p-Fluorophenyl)-2-(2'-pyridyl)ethene obtained from the condensation reaction of 2-picoline and p-fluorophenylaldehyde under catalyst- and solvent-free conditions. Res. Chem. Intermed. 2007, 33, 623–629. [Google Scholar] [CrossRef]
- Castro, M.E.; Percino, M.J.; Chapela, V.M.; Cerón, M.; Soriano, G.; Melendez, F. Theoretical Study of the UV/Vis Absorption Spectra of Styrylpyridine Compounds Using TD-DFT Calculations. J. Mol. Model. 2013, 19, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Percino, M.J.; Chapela, V.M.; Perez-Gutierrez, E.; Ceron, M.; Soriano, G. Synthesis, optical and spectroscopic characterization of substituted 3-phenyl-2-arylacrylonitriles. Chem. Pap. 2011, 65, 42–51. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Cerón, M.; Soriano-Moro, G.; Castro, M.E. Synthesis and molecular structure of the 1-phenyl-2-(2-pyridyl)ethanol intermediate obtained from the condensation reaction of 2-picoline and benzaldehyde. Res. Chem. Intermed. 2013. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Rintoul, L.; Micallef, A.S.; Bottle, S.E. The vibrational group frequency of the N-O• stretching band of nitroxide stable free radicals. Spectrochim. Acta Part A 2008, 70, 713–717. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Solomon, P.H. Infrared Absorption Spectroscopy, 2nd ed.; Holden-Day Inc.: Oakland, CA, USA, 1977. [Google Scholar]
- Williams, D.H.; Fleming, I. Spectroscopic Methods in Organic Chemistry, 3rd ed.; McGraw-Hill: Maidenhead, UK, 1980. [Google Scholar]
- Daku, L.M.L.; Linares, J.; Boillot, M.-L. Ab Initio Static and Molecular Dynamics Study of 4-Styrylpyridine. ChemPhysChem 2007, 8, 1402–1416. [Google Scholar] [CrossRef] [PubMed]
- Daku, L.M. L.; Linares, J.; Boillot, M.-L. Ab initio static and molecular dynamics study of the absorption spectra of the 4-styrylpyridine photoswitch in its cis and trans forms. Phys. Chem. Chem. Phys. 2010, 12, 6107–6123. [Google Scholar] [CrossRef] [PubMed]
- Zhadanov, R.I.; Golubev, V.A.; Rozantsev, E.G. Synthesis and structure of 1-oxopiperidinium tribromides. Izv. Akad. Nauk SSSR. Ser. Khim. 1970, 1, 186–187. [Google Scholar]
- Dane, E.L.; Maly, T.; Debelouchina, G.T.; Griffin, R.G.; Swager, T.M. Synthesis of a BDPA-TEMPO biradical. Org. Lett. 2009, 11, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXTL/PC User Manual; Siemens Analytical X-ray Instruments Inc.: Madison, WI, USA, 1990. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soriano-Moro, G.; Percino, M.J.; Sánchez, A.L.; Chapela, V.M.; Cerón, M.; Castro, M.E. X-ray Structures of Precursors of Styrylpyridine-Derivatives Used to Obtain 4-((E)-2-(Pyridin-2-yl)vinyl)benzamido-TEMPO: Synthesis and Characterization. Molecules 2015, 20, 5793-5811. https://doi.org/10.3390/molecules20045793
Soriano-Moro G, Percino MJ, Sánchez AL, Chapela VM, Cerón M, Castro ME. X-ray Structures of Precursors of Styrylpyridine-Derivatives Used to Obtain 4-((E)-2-(Pyridin-2-yl)vinyl)benzamido-TEMPO: Synthesis and Characterization. Molecules. 2015; 20(4):5793-5811. https://doi.org/10.3390/molecules20045793
Chicago/Turabian StyleSoriano-Moro, Guillermo, María Judith Percino, Ana Laura Sánchez, Víctor Manuel Chapela, Margarita Cerón, and María Eugenia Castro. 2015. "X-ray Structures of Precursors of Styrylpyridine-Derivatives Used to Obtain 4-((E)-2-(Pyridin-2-yl)vinyl)benzamido-TEMPO: Synthesis and Characterization" Molecules 20, no. 4: 5793-5811. https://doi.org/10.3390/molecules20045793