
Enantioselective Synthesis of cis-Decalins Using Organocatalysis and Sulfonyl Nazarov Reagents



Abstract
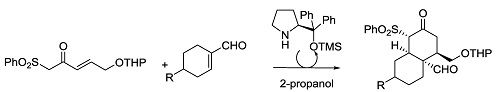
: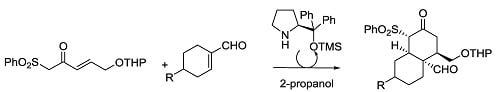
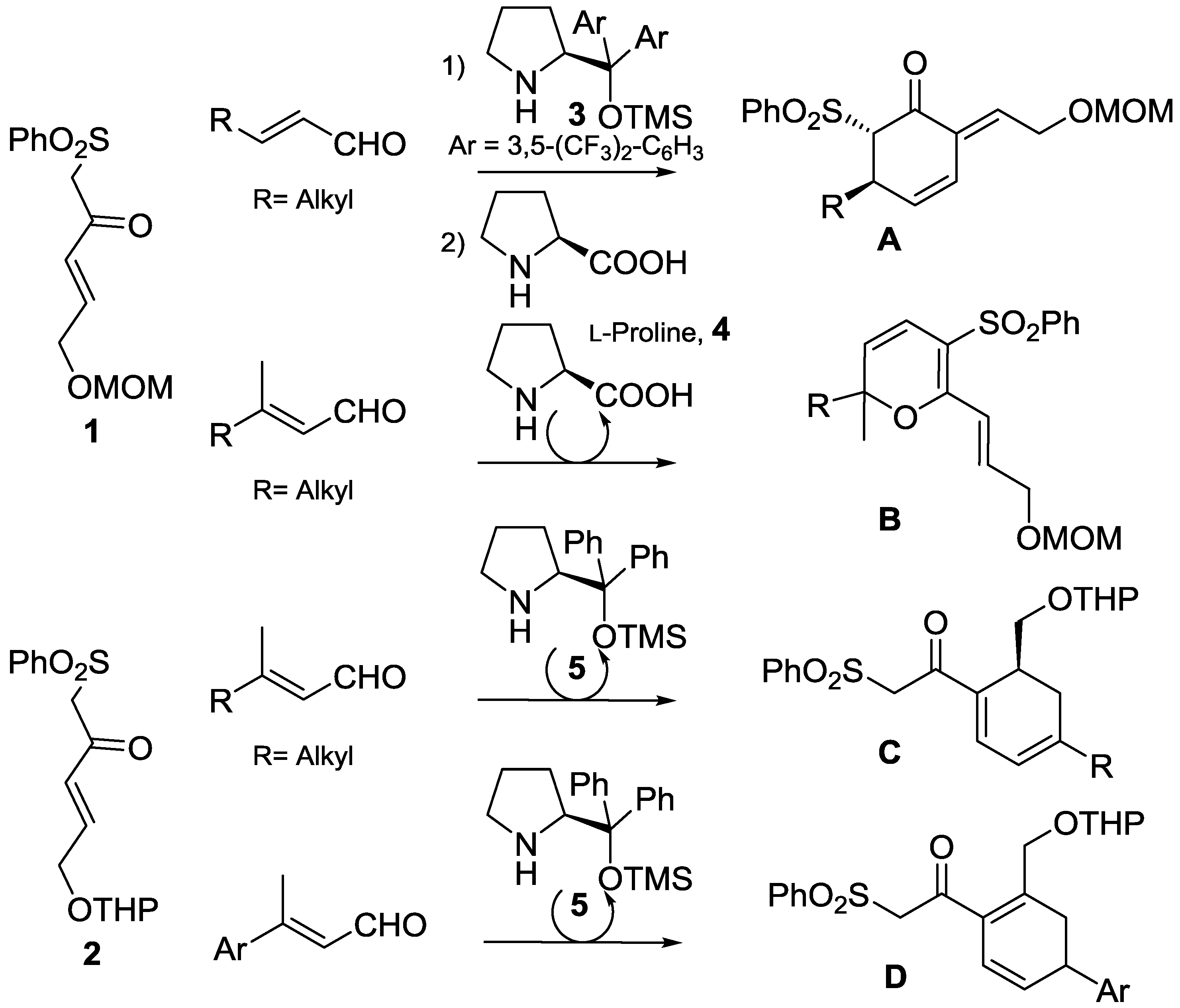
1. Introduction

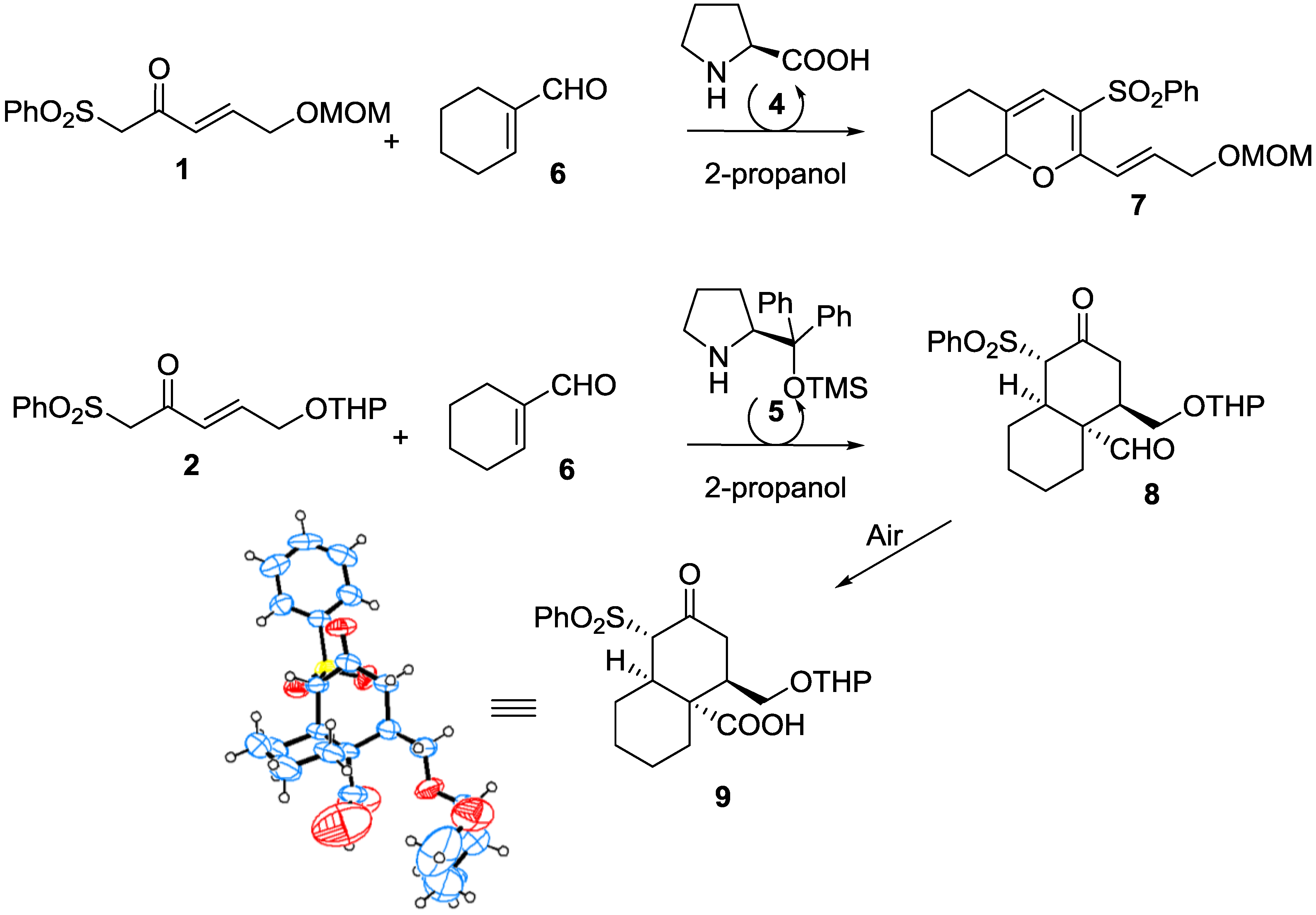
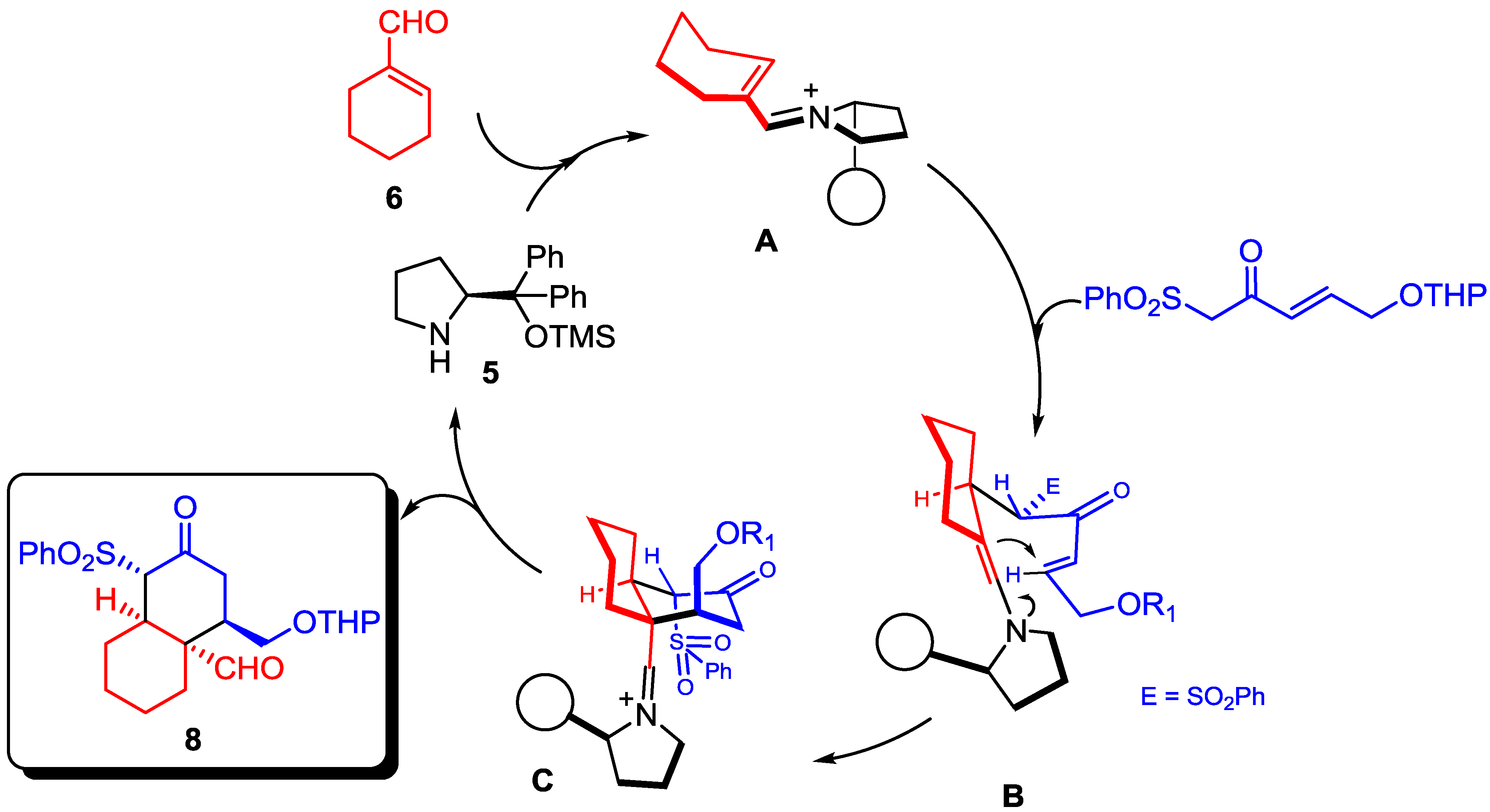
2. Results and Discussion


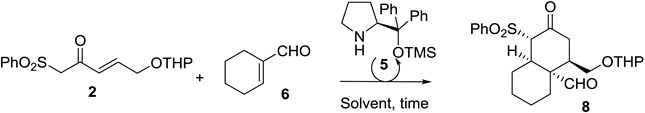
| Entry [a] | 2/6 ratio. | Solvent | T (h) [b] | Yield [%] [c] | ee |
|---|---|---|---|---|---|
| 1 | 2/1 | n-Hexane | 72 | S.M. | -- |
| 2 | 2/1 | Et2O | 72 | S.M. | -- |
| 3 | 2/1 | THF | 72 | S.M. | -- |
| 4 | 2/1 | MeOH | 72 | 20 | ND |
| 5 | 2/1 | EtOH | 72 | 52 | 96 |
| 6 [d] | 2/1 | EtOH | 48 | 53 | 85 |
| 7 | 2/1 | 2-propanol | 72 | 35 | ND |
| 8 | 2/1 | H2O | 48 | 6 | ND |
| 9 | 2/1 | NO SOLVENT | 72 | 18 | ND |
| 10 | 1/1 | EtOH | 72 | 30 | ND |
| 11 | 1/2 | EtOH | 72 | 42 | ND |
| Entry [a] | Catalys 5 (%) | Yield [%] [b] | ee |
|---|---|---|---|
| 1 | 0 | S.M. | -- |
| 2 | 5 | 28 | ND |
| 3 | 10 | 29 | ND |
| 4 | 20 | 52 | 96 |
| 5 | 50 | 60 | 96 |
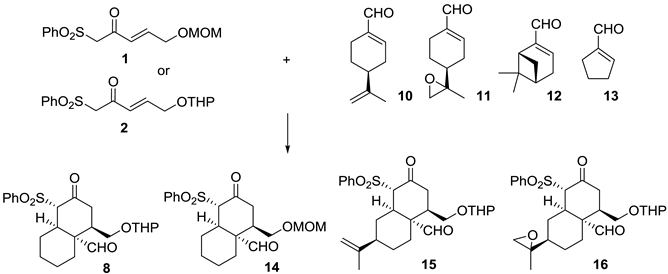
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry [a] | Cyclic Enal. | Product | T (h) [b] | Yield [%] [c] | ee [d] |
|---|---|---|---|---|---|
| 1 | 6 | 8 | 72 | 51 | 96 |
| 2 [e] | 6 | ent-8 | 72 | 51 | -96 |
| 3 [f,g] | 6 | 14 | 48 | 50 | 96 |
| 4 | 10 | 15 | 48 | 63 | 90 |
| 5 | 10 | 15 | 96 | 15 | N.D. |
| 6 [g] | 10 | 15 | 48 | 85 | 90 |
