2.1. Quantification of Modifiable Functional Groups of the S-Layer Protein SlfB by Potentiometric Titration
The gene sequence and amino acid composition of SlfB has been previously determined [
19]. SlfB with a molecular weight of 126 kDa contains 62 aspartate, 63 glutamate, 88 lysine, 15 arginine, and 37 tyrosine residues. Therefore, a total amount of 125 COOH-groups, 103 NH
2-groups, and 37 OH-groups are theoretically available for modifications. However, due to protein folding, surface charges, and steric hindrance, it can be expected that only a small amount of these groups is really accessible for linkage with molecules. In this work, potentiometric titration was performed in order to elucidate the amount of modifiable groups, and to evaluate the potential of S-layer proteins for the introduction of functional molecules in high densities. The potentiometric titration was performed as described in
Section 3.1. and results are given in the following
Table 1. The experiments were done repeatedly with solutions of protein monomers and polymers. The pKa values and site densities of monomers and polymers were almost the same, indicating that in both cases the same amounts of COOH- and NH
2-groups are theoretically available for modification.
Table 1.
Determined pKa-values of SlfB and modification rates evaluated by potentiometric titration; * can be assigned to NH2- or OH-groups.
Table 1.
Determined pKa-values of SlfB and modification rates evaluated by potentiometric titration; * can be assigned to NH2- or OH-groups.
| pKa-Value | Site Density (molfunctional group/molSlfB) | Ratio of Functional Group in the Protein |
|---|
| 3.98 ± 0.02
| 28 ± 1.0 | 22.4% ± 0.8% | COOH |
| 5.65 ± 0.06
| 8.5 ± 0.7 | 6.8% ± 0.6% | COOH |
| 7.76 ± 0.06
| 12 ± 1.0 | 11.7% ± 1.0% | NH2 |
| 9.49 * ± 0.04
| 57 ± 4.0 | 55.3% ± 3.9% | NH2 or OH |
The results show four different pKa values: 3.98, 5.65, 7.76, and 9.49. The pKa-values of 3.98 and 5.65 can be assigned to glutamic and aspartic acid, respectively, the pKa-value of 7.76 can be allocated to lysine or arginine, and the pKa-value of 9.49 cannot be precisely assigned. This pKa-value could be the result of titrated NH
2- or OH-groups. The hydroxyl group of tyrosine has a theoretical pKa of 10.10 and is amenable to titration. SlfB contains 3.1 mol % of tyrosine, and therefore, is also characterized by potentially free OH-groups. In summary, the amount of modifiable COOH-groups was calculated to be 36.5 mol per mol protein, while the amount of NH
2-groups was determined to be 12 mol per mol protein when the pKa-value of 9.49 is not considered and 69 mol per mol protein when it is applied to these groups. This means SlfB possesses 36.5 free COOH- and either 12 or 69 free NH
2-groups theoretically available for modification if the polymeric structure of the protein is stable. It can be assumed that all other COOH- and NH
2-groups that could be not titrated are blocked via intramolecular and intermolecular interactions of the monomer and the polymer, respectively. A theoretical modification rate of at least 48 mol∙mol
SlfB−1 is therefore possible. SlfB exhibits a p4-symmetry with a length of 13 nm [
20] that is consistent with an area of 169 nm². Provided that all available COOH- and NH
2-groups are modified, then 192 mol of the desired molecules can be linked to an area of 169 nm
2. However, these calculations do not consider steric hindrance and charge effects. Therefore, it can be expected that the real amount is much lower.
2.2. Modification of SlfB with Tryptophan
For a more detailed analysis, in another experiment the amino acid tryptophan was linked to the COOH- and NH
2-groups of SlfB. This experiment was performed in order to determine a more realistic modification rate. Tryptophan was chosen because of its small size, its minor charge effects due to its neutral character, and its aromatic residual, which allows for the direct quantification of bound tryptophan with UV/VIS-measurements. S-layer proteins were modified with the amino acid tryptophan at their COOH- and NH
2-groups as described in
Section 3.2. These results are shown in the following
Table 2.
