Abstract
Heparin and heparan sulphate (H/HS) are important members of the glycosaminoglycan family of sugars that regulate a substantial number of biological processes. Such biological promiscuity is underpinned by hetereogeneity in their molecular structure. The degree of O-sulfation, particularly at the 6-position of constituent d-GlcN units, is believed to play a role in modulating the effects of such sequences. Synthetic chemistry is essential to be able to extend the diversity of HS-like fragments with defined molecular structure, and particularly to deconvolute the biological significance of modifications at O6. Here we report a synthetic approach to a small matrix of protected heparin-type oligosaccharides, containing orthogonal d-GlcN O-6 protecting groups at programmed positions along the chain, facilitating access towards programmed modifications at specific sites, relevant to sulfation or future mimetics.
1. Introduction
Heparin and heparan sulphate (H/HS) are ubiquitous biological oligosaccharides which regulate many important signalling processes, especially within the extracellular matrix [1,2,3,4,5]. H/HS oligosaccharides consist of repeating glucosamine and uronic acid monomers and are highly polydisperse, varying not only in the backbone constitution (l-iduronic and/or d-glucuronic acid residues) and length (including the biologically effective region), but also in a variety of sulfation patterns. Sulfation is obligatory on O-2, where l-IdoA is installed, but can be present on combinations of glucosamine N and O-6 sites. In native sequences, sulfation is typically organized into high- or lower-sulfation domains, and, along with the ratio of l-IdoA/D-GlcA, correlates to the heparin/heparan sulfate designation. Sequences which contain l-iduronic acid have attracted particular interest due to the evidence of many key bio-effector sequences being l-IdoA enriched (higher-sulphated, l-idoA heparin-like structure shown in Figure 1). Understanding structure/function relationships of H/HS chains that modulate such processes is a major challenge in chemical biology.
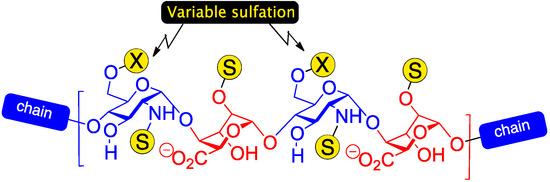
Figure 1.
Higher sulfation heparin-type region with varying levels of GlcN O-6 sulfation.
The extensive microhetereogeneity of these oligosaccharides ensures that methods for synthesis of diverse, defined H/HS fragments are central to revealing structure-functional information in their chemical biology [6,7] and synthetic interest in solution, solid phase and utilizing enzymatic methods has been expanding [8,9,10,11,12,13,14,15,16,17,18,19,20,21].
Homogenous synthetic O-6-sulfation levels have been shown to affect the binding preferences of GAGs to target proteins [7,22,23], and recent examples have evidenced that even disaccharides can exhibit differential recognition based on sulfation patterns [24,25]. It is not yet clear how such binding effects translate to biological effects, nor how biological effects may be affected by defined regioselectivity of O-6 sulfation. There is, however, significant evidence that the overall level of glucosamine-6-O-sulfation, or de-6-O-sulfation, of D-GlcN, (Figure 1) is of central importance to effecting differential H/HS biological signalling events mediated by several HS-dependent cytokines (including CXCL8 [26,27], CXCL12 [28], FGF2 [29,30,31] and VEGF [28,30,31]). Many biological studies have employed enzymatically-generated heterogeneous materials as such native isolation only provides access to heterogeneous mixtures, differentiated by average sulfation levels. Interest in medium length structurally-defined heparanoids has been emboldened by the success of fondaparinux and activity of recently-reported longer synthetic mixed heparins [32]. This strongly suggests that evaluating programmed site-specific sulfation diversity is a high value target area. More ready availability of O-6 regiochemically differentiated fragments to underpin access towards such systems is required, and of particular utility would be medium length targets where terminal sulfate clustering or separation could be delivered (Figure 2).
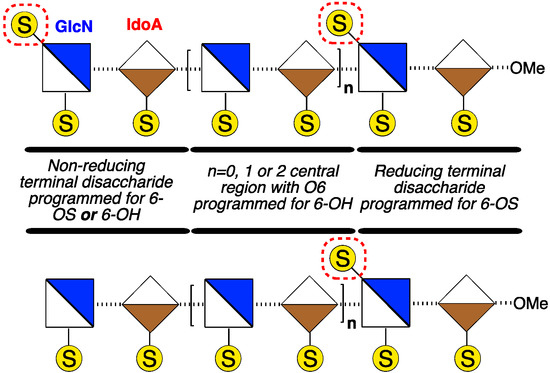
Figure 2.
Strategic sequences with reducing terminus 6-O-sulfated (6-OS), central sequence de-6-O-sulfated (6-OH) and non-reducing terminus programmable for 6-OS or 6-OH.
To access such a series of mono or bis-terminus-modified oligosaccharides requires application of two types of disaccharide donor module, bearing orthogonal protections of O-6 groups to provide correlation to sites for ultimate 6-O-de-sulfation or 6-O-sulfation respectively, in any target H/HS sequences (Figure 3).
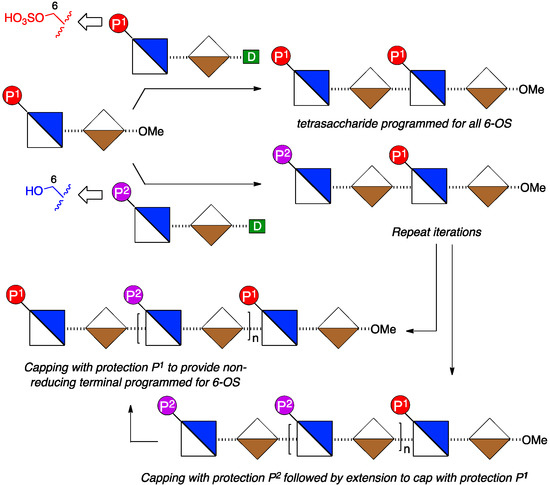
Figure 3.
Strategy with fixed reducing terminal O6 protection to provide a matrix of oligosaccharides with single programmed reducing terminal unit or both terminal units programmed for 6-OS by choice of protecting group (d = glycosyl donor group).
This, combined with a single reducing terminal disaccharide module could then be employed to access a ladder of all mono- and bis-terminal unit differentiated oligosaccharides.