| 7 [e] | 10 | ent-15 | 48 | 4 | N.D. |
| 8 [e] | 10 | -- | 72 | -- | N.D. |
| 9 [h] | 10 | ent-15 | 96 | 4 | N.D. |
| 10 | 11 | 16 | 48 | 22 | N.D. |
| 11 [i] | 11 | 16 | 48 | 39 | -- [j] |
| 12 [g,i] | 11 | 16 | 48 | 30 | N.D. |
| 13 | 12 | S.M. | 96 | -- | -- |
| 14 [e] | 12 | S.M. | 120 | -- | -- |
| 15 | 13 | S.M. [k] | 48 | -- | -- |
| 16 | 13 | S.M. [k] | 72 | -- | -- |
| 17 | 13 | S.M. [k] | 96 | -- | -- |
| 18 | 13 | S.M. [k] | 120 | -- | -- |
| 19 [g] | 13 | S.M. [k] | 120 | -- | -- |

3. Experimental Section
3.1. General
3.2. General Procedure for the Synthesis of Chiral cis-Decalins 8, 14‒16
3.2.1. (1S,4R,4aS,8aR)-2-Oxo-1-(phenylsulfonyl)-4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)-deca-hydronaphthalene-4a-carbaldehyde (8)
3.2.2. (1R,4S,4aR,8aS)-2-Oxo-1-(phenylsulfonyl)-4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)deca-hydronaphthalene-4a-carbaldehyde (ent-8)
3.2.3. (1S,4R,4aS,8aR)-4-((Methoxymethoxy)methyl)-2-oxo-1-(phenylsulfonyl)octahydronaphthalene-4a(2H)-carbaldehyde (14)
3.2.4. (1S,4R,4aS,7R,8aR)-2-Oxo-1-(phenylsulfonyl)-7-(prop-1-en-2-yl)-4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)decahydronaphthalene-4a-carbaldehyde (15)
3.2.5. (1S,4R,4aS,7R,8aR)-7-(2-Methyloxiran-2-yl)-2-oxo-1-(phenylsulfonyl)-4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)decahydronaphthalene-4a-carbaldehyde (16)
3.2.6. X-ray Crystal Data for Compounds 9