Table 2.
Total amount of modified COOH and NH2-groups with tryptophan.
Table 2.
Total amount of modified COOH and NH2-groups with tryptophan.
| Modification of | Modification Rate | Molar Loading (molTrp∙molSlfB−1) |
|---|
| NH2 | 26.4% to 56.7% | 27.2 to 58.4 |
| COOH
| 21.2% to 53.4% | 26.5 to 66.7 |
The total amount of theoretically available COOH- and NH
2-groups is 125 and 103, respectively. The table shows that up to one half of the COOH- or NH
2-groups of SlfB were modified with tryptophan, which corresponds to a molar load of 67 tryptophan on the COOH- and 58 tryptophan on the NH
2-groups. This equates to a density of up to 1.58 modifiable COOH-groups per nm
2 and up to 1.38 modifiable NH
2-groups per nm
2. Lower rates were determined with potentiometric titration: 36.5 mol COOH-groups could be titrated and can be claimed to be “available for modification”, corresponding to a density of 0.86 COOH-groups per nm
2. In case of NH
2-groups, the number of possible titrated groups was determined to be 12 mol and 69 mol, respectively (
Table 1).
It is noted that some carboxylic groups might be deprotonated at pH 3 before starting the titration. These deprotonated groups are not detected by the titration, but they are available for modification, explaining the different values. It is also noted that the complete modification of all COOH- and NH
2-groups of the protein is not possible. Due to the structure of the protein, interactions of different functional groups of the protein, and steric hindrance, not all groups are accessible for modification with tryptophan. Furthermore, some intramolecular reactions of EDC-activated COOH-groups may occur. However, regarding these aspects and limitations, the obtained modification rate is quite high. In a former publication Weigert
et al. [
21] performed a complex procedure comprising several modification and extraction steps in order to determine the number of free carboxyl groups exposed in the surface of the S-layer lattice of
L. sphaericus CCM2120. They calculated a charge density of 1.6 carboxyl groups per nm
2 that is in the range of the values obtained in the present work. However, these researchers investigated the available carboxyl groups of S-layers on cell wall fragments, not of isolated protein polymers or monomers. The methods described in the present work allow a much easier and faster determination of available COOH- as well as of NH
2-groups.
Experiments in the present study were also performed with protein polymers and monomers. The modification rates of NH2-groups were found to be similar for both, whereas the determined amount of modifiable COOH-groups was nearly twice as much for the monomers in comparison to the polymers. These differences can be expected considering that for the polymers the carboxyl groups necessary for exhibiting the polymeric structure are blocked by the binding of Ca2+. Conversely, it has been reported that the modification of monomers disturbs the self-assembly properties of the proteins, probably due to the occupation of Ca-binding sites. Therefore, the present study concentrates on S-layers in their polymeric form.
The results showed that at least 20% of the calculated functional groups were modifiable on SlfB. In other work SlfB was modified with FRET-pairs and showed modification rates of only 0.15–0.6 mol
dye∙mol
SlfB−1 [
15]. Although the reported modification rates are not in conflict with those reported here, they prove the influence of molecule size and charge effects on the modification rates. Commercial available fluorescence dyes such as HiLyte
® dyes are well-known, highly-negative charged molecules due to their elevated chloride content. Furthermore, the molecular size of the fluorescence dyes is nearly 1000 g∙mol
−1, five times higher than that of tryptophan with 204 g∙mol
−1. In a previous study from Küpcü, S-layer proteins were modified with either enzymes or human IgG with the help of EDC [
13,
17]. In another work, Jahn-Schmid
et al. conjugated the allergen Bet v1 to different kinds of S-layer products [
18]. In these studies similar modification rates with fluorescence dyes could be reached [
13].