2. Results and Discussion
As part of a program to develop efficient and scalable access to a variety of H/HS oligosaccharide fragments, we have developed effective methods to access heparin-like per-6-O-sufated [33] and per-6-O-desulfated [30,34,35] species. These syntheses underscore the efficacy of d-GlcN-l-IdoA-SPh donors 1 and 2 as efficient glycosyl building blocks in the construction of homogeneously O-6-derivatized oligomers up to dodecasaccharide. The d-GlcN 6-OH protecting groups (OBn or OAc) determine the fate of O-6 (either OBn = 6-OH or OAc = 6-OS), through common sulfation and deprotection steps (Figure 4). Here we extend this work to employ a modular approach to prepare protected H/HS-like precursor fragments containing regio-differentiated protections of sequence-specific O-6 sites, relevant to different H/HS fragments with programmed site-specific sulfations, or for the introduction of other region-defined mimetics. We demonstrate application of this to deliver five novel single-terminal and double-terminal differentiated tetra-, hexa- and octasaccharides across this matrix.
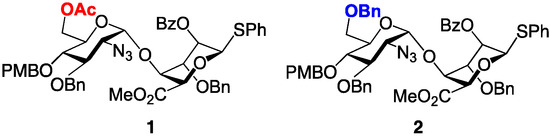
Figure 4.
Disaccharide modules 1 and 2 with alternate d-GlcN 6-position protecting groups, programmed for access to 6-OH/6-OS final compounds.
The homologation matrix for all oligosaccharides thus began with OMe-capped disaccharide 3, the gram scale synthesis of which we have previously reported [36]. Ceric ammonium nitrate mediated removal of the PMB group provided the required novel disaccharide acceptor 4 in high yield (Scheme 1).
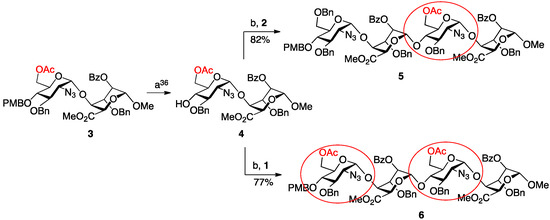
Scheme 1.
Synthesis of terminally-differentiated tetrasaccharides. (a) Ceric ammonium nitrate (CAN), MeCN/H2O; (b) N-iodosuccinimide (NIS), AgOTf, DCM.
This material embeds O-6 protection in the reducing terminal disaccharide unit and thus allies deprotection and sulfation of d-GlcN O-6 in any derived oligosaccharide targets (in addition to the obligatory O-2 sulfation of the l-iduronic acid residue), whilst enabling potential controlled modification for new regiodefined mimetics.
Disaccharide 4 was then employed to illustrate parallel synthesis of tetra- and hexasaccharides, towards a target octasaccharide with double terminal O-6 acylated units. The first cycle of iteration illustrated use of acceptor 4 in two separate glycosylation reactions, firstly with donor module 1 (to install a second 6-OAc group) and secondly with donor 2 (to install a 6-OBn protecting group). Both glycosylations proceeded in good to excellent yields, affording the two new alpha-linked tetrasaccharides 5 and 6 in 82% and 77% yields, respectively.
To progress towards the next homologation targets with the same alternatives of non-reducing 6-OAc or 6-OBn units, tetrasaccharide 5 was subjected to the same two parallel glycosylations, again using either of the two disaccharide donor modules, 1 or 2. Thus, 5 was first deprotected at the non-reducing end 4-position with CAN and then separately glycosylated with 1 and 2 to yield hexasaccharides 7 and 8 in very good to excellent yields (70% for 7, 96% for 8). This afforded the second tier of species with different O-6 protecting group patterns, encoding accessibility to H/HS species with d-GlcN 6OS-6OH-6OS or 6OH-6OH-6OS sulfations.
Finally, the mono-6-OAc hexasaccharide 8 was elaborated by a further cycle of iteration to octasaccharide 9, providing a third oligosaccharide length with differentiated O-6 protecting groups encoding for ultimate sulfation at both the non-reducing and reducing termini. This iteration proceeded with similar efficiency to the prior cycles, with 8 deprotected via the established conditions in 78% yield and the resulting acceptor hexasaccharide glycosylated with iterative donor 1 to afford 9 in 66% yield (Scheme 2).
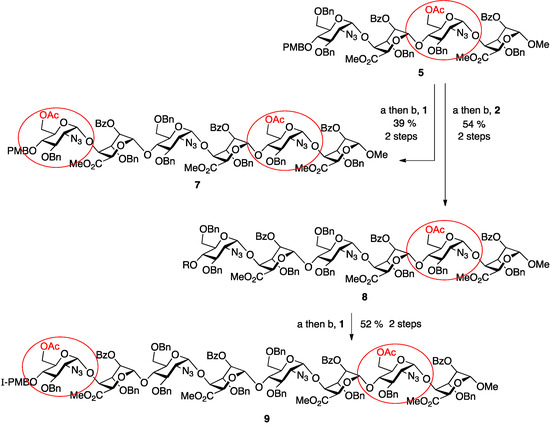
Scheme 2.
Synthesis of ladder of mono- or bis-terminally-differentiated hexasaccharides 7 and 8 and octasaccharide 9. (a) CAN, MeCN/H2O; (b) NIS, AgOTf, DCM.
Of note during this final iteration was the need to raise the number of molar equivalents of glycosylation activator, NIS, from 5 (in prior iterations), to 7 for formation of 9. This resulted in effective glycosylation, but also a concomitant iodination of the electron rich PMB ring, as observed by ESI-MS (m/z = 1612.0377, 100%, z = 2 and no evidence of the non-iodinated species). This is a useful methodological note for SPh/NIS mediated glycosylations of substrates containing electron rich aromatic rings, but does not detract from the overall utility of the method, as the iodinated PMB group would necessarily be subsequently removed, either for any further iteration, or under final oligosaccharide deprotection procedures.
During these iterative steps both the disaccharide donors 1 or 2, delivered glycosylation yields close to or above 70%, though in general the 6-OAc systems performed to give slightly lower yields than the 6-OBn analogues. This result is in keeping with our [33,35] and others [19,20] previously established routes using 6-O-ether/acetal vs. 6-O-ester type disaccharide building blocks to effect oligosaccharide syntheses.
For hexa- and octasacccharide systems 7 and 9, 1H-NMR data indicated that all the l-iduronate unit conformations are dependent on location in the sequence. The 3JH1-H2 coupling constants are <6.0 Hz for all l-IdoA residues of 7 and 9 (Figure 5). Figure 5 illustrates a clear resolution of each constituent l-IdoA H1 signal allowing the result of an iterative oligosaccharide homologation process (i.e., transformation of 7 to 9) to be readily correlated with such clear spectral dispersion. This dispersion also facilitates conformational analysis, the differential couplings for the reducing terminal l-IdoA H1 compared to the internal and non-reducing terminal l-IdoA H1 signals is consistent with the former being predominantly 1C4, whilst the larger couplings for the remaining l-IdoA units would be consistent with more significantly contributions from the 2So skew-boat [14,37].