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dalko, P.I.; Moisan, L. Enantioselective organocatalysis. Angew. Chem. Int. Ed. Engl. 2001, 113, 3726–3748. [Google Scholar] [CrossRef]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis. From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- List, B. Asymmetric Organocatalysis; Springer-Verlag: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Waser, M. Asymmetric Organocatalysis in Natural Product Syntheses; Springer-Verlag: Wien, Austria, 2012. [Google Scholar]
- Merritt, A.T.; Ley, S.V. Clerodane diterpenoids. Nat. Prod. Rep. 1992, 9, 243–287. [Google Scholar] [CrossRef]
- Okino, T.; Yoshimura, E.; Hirota, H.; Fusetani, N. Antifouling kalihinenes from the marine sponge Acanthella cavernosa. Tetrahedron Lett. 1995, 36, 8637–8640. [Google Scholar] [CrossRef]
- Iwagawa, T.; Kaneko, M.; Okamura, H.; Nakatani, M.; van Soest, R.W.M. New alkaloids from the Papua New Guinean sponge Agelas nakamurai. J. Nat. Prod. 1998, 61, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Hirose, Y. Stereochemistry of (+)-α-ylangene. Tetrahedron Lett. 1969, 10, 1601–1604. [Google Scholar] [CrossRef]
- Tada, M.; Moriyama, Y.; Tanahashi, Y.; Takahashi, T. Furanoeremophilane-6β,10β-diol and Its Derivatives. New Furanosesquiterpenes from Ligularia japonica LESS. Bull. Chem. Soc. Jpn. 1974, 47, 1999–2002. [Google Scholar] [CrossRef]
- Kulkarni, K.S.; Paknikar, S.K.; Bhattacharyya, S.C. Terpenoids—XLVIII: Structure and stereochemistry of hydroxyvaleranone and acetylhydroxyvaleranone. Tetrahedron 1964, 20, 1289–1300. [Google Scholar] [CrossRef]