2.3. Inserting SH-Groups to SlfB
Sulfhydryl (SH-) groups are present in cysteine containing proteins and fulfill important functions, e.g., in protein folding or in the active site of enzymes. Unbound or reduced sulfhydryl groups are useful targets for protein conjugation and labeling. Crosslinking via SH-groups is regarded as more selective and precise than labeling via primary amines. However, SlfB, as most bacterial S-layers, does not contain any cysteines, thus prohibiting the use of natural protein-own SH-groups for modification. The lack of SH groups necessitates the introduction of such groups using sulfhydryl-addition reagents such as SATA (
N-succinimidyl
S-acetylthioacetate) (
Section 3.3.). In this work the suitability of S-layer proteins for such modifications is investigated.
The amount of introduced SH-groups was determined by Ellman’s reagent, and a qualitative determination was completed by means of native-PAGE (native polyacrylamide gel electrophoresis) as described in
Section 3.3. The results show that the number of introduced sulfhydryl groups increases with increasing amounts of used crosslinker SATA (see
Table 3 and
Figure 1). A modification rate of 10 mol
SH∙mol
SlfB−1 could be reached in the case of a 10 and 20 molar excess of SATA. In a native-PAGE the mobility of modified SlfB increases with the amount of generated SH-groups. This is caused by the enrichment of negatively charged groups, and hence, proves the introduction of the new functional SH-group. In
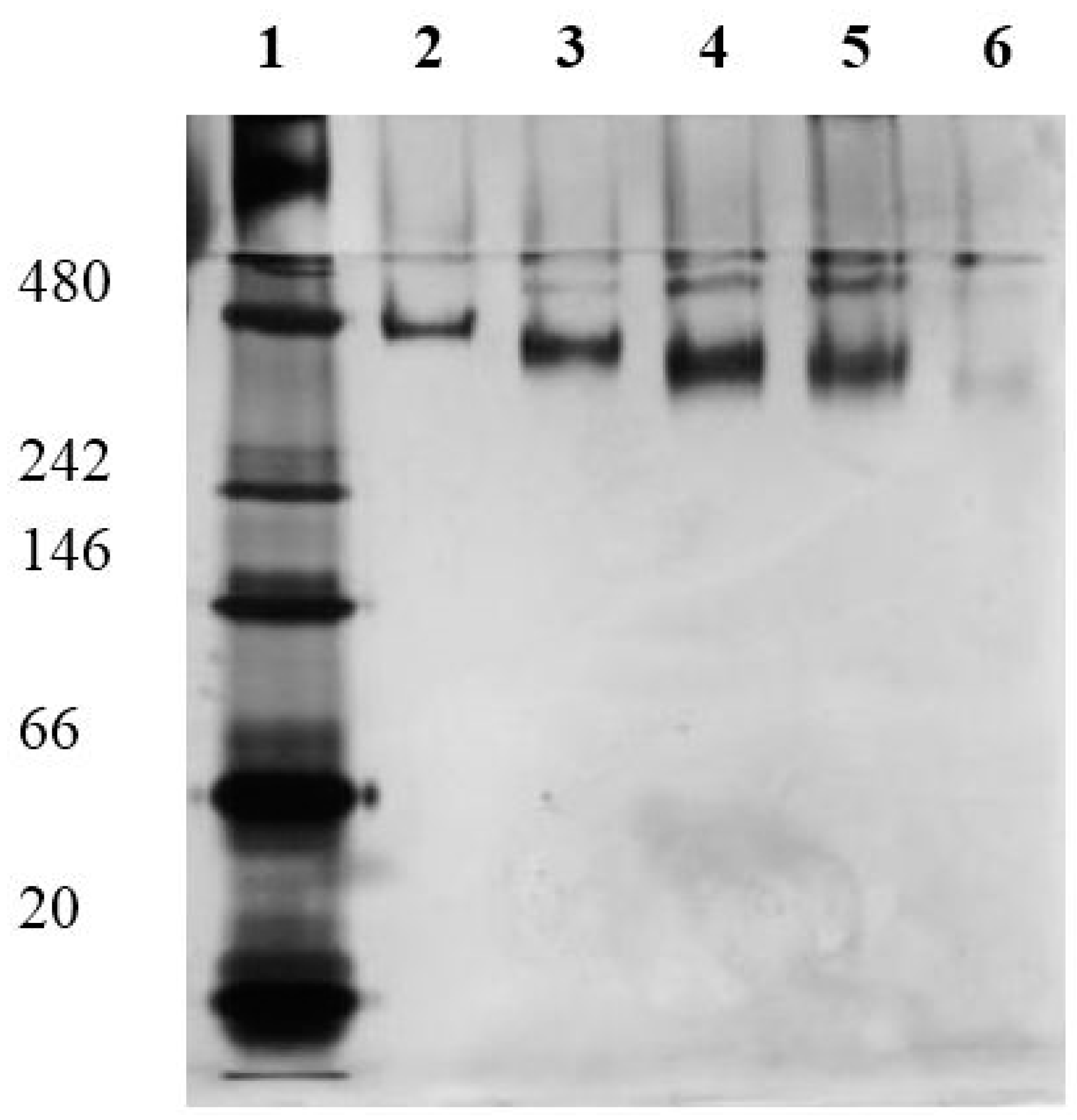
Figure 1 the results of the performed native-PAGE are shown. The native SlfB shows an estimated molecular weight of 480 kDa, which corresponds to one S-layer unit cell (p4-symmetry). If this protein is modified with different amounts of SH-groups, the protein shows more mobility resulting in an apparently lower molecular weight. A comparably performed SDS-PAGE did not show any size reducing effects of the protein if modified with SH-groups (data not shown). The higher mobility of SH-modified SlfB proves the successful linking of SH-groups to the protein.