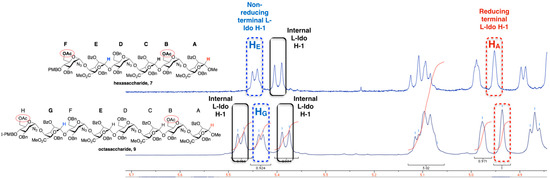
Figure 5.
1H-NMR signals (400 MHz) for iduronate anomeric regions of hexasaccharide 7 and octasaccharide 9.
3. Experimental Section
General Information
Reagents and solvents were purchased from Sigma-Aldrich (Gillingham, UK) or Alfa Aesar (Heysham, UK). All reactions were carried out under an atmosphere of dry nitrogen, unless mentioned otherwise. 1H-NMR spectra were recorded on a Bruker Advance Ultrashield DPX 400 (400 MHz) and 13C-NMR spectra were recorded at 100 MHz on Bruker 300 and 400 DPX spectrometer. Melting points were obtained using a Stuart Scientific SMP1 melting point apparatus. Low-resolution mass spectra were recorded on a Micro Mass Trio 200 spectrometer (Wilmslow, UK) while high-resolution mass spectra were measured on a Kratos Concept IS spectrometer (Manchester, UK). Elemental analyses were performed using a Thermo Flash 2000 Organic Elemental Analyzer (ThermoFisher scientific, Warrington, UK) for CHN analysis. Flash chromatography was conducted using Merck (Nottingham, UK) silica gel 60 (particle size 40–60 μm). Analytical TLC were performed on Merck 60 F254 aluminium-backed plates containing a 254 nm fluorescent indicator. Optical rotations were measured at 589 nm in a 1 dm cell using an Optical Activity AA1000 polarimeter.
Methyl 6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranoside) uronate (4). Ceric (IV) ammonium nitrate (770 mg, 1.4 mmol) was added to a solution of 3 (600 mg, 0.7 mmol) in acetonitrile and water (17 mL, 8:1, v/v). The mixture was stirred for 1 h at ambient temperature and then diluted with DCM (150 mL), washed with saturated aq. NaHCO3 (100 mL) and saturated aqueous NaCl (50 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The product was purified by column chromatography (EtOAc/hexane 1:3), yielding 4 (470 mg, 0.64 mmol, 91%) as a foam. Rf 0.31 (EtOAc/hexane 2:3); = +8.6 (c = 0.26, CH2Cl2); 1H-NMR (400 MHz; CDCl3) δ 8.18–8.15 (m, 2H, ArH), 7.52–7.17 (m, 13H, ArH), 5.14–5.13 (brs, 1H, H2IdoA), 5.11–5.10 (brs, 1H, H1IdoA), 4.94 (d, 1H, J = 11.9 Hz, CH2Ph), 4.86 (d, J = 2.0 Hz, 1H, H5IdoA), 4.77 (d, 1H, J = 11.9 Hz, CH2Ph), 4.67 (d, J = 3.4 Hz, 1H, H1GlcN), 4.64 (dd, J = 12.6, 2.8 Hz, 1H, H6aGlcN), 4.29 (d, 1H, J = 10.6 Hz, CH2Ph), 4.17 (dd, J = 12.5, 2.2 Hz, 1H, H6bGlcN), 4.16–4.12 (m, 1H, H4IdoA), 4.03–4.00 (m, 3H, CH2Ph, H3IdoA), 3.46 (dt, J = 10.0, 2.4 Hz, 1H, H5GlcN), 3.81 (s, 3H, C(O)OCH3), 3.51 (s, 3H, OCH3), 3.45 (dd, J = 10.0, 8.8 Hz, 1H, H4GlcN), 3.33 (dd, J = 10.0, 9.0 Hz, 1H, H3GlcN), 3.18 (dd, J = 10.1, 3.6 Hz, 1H, H2GlcN), 2.10 (s, 3H, C(O)CH3); 13C-NMR (100 MHz; CDCl3) δ 172.3, 169.8, 165.6, 137.9, 137.5, 133.5, 130.1, 129.7, 128.9, 128.6, 128.6, 128.3, 128.1, 128.0, 100.5, 99.7, 79.3, 76.0, 75.0, 72.8, 72.6, 71.3, 70.4, 67.9, 67.1, 63.3, 62.7, 56.4, 52.5, 21.0; HRMS (FT MS NSI+) m/z calcd for C37H45N4O13 [M+NH4]+ 753.2978, found 753.2980.
Methyl (2-azido-3,6-di-O-benzyl-2-deoxy-4-O-p-methoxybenzyl-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranosyl) uronate)-(1→4)-6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranoside) uronate (5). Acceptor 4 (770 mg, 1.05 mmol) and donor 2 (1.4 g, 1.42 mmol) were mixed together, evaporated from dry toluene (3 × 20 mL) and dried under high vacuum for 1 h. The foam was dissolved in dry DCM (20 mL) and powdered molecular sieves (4 Å, 650 mg) added. The mixture was cooled to 0 °C and NIS (1.18 g, 5.25 mmol) added. The mixture was stirred for a further 15 min at this temperature and a catalytic amount (small spatula tip) of AgOTf was then added. The mixture was kept under nitrogen at 0 °C for another 30 min and was then quenched by addition of saturated aqueous Na2S2O3 and saturated aqueous NaHCO3 (10 mL, 1:1, v/v). The suspension was filtered through Celite®, the phases separated and the organic layer washed with saturated aqueous NaCl (10 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The mixture was purified by column chromatography (EtOAc/hexane, 1:4→2:3), yielding 5 (1.3 g, 0.81 mmol, 77%). Rf 0.31 (EtOAc/hexane 1:2); 1H-NMR (400 MHz; CDCl3) δ 8.14–8.09 (m, 4H, Bz-ArH), 7.47–7.13 (m, 33H, ArH), 7.07 (d, 1H, J = 8.7 Hz, PMB), 6.84 (d, 1H, J = 8.7 Hz, PMB), 5.46 (d, 1H, J = 4.8 Hz, H1'IdoA), 5.18–5.16 (m, 1H, H2'IdoA), 5.07 (brs, 1H, H1IdoA), 5.05 (brs, 1H, H2IdoA), 4.99 (d, 1H, J = 3.5 Hz, H1GlcN), 4.91 (d, 1H, J = 11.8 Hz, CH2Ph), 4.78–4.74 (m, 4H, H5’IdoA, H5IdoA, 2 × CH2Ph), 4.67–4.36 (m, 10H, H1GlcN, H4’IdoA, H4’IdoA, 7 × CH2Ph), 4.29–4.27 (m, 2H, CH2Ph), 4.13–4.06 (m, 2H, H3’IdoA, H3IdoA), 3.93–3.92 (m, 1H, H5’GlcN), 3.88–3.80 (s, 3H, PMBOCH3), 3.76–3.59 (m, 12H, H3’GlcN, H3’GlcN, H4’GlcN, H4’GlcN, H5GlcN, 2 × H6’GlcN, 2 × H6GlcN, OCH3), 3.47 (s, 6H, C(O)OCH3), 3.24 (dd, 1H, J = 10.1, 3.6 Hz, H2’GlcN), 3.19–3.15 (m, 1H, H2GlcN), 2.09 (s, 3H, C(O)CH3); 13C-NMR (100 MHz; CDCl3) δ 170.8, 169.7, 169.6, 169.5, 165.7, 165.3, 159.4, 137.9, 137.4, 133.5, 132.5, 130.5, 130.1, 130.0, 129.6, 128.9, 128.8, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.5, 113.8, 100.4, 99.4, 99.0, 98.6, 79.9, 78.7, 76.1, 75.8, 75.0, 74.8, 74.4, 74.1, 73.6, 72.6, 69.8, 68.2, 67.1, 63.6, 63.5, 56.3, 55.4, 52.3, 52.0, 20.8; HRMS (FT MS NSI+) m/z calcd for C86H94N7O25 [M+NH4]+ 1624.6294, found 1624.6299.