- Singh, V.; Iyer, S.R.; Pal, S. Recent approaches towards synthesis of cis-decalins. Tetrahedron 2005, 61, 9197–9231. [Google Scholar] [CrossRef]
- Lavallée, J.-F.; Deslongchamps, P. Synthesis of cis-decalin via Diels-Alder and double Michael cycloaddition with substituted Nazarov reagent. Tetrahedron Lett. 1988, 29, 5117–5118. [Google Scholar] [CrossRef]
- Audran, G.; Brémond, P.; Feuerstein, M.; Marque, S.R.A.; Santelli, M. Nazarov reagents and their use in organic synthesis. Tetrahedron 2013, 69, 8325–8348. [Google Scholar] [CrossRef]
- Peña, J.; Antón, A.B.; Moro, R.F.; Marcos, I.S.; Garrido, N.M.; Díez, D. Tandem catalysis for the synthesis of 2-alkylidene cyclohexenones. Tetrahedron 2011, 67, 8331–8337. [Google Scholar] [CrossRef]
- Peña, J.; Moro, R.F.; Basabe, P.; Marcos, I.S.; Díez, D. Solvent free l-proline-catalysed domino Knoevenagel/6π-electrocyclization for the synthesis of highly functionalised 2H-pyrans. RSC Adv. 2012, 2, 8041–8049. [Google Scholar] [CrossRef]
- Peña, J.; Moro, R.F.; Basabe, P.; Marcos, I.S.; Díez, D. Highly functionalised cyclohexa-1,3-dienes by sulfonyl Nazarov reagents. Tetrahedron 2014, 70, 4386–4394. [Google Scholar] [CrossRef]
- Trabocchi, A. Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery, and Chemical Biology; John Wiley and Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Sheldrich, G.M. SHELXTLTM. Structure Determination Software Suit, Version 6.14; Bruker AXS, Inc.: Madison, WI, USA, 2003.