Methyl (6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-4-O-p-methoxybenzyl-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranosyl) uronate)-(1→4)-6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranoside) uronate (6). Acceptor 4 (167 mg, 0.23 mmol) and donor 1 (286 mg, 0.31 mmol) were mixed together, evaporated from dry toluene (3 × 5 mL) and dried under high vacuum for 1 h. The foam was dissolved in dry DCM (5 mL) and powdered molecular sieves (4 Å, 125 mg) added. The mixture was cooled to 0 °C and NIS (255 mg, 1.14 mmol) added. The mixture was stirred for a further 15 min at this temperature and a catalytic amount (small spatula tip) of AgOTf was then added. The mixture was kept under nitrogen at 0 °C for another 30 min and was then quenched by addition of saturated aqueous Na2S2O3 and saturated aqueous NaHCO3 (15 mL, 1:1, v/v). The suspension was filtered through Celite®, the phases separated and the organic layer washed with saturated aqueous NaCl (10 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The mixture was purified by column chromatography (DCM/EtOAc 7:1), yielding 6 (270 mg, 0.23 mmol, 77%) as a foam. Rf 0.13 (EtOAc/hexane 1:2);
= −1.5 (c = 0.31, CH2Cl2); 1H-NMR (400 MHz; CDCl3) δ 8.10–8.05 (m, 4H, Bz-ArH), 7.46–7.14 (m, 28H, ArH), 6.84 (d, J = 8.7 Hz, 1H, PMB), 5.43 (d, J = 4.4 Hz, 1H, H1’IdoA), 5.11 (t, J = 4.8 Hz, 1H, H2’IdoA), 5.03–5.02 (brs, 1H, H2IdoA), 5.02–5.01 (brs, 1H, H1IdoA), 4.89–4.86 (m, 3H, H1GlcN, 2 × CH2Ph), 4.75 (d, J = 2.0 Hz, 1H, H5IdoA), 4.73–4.21 (m, 15H, H1’GlcN, H5’IdoA, H5’GlcN, H6a’GlcN, H6aGlcN, H3’IdoA, H4’IdoA, 8 × CH2Ph), 4.09–3.43 (m, 11H, H3GlcN, H3’GlcN, H4GlcN, H4’GlcN, H5GlcN, H6b’GlcN, H6bGlcN, H3IdoA, H4IdoA, 2 × CH2Ph), 3.77 (s, 3H, C(O)OCH3), 3.59 (s, 3H, PMBOCH3), 3.56 (s, 3H, OCH3), 3.41 (s, 3H, C(O)OCH3), 3.17 (dd, J = 10.2, 3.5 Hz, 1H, H2GlcN), 3.13 (dd, J = 10.2, 3.6 Hz, 1H, H2’GlcN), 2.06 (s, 3H, C(O)CH3), 1.94 (s, 3H, C(O)CH3); 13C-NMR (100 MHz; CDCl3) δ 170.8, 170.6, 169.6, 169.5, 165.6, 165.2, 159.5, 137.7, 137.6, 137.4, 137.2, 133.5, 133.5, 129.9, 128.5, 128.5, 128.4, 128.3, 128.2, 128.1, 127.9, 113.9, 100.4, 99.2, 99.1, 98.4, 80.0, 78.7, 77.4, 77.0, 75.9, 75.6, 75.4, 75.0, 74.7, 74.7, 74.6, 74.0, 72.4, 72.1, 70.4, 70.2, 70.1, 69.6, 68.0, 67.0, 63.6, 63.4, 62.2, 61.8, 56.2, 55.3, 52.3, 52.1, 20.9, 20.8; FT MS NSI+ m/z calcd for C81H90N7O26 [M+NH4]+ 1576.5930, found 1576.5906.
Methyl (6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-4-O-p-methoxybenzyl-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-L-idopyranosyl) uronate)-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranosyl) uronate)-(1→4)-6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranoside) uronate (7). Ceric (IV) ammonium nitrate (818 mg, 1.50 mmol) was added to a solution of 5 (1.2 g, 0.75 mmol) in acetonitrile and water (22 mL, 8:1, v/v). The mixture was stirred for 1 h at ambient temperature, whereupon TLC (EtOAc/hexane, 1:2) showed the reaction to be complete. The solution was diluted with DCM (150 mL), washed with saturated aqueous NaHCO3 (100 mL) and saturated aqueous NaCl (50 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The product was purified by column chromatography (DCM/EtOAc, 30:1), yielding the desired acceptor tetrasaccharide (600 mg, 0.40 mmol, 56%) which was used immediately in the next step. Rf = 0.21 (EtOAc/hexane 1:2); HRMS (FT MS NSI+) m/z calcd for C78H82N6O24 [M+NH4]+ 1504.5719, found 1504.5714. The above acceptor (238 mg, 0.16 mmol) and donor 1 (202 mg, 0.35 mmol) were mixed together, evaporated from dry toluene (3 × 5 mL) and dried under high vacuum for 1 h. The foam was dissolved in dry DCM (4 mL) and powdered molecular sieves (4 Å, 200 mg) added. The mixture was cooled to 0 °C and NIS (180 mg, 0.80 mmol) added. The mixture was stirred for a further 15 min at this temperature and a catalytic amount (small spatula tip) of AgOTf was then added. The mixture was kept under nitrogen at 0 °C for another 30 min and was then quenched by addition of saturated aqueous Na2S2O3 and saturated aqueous NaHCO3 (15 mL, 1:1, v/v). The suspension was filtered through Celite®, the phases separated and the organic layer washed with saturated aqueous NaCl (10 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The crude mixture was purified by column chromatography (EtOAc/hexane, 7:13) to yield 7 (260 mg, 0.11 mmol, 70%). Rf = 0.1 (EtOAc/hexane 1:2); 1H-NMR (400 MHz; CDCl3) δ 8.08–8.02 (m, 4H, Bz-ArH), 7.89–7.86 (m, 2H, ArH), 7.50–6.94 (m, 46H, ArH), 6.81–6.79 (m, 2H, ArH), 5.44 (d, 1H, J = 3.9 Hz, H1IdoA), 5.40 (d, 1H, J = 5.8 Hz, H1IdoA), 5.12–5.08 (m, 2H, H2IdoA), 4.99 (brs, 1H, H2IdoA), 4.95 (brs, 1H, H1IdoA), 4.89 (d, 1H, J = 3.8 Hz, H1GlcN), 4.84–4.82 (m, 2H, H1GlcN, CH2Ar), 4.74–4.66 (m, 7H, H5IdoA, 6 × CH2Ar), 4.63–4.59 (m, 3H, CH2Ar, 2 × H5IdoA), 4.47–4.46 (m, 3H, 2 × CH2Ar, H1GlcN), 4.41 (d, 1H, J = 2.5 Hz, H4IdoA), 4.38–4.37 (m, 1H, H4IdoA), 4.34 (s, 1H, H4IdoA), 4.29–3.87 (m, 11H, 6 × H6GlcN, 5 × CH2Ar), 3.84–3.81 (m, 3H, H3doA), 3.74 (s, 3H, OCH3), 3.72–3.66 (m, 4H, CH2Ar, H5GlcN × 3), 3.62–3.52 (m, 5H, 3 × H4GlcN, 2 × H3GlcN), 3.48 (s, 3H, OCH3), 3.42 (s, 3H, OCH3), 3.38 (s, 3H, OCH3), 3.37–3.35 (m, 4H, OCH3, H3GlcN), 3.24 (dd, 1H, J = 10.3, 3.7 Hz, H2-GlcN), 3.15 (dd, 1H, J = 10.3, 3.4 Hz, H2-GlcN), 3.09 (dd, 1H, J = 10.2, 3.6 Hz, H2-GlcN), 2.02 (s, 3H, C(O)CH3), 1.90 (s, 3H, C(O)CH3); 13C-NMR (100 MHz; CDCl3) δ 170.8, 170.6, 169.7, 169.5, 169.2, 165.6, 165.3, 165.2, 159.5, 137.8, 137.7, 137.6, 137.4, 137.3, 137.3, 133.6, 133.5, 130.0, 129.9, 129.8, 129.6, 129.3, 129.2, 128.9, 128.8, 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.8 127.6, 127.5, 127.4, 113.9, 100.4, 99.5, 99.2, 98.7, 98.3, 98.0, 80.0, 78.3, 78.2, 77.4, 77.2, 77.0, 76.4, 75.9, 75.8, 75.4, 75.3, 75.2, 75.0, 74.6, 74.4, 74.3, 74.2, 73.7, 73.6, 72.4, 72.3, 71.7, 71.5, 71.2, 70.4, 70.1, 70.0, 69.7, 67.9, 67.5, 67.0, 63.5, 63.4, 63.1, 62.3, 61.7, 56.5, 56.2, 55.4, 52.2, 52.1, 51.7, 29.7, 29.7, 20.9, 20.8; HRMS (FT MS NSI+) m/z calcd for C122H132N11O37 [M+2NH4]2+ 1173.4522, found 1173.4522.
Methyl (2-azido-3,6-di-O-benzyl-2-deoxy-4-O-p-methoxybenzyl-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-l-idopyranosyl) uronate)-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-α-d-gluco-pyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranosyl) uronate)-(1→4)-6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-L-ido-pyranoside) uronate (8). See 7 for PMB deprotection of 5. Acceptor (320 mg, 0.22 mmol) and donor 2 (295 mg, 0.30 mmol) were mixed together, evaporated from dry toluene (3 × 5 mL) and dried under high vacuum for 1 h. The foam was dissolved in dry DCM (6 mL) and powdered molecular sieves (4 Å, 250 mg) added. The mixture was cooled to 0 °C and NIS (240 mg, 1.07 mmol) added. The mixture was stirred for a further 15 min at this temperature and a catalytic amount (small spatula tip) of AgOTf was then added. The mixture was kept under nitrogen at 0 °C for another 30 min and was then quenched by addition of saturated aqueous Na2S2O3 and saturated aqueous NaHCO3 (15 mL, 1:1, v/v). The suspension was filtered through Celite®, the phases separated and the organic layer washed with saturated aqueous NaCl (10 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The crude mixture was purified by column chromatography (EtOAc/hexane, 3:7), to yield 8 (487 mg, 0.21 mmol, 96%). Rf = 0.48 (EtOAc/hexane 2:3); 1H-NMR (400 MHz; CDCl3) δ 8.15–8.09 (m, 4H, Bz-ArH), 7.95–7.93 (m, 2H, Bz-ArH), 7.58–7.27 (m, 33H, ArH), 7.23–7.19 (m, 12H, ArH), 7.10–7.03 (m, 6H, ArH), 6.84–6.82 (m, 2H, PMB), 5.52–5.51 (m, 1H, H1IdoA), 5.47 (d, 1H, J = 6.0 Hz, H1IdoA), 5.20–5.17 (m, 2H, H2IdoA), 5.07 (brs, 1H, H1IdoA), 5.02 (brs, 1H, H2IdoA), 4.98–4.95 (m, 2H, H1GlcN), 4.90 (d, 1H, J = 11.9 Hz, CH2Ph), 4.81–4.20 (m, 24H, H1GlcN, 3 ×H5IdoA, H3IdoA, 6 × H6GlcN, 11 × CH2Ph), 4.08–4.01 (m, 4H, 3 × H4IdoA, CH2Ph), 3.96–3.55 (m, 18H, 5 × CH2Ph, H3GlcN, 3 × H4GlcN, 3 × H5GlcN, C(O)OCH3, PMBOCH3), 3.46–3.37 (m, 11H, 2 × C(O)OCH3, OCH3, 2 × H3GlcN), 3.33–3.29 (m, 1H, H2GlcN), 3.27–3.23 (m, 1H, H2GlcN), 3.16 (dd, 1H, J = 10.2, 3.6 Hz, H2GlcN), 2.10–2.07 (s, 3H, C(O)CH3); 13C-NMR (100 MHz; CDCl3) δ 170.7, 169.7, 169.5, 169.2, 165.6, 165.2, 137.9, 137.8, 137.7, 137.4, 137.3, 137.2, 133.5, 130.0, 129.9, 129.8, 129.5, 129.4, 129.3, 129.2, 128.8, 128.7, 128.4, 128.4, 128.3, 128.1, 128.0, 128.0, 127.9, 127.8, 127.7, 127.6, 127.4, 127.3, 113.7, 100.3, 99.6, 99.1, 98.7, 98.2, 98.0, 79.8, 78.3, 75.9, 75.7, 75.2, 74.9, 74.6, 74.5, 74.4, 74.3, 74.2, 73.6, 73.5, 72.4, 71.8, 71.7, 71.4, 71.3, 70.7, 69.7, 67.9, 67.7, 67.1, 63.4, 63.3, 61.6, 56.2, 55.3, 52.1, 51.9, 51.7, 20.9; HRMS (FT MS NSI+) m/z calcd for C127H139N11O36 [M+2NH4]2+ 1197.4704, found 1197.4706.