- Ravindar, K.; Caron, P.-Y.; Deslongchamps, P. Anionic polycyclization entry to tricycles related to quassinoids and terpenoids: A stereocontrolled total synthesis of (+)-cassaine. J. Org. Chem. 2014, 79, 7979–7999. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 8 and 14 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña, J.; Silveira-Dorta, G.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Sanz, F.; Díez, D. Enantioselective Synthesis of cis-Decalins Using Organocatalysis and Sulfonyl Nazarov Reagents. Molecules 2015, 20, 6409-6418. https://doi.org/10.3390/molecules20046409
Peña J, Silveira-Dorta G, Moro RF, Garrido NM, Marcos IS, Sanz F, Díez D. Enantioselective Synthesis of cis-Decalins Using Organocatalysis and Sulfonyl Nazarov Reagents. Molecules. 2015; 20(4):6409-6418. https://doi.org/10.3390/molecules20046409
Chicago/Turabian StylePeña, Javier, Gastón Silveira-Dorta, Rosalina F. Moro, Narciso M. Garrido, Isidro S. Marcos, Francisca Sanz, and David Díez. 2015. "Enantioselective Synthesis of cis-Decalins Using Organocatalysis and Sulfonyl Nazarov Reagents" Molecules 20, no. 4: 6409-6418. https://doi.org/10.3390/molecules20046409
APA StylePeña, J., Silveira-Dorta, G., Moro, R. F., Garrido, N. M., Marcos, I. S., Sanz, F., & Díez, D. (2015). Enantioselective Synthesis of cis-Decalins Using Organocatalysis and Sulfonyl Nazarov Reagents. Molecules, 20(4), 6409-6418. https://doi.org/10.3390/molecules20046409