Methyl (6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-4-O-p-methoxy-m-iodobenzyl-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranosyl) uronate)-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranosyl) uronate)-(1→4)-2-azido-3,6-di-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranosyl) uronate)-(1→4)-6-O-acetyl-2-azido-3-O-benzyl-2-deoxy-α-d-glucopyranosyl-(1→4)-(methyl 2-O-benzoyl-3-O-benzyl-α-l-idopyranoside) uronate (9). Ceric (IV) ammonium nitrate (210 mg, 0.38 mmol) was added to a solution of 8 (450 mg, 0.19 mmol) in acetonitrile and water (4 mL, 8:1, v/v). The mixture was stirred for 1 h at ambient temperature, whereupon TLC (EtOAc/hexane, 2:3) showed the reaction to be complete. The solution was diluted with DCM (100 mL), washed with saturated aqueous NaHCO3 (50 mL) and saturated aqueous NaCl (50 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The product was purified by column chromatography (DCM/EtOAc, 12:1), yielding the desired acceptor hexasaccharide (330 mg, 0.35 mmol, 78%) which was used immediately in the next step. Rf = 0.38 (EtOAc/hexane, 2:3); HRMS (FT MS NSI+) m/z calcd for C119H131N11O35 [M+2NH4]2+ 1137.4416, found 1137.4416. The above acceptor (294 mg, 0.13 mmol) and donor 1 (166 mg, 0.18 mmol) were mixed together, evaporated from dry toluene (3 × 5 mL) and dried under high vacuum for 1 h. The foam was dissolved in dry DCM (4 mL) and powdered molecular sieves (4 Å, 200 mg) added. The mixture was cooled to 0 °C and NIS (200 mg, 0.89 mmol) added. The mixture was stirred for a further 15 min at this temperature and a catalytic amount (small spatula tip) of AgOTf was then added. The mixture was kept under nitrogen at 0 °C for another 30 min and was then quenched by addition of saturated aqueous Na2S2O3 and saturated aqueous NaHCO3 (15 mL, 1:1, v/v). The suspension was filtered through Celite®, the phases separated and the organic layer washed with saturated aqueous NaCl (10 mL). The organic phase was then dried over MgSO4 and solvent removed in vacuo. The crude product was purified by column chromatography (EtOAc/hexane, 2:7), to yield 9 (265 mg, 0.09 mmol, 66%). Rf = 0.40 (EtOAc/hexane 2:3); 1H-NMR (400 MHz; CDCl3) δ 8.07 (d, J = 7.2 Hz, 2H, ArH), 8.02 (d, J = 6.6 Hz, 2H, ArH), 7.89 (d, J = 7.3 Hz, 2H, ArH), 7.84 (d, J = 7.3 Hz, 2H, ArH), 7.56 (d, J = 2.0 Hz, 1H, ArH), 7.52–6.88 (m, 68H, ArH), 6.68 (d, J = 8.5 Hz, 1H, ArH), 5.47 (d, J = 5.1 Hz, 1H, H1IdoA), 5.43 (d, J = 3.5 Hz, 1H, H1IdoA), 5.38 (d, J = 5.9 Hz, 1H, H1IdoA), 5.12–5.07 (m, 3H, H2IdoA), 4.98 (s, 1H, H2IdoA), 4.94 (s, 1H, H1IdoA), 4.88–4.80 (m, 4H, 3 × H1GlcN, CH2Ar), 4.73–4.58 (m, 12H, 11 × CH2Ar, H5IdoA), 4.51–4.11 (m, 18H, H1GlcN, 4 × H4IdoA, 3 × H5IdoA, 10 × CH2Ar), 3.99–3.93 (m, 6H, 2 × H6-GlcN 4 × H3IdoA), 3.86–3.66 (m, 9H, 6 × H6-GlcN, OCH3), 3.56–3.21 (m, 29H, 4 × H5-GlcN, 4 × H3-GlcN, 4 × H4-GlcN, 5 × OCH3, 2 × H2-GlcN), 3.14 (dd, J = 10.2, 3.4 Hz, 1H, H2-GlcN), 3.07 (dd, J = 10.2, 3.5 Hz, 1H, H2-GlcN), 2.01 (s, 3H, C(O)CH3), 1.90 (s, 3H, C(O)CH3); 13C-NMR (100 MHz; CDCl3) δ 177.3, 170.8, 170.5, 169.7, 169.5, 169.3, 169.2, 165.5, 165.2, 165.2, 157.9, 139.3, 137.8, 137.8, 137.7, 137.6, 137.5, 137.3, 133.6, 133.5, 131.9, 130.0, 129.9, 129.9, 129.8, 129.7, 129.6, 129.5, 129.3, 129.2, 129.2, 128.8, 128.7, 128.5, 128.5, 128.4, 128.4, 128.4, 128.3, 128.2, 128.2, 128.1, 128.1, 128.0, 127.9, 127.9, 127.9, 127.8, 127.8, 127.6, 127.5, 127.5, 127.4, 127.3, 127.2, 110.7, 100.3, 99.5, 99.1, 98.7, 98.3, 98.0, 97.9, 80.0, 78.2, 78.02, 77.4, 77.0, 76.5, 75.9, 75.9, 75.7, 75.4, 75.2, 75.0, 74.9, 74.6, 74.5, 74.4, 74.2, 73.7, 73.6, 73.6, 72.4, 72.2, 71.8, 71.4, 70.6, 70.0, 69.7, 69.4, 67.8, 67.5, 67.3, 66.9, 63.5, 63.3, 63.2, 62.8, 62.1, 61.6, 56.4, 56.3, 56.2, 55.3, 52.2, 52.1, 51.7, 51.6, 29.8, 29.6; HRMS (FT MS NSI+) m/z calcd for C163H175N14O48I [M+2NH4]2+ 1,612.0375, found 1,612.0377.
4. Conclusions
A modular synthetic access to differentially protected H/HS-like oligosaccharides is demonstrated using two d-GlcN-l-IdoA modules with differentiated d-GlcN O-6 protecting groups, suitable for ultimate installation of either a 6-OH or 6-OS moiety. This is applied to generate a ladder of tetrasaccharide, hexasaccharide and octasaccharide systems which retain a common reducing end 6-OAc, either as the only acylated O-6, or combined with double-terminal units to provide oligosaccharides withterminal d-GlcN units both bearing O-6 acylations. High stereochemical integrity in synthesis is evidenced by NMR spectra, which allow ready comparisons of differentiated l-IdoA H-1 across the series. This approach should facilitate wider access to medium length heparin-like oligosaccharides with ready programming of site-specific changes at O-6 sites along different backbones.
Supplementary Materials
Supplementary materials (1D and 2D NMR). Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/04/6167/s1.
Acknowledgments
The CRUK [C2075/A9106], MRC [G0601746 and G902173] are thanked for project grant funding. EPSRC for NMR instrumentation (GR/L52246) and the EPSRC National Mass Spectrometry Service, Swansea, are also thanked for mass spectroscopic analyses.
Author Contributions
M.B. performed the syntheses, contributed to experimental writing, data collection and analyses. C.E.D. and G.J.M. contributed additional data and analysis to experimental. G.J.M. collated and produced the Supplementary data file. S.U.H. contributed to experimental data and collation. G.J.M. and J.M.G. wrote the manuscript and edited the S.I. data. G.C.J. and J.M.G. designed the research. All authors approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Casu, B.; Naggi, A.; Torri, G. Heparin-derived heparan sulfate mimics to modulate heparan sulfate-protein interaction in inflammation and cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, P.H.; Werz, B. Synthesis and medical applications of oligosaccharides. Nature 2007, 446, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Zulueta, M.M.L.; Lin, S.-Y.; Hu, Y.-P.; Hung, S.-C. Synthetic heparin and heparan sulfate oligosaccharides and their protein interactions. Curr. Opin. Chem. Biol. 2013, 17, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Kjellén, L. Pathophysiology of heparan sulphate: Many diseases, few drugs. J. Int. Med. 2013, 273, 555–571. [Google Scholar] [CrossRef]
- De Paz, J.-L.; Moseman, A.; Noti, C.; Polito, L.; von Andrian, U.H.; Seeberger, P.H. Profiling Heparin-Chemokine Interactions Using Synthetic Tools. ACS Chem. Biol. 2007, 2, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Noti, C.; Seeberger, P.H. Chemical Approaches to Define the Structure-Activity Relationship of Heparin-like Glycosaminoglycans. Chem. Biol. 2005, 12, 731–756. [Google Scholar] [CrossRef] [PubMed]
- Dulaney, S.B.; Huang, X. Strategies in Synthesis of Heparin/Heparan Sulfate Oligosaccharides. Adv. Carbohydr. Chem. Biochem. 2012, 67, 95–136. [Google Scholar] [PubMed]
- Zong, C.; Venot, A.; Dhamale, O.; Boons, G.-J. Fluorous Supported Modular Synthesis of Heparan Sulfate Oligosaccharides. Org. Lett. 2013, 15, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Guedes, N.; Czechura, P.; Echeverria, B.; Ruiz, A.; Michelena, O.; Martin-Lomas, M.; Reichardt, N.-C. Toward the Solid-Phase Synthesis of Heparan Sulfate Oligosaccharides: Evaluation of Iduronic Acid and Idose Building Blocks. J. Org. Chem. 2013, 78, 6911–6934. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Masuko, S.; Takieddin, M.; Xu, H.; Liu, R.; Jing, J.; Mousa, S.A.; Linhardt, R.J.; Liu, J. Chemoenzymatic Synthesis of Homogeneous Ultralow Molecular Weight Heparins. Science 2011, 334, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Codée, J.D.C.; Stubba, B.; Schiattarella, M.; Overkleeft, H.S.; van Boeckel, C.A.A.; van Boom, J.H.; van der Marel, G.A.A. Modular Strategy Toward the Synthesis of Heparin-like Oligosaccharides Using Monomeric Building Blocks in a Sequential Glycosylation Strategy. J. Am. Chem. Soc. 2005, 127, 3767–3773. [Google Scholar] [CrossRef] [PubMed]
- Poletti, L.; Lay, L. Chemical Contributions to Understanding Heparin Activity: Synthesis of Related Sulfated Oligosaccharides. Eur. J. Org. Chem. 2003, 16, 2999–3024. [Google Scholar] [CrossRef]
- De Paz, J.-L.; Angulo, J.; Lassaletta, J.M.; Nieto, P.M.; Horcajo, M.-R.; Lozano, R.M.; Gallego, G.G.; Martín-Lomas, M. The activation of fibroblast growth factors by heparin: Synthesis, structure, and biological activity of heparin like oligosaccharides. ChemBioChem 2001, 2, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, Y.; Yang, B.; Tiruchinapally, G.; Sun, B.; Liu, R.; Liu, S.D.J.; Huang, X. Preactivation-Based, One-Pot Combinatorial Synthesis of Heparin-like Hexasaccharides for the Analysis of Heparin-Protein Interactions. Chem. Eur. J. 2010, 16, 8365–8375. [Google Scholar] [CrossRef] [PubMed]
- De Paz, J.L.; Martín-Lomas, M. Synthesis and Biological Evaluation of a Heparin-Like Hexasaccharide with the Structural Motifs for Binding to FGF and FGFR. Eur. J. Org. Chem. 2005, 2005, 1849–1858. [Google Scholar] [CrossRef]
- Schwörer, R.; Zubkova, O.V.; Turnbull, J.E.; Tyler, P.C. Synthesis of a Targeted Library of Heparan Sulfate Hexa- to Dodecasaccharides as Inhibitors of β-Secretase: Potential Therapeutics for Alzheimer’s Disease. Chem. Eur. J. 2013, 19, 6817–6823. [Google Scholar] [CrossRef] [PubMed]
- Arungundram, S.; Al-Mafraji, K.; Asong, J.; Leach, F.E.; Amster, I.J.; Venot, A.; Turnbull, J.E.; Boons, G.-J. Modular Synthesis of Heparan Sulfate Oligosaccharides for Structure-Activity Relationship Studies. J. Am. Chem. Soc. 2009, 131, 17394–17405. [Google Scholar] [CrossRef] [PubMed]
- Poletti, L.; Fleischer, M.; Vogel, C.; Guerrini, M.; Torri, G.; Lay, L. A Rational Approach to Heparin-Related Fragments—Synthesis of Differently Sulfated Tetrasaccharides as Potential Ligands for Fibroblast Growth Factors. Eur. J. Org. Chem. 2001, 14, 2727–2734. [Google Scholar] [CrossRef]
- De Paz, J.-L.; Ojeda, R.; Reichardt, N.; Lomas, M.-M. Some Key Experimental Features of a Modular Synthesis of Heparin-Like Oligosaccharides. Eur. J. Org. Chem. 2003, 17, 3308–3324. [Google Scholar] [CrossRef]
- Tiruchinapally, G.; Yin, Z.; el Dakdouki, M.; Wang, Z.; Huang, X. Divergent Heparin Oligosaccharide Synthesis with Preinstalled Sulfate Esters. Chem.-Eur. J. 2011, 17, 10106–10112. [Google Scholar] [CrossRef] [PubMed]
- Gama, C.I.; Tully, S.E.; Sotogaku, N.; Clark, P.M.; Rawat, M.; Vaidehi, N.; Goddard, W.A.; Nishi, A.; Hsieh-Wilson, L.C. Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2006, 2, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-P.; Lin, S.-Y.; Huang, C.-Y.; Zulueta, M.M.L.; Liu, J.-Y.; Chang, W.; Hung, S.-C. Synthesis of 3-O-sulfonated heparan sulfate octasaccharides that inhibit the herpes simplex virus type 1 host-cell interaction. Nat. Chem. 2011, 3, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-P.; Zhong, Y.-Q.; Chen, Z.-G.; Chen, C.-Y.; Shi, Z.; Zulueta, M.M.L.; Ku, C.-C.; Lee, P.-Y.; Wang, C.-C.; Hung, S.-C. Divergent synthesis of 48 heparan sulfate-based disaccharides and probing the specific sugar−fibroblast growth factor-1 interaction. J. Am. Chem. Soc. 2012, 134, 20722–20727. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Ho, I.-H.; Ku, C.-C.; Zhong, Y.-Q.; Hu, Y.-P.; Chen, Z.-G.; Chen, C.-Y.; Lin, W.-C.; Zulueta, M.M.L.; Hung, S.-C.; et al. Interactions That Influence the Binding of Synthetic Heparan Sulfate Based Disaccharides to Fibroblast Growth Factor-2. ACS Chem. Biol. 2014, 9, 1712–1717. [Google Scholar]
- Handel, T.M.; Johnson, Z.; Crown, S.E.; Lau, E.K.; Proudfoot, A.E. Regulation of protein function by glycosylaminoglycans–as exemplified by chemokines. Annu. Rev. Biochem. 2005, 74, 385–410. [Google Scholar] [CrossRef] [PubMed]
- Spillmann, D.; Witt, D.; Lindahl, U. Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 1998, 273, 15487–15493. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, K.; Morimoto-Tomita1, M.; Bistrup, A.; Li, J.; Lyon, M.; Gallagher, J., 3rd; Werb, Z.; Rosen, S.D. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006. [CrossRef]
- Jastrebova, N.; Vanwildemeersch, M.; Rapraeger, A.C.; Giménez-Gallego, G.; Lindahl, U.; Spillmann, D. Heparan Sulfate-related Oligosaccharides in Ternary Complex Formation with Fibroblast Growth Factors 1 and 2 and Their Receptors. J. Biol. Chem. 2006, 281, 26884–26892. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.L.; Hansen, S.U.; Baráth, M.; Rushton, G.; Gardiner, J.M.; Avizienyte, E.; Jayson, G.C. Synthetic heparan sulfate oligosaccharides inhibit endothelial cell function essential for angiogenesis. PLoS ONE 2010, 5, e11644. [Google Scholar] [CrossRef] [PubMed]
- Ferreras, C.; Rushton, G.; Cole, C.; Babur, M.; Telfer, B.A.; van Kuppevelt, T.H.; Gardiner, J.M.; Williams, K.J.; Jayson, G.C.; Avizienyte, E. Endothelial heparan sulfate 6-O-sulfation levels regulate angiogenic responses of endothelial cells to fibroblast growth factor 2 and vascular endothelial growth factor. J. Biol. Chem. 2012, 287, 36132–36146. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cai, C.; Chandarajoti, K.; Hsieh, P.-H.; Li, L.; Pham, T.-Q.; Sparkenbaugh, E.M.; Sheng, J.; Key, N.S.; Pawlinski, R.; et al. Design of homogeneous and reversible low molecular weight heparins. Nat. Chem. Biol. 2014, 10, 248–252. [Google Scholar]
- Miller, G.J.; Hansen, S.U.; Avizienyte, E.; Rushton, G.; Cole, C.; Jayson, G.C.; Gardiner, J.M. Efficient chemical synthesis of heparin-like octa-, deca- and dodecasaccharides and inhibition of FGF2- and VEGF165-mediated endothelial cell functions. Chem. Sci. 2013, 4, 3218–3222. [Google Scholar] [CrossRef]
- Hansen, S.U.; Miller, G.J.; Cole, C.; Rushton, G.; Avizienyte, E.; Jayson, G.C.; Gardiner, J.M. Tetrasaccharide iteration synthesis of a heparin-like dodecasaccharide and radiolabelling for in vivo tissue distribution studies. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Hansen, S.U.; Miller, G.J.; Jayson, G.C.; Gardiner, J.M. First Gram-Scale Synthesis of a Heparin-Related Dodecasaccharide. Org. Lett. 2013, 15, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.J.; Hansen, S.U.; Baráth, M.; Johannessen, C.; Blanch, E.W.; Jayson, G.C.; Gardiner, J.M. Synthesis of a heparin-related GlcN-IdoA sulfation-site variable disaccharide library and analysis by Raman and ROA spectroscopy. Carbohydr. Res. 2014, 400, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Torri, G.; Casu, B.; Gatti, G.; Jacquinet, J.C.; Sinay, P.; Petitou, M.; Choay, J. Evidence for conformational equilibrium of the sulfated l-iduronate residue in heparin and in synthetic heparin mono- and oligo-saccharides: NMR and force-field studies. J. Am. Chem. Soc. 1986, 108, 6773–6778. [Google Scholar] [CrossRef]
- Sample Availability: For availability of samples of compounds contact the corresponding author.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).