Table 3.
Modification rate of SlfB modified with SATA; determined by Ellmann’s reagent; * native SlfB was used as a reference.
Table 3.
Modification rate of SlfB modified with SATA; determined by Ellmann’s reagent; * native SlfB was used as a reference.
| Molar Excess of SATA to SlfB | Modification Rate (molSH∙molSlfB−1) |
|---|
| Reference *
| 1.5 |
| 2-fold
| 3.8 |
| 4-fold
| 3.7 |
| 10-fold
| 10.0 |
| 20-fold
| 9.5 |
Figure 1.
Native PAGE of SlfB modified with different molar excess of SATA; lane 1—molecular weight standard with 480, 242, 66, and 20 kDa; lane 2—native SlfB; lane 3–6—SlfB modified with a 2-, 4-,10-, and 20-fold molar excess of SATA.
Figure 1.
Native PAGE of SlfB modified with different molar excess of SATA; lane 1—molecular weight standard with 480, 242, 66, and 20 kDa; lane 2—native SlfB; lane 3–6—SlfB modified with a 2-, 4-,10-, and 20-fold molar excess of SATA.
Principally, both methods are suitable for the determination of SH-groups. However, detection with the help of Ellman’s reagent needs at least 1 mg of protein per sample, whereas in case of native-PAGE only a few micrograms of protein are needed. A disadvantage of the native-PAGE is that only a qualitative check of the success of modification can be performed. For the use of the Ellman’s reagent method, a primary disadvantage is a possibly high inaccurateness as seen in this study. Although SlfB contains no cysteine, with Ellman’s reagent a content of 1.5 molSH∙molSlfB−1 could be detected, demonstrating that a high background signal has to be considered and measured modification rates have to be interpreted carefully.
Principally, crosslinking with SATA uses the same amino groups of SlfB as EDC crosslinking with tryptophan. However, the modification rate with SATA was much lower than the modification rate with tryptophan (
Table 2). These differences can be explained by the relatively large structure and molecular weight of the crosslinker SATA. As discussed for fluorescent dyes, steric hindrance effects can result in a lower modification rate.
Besides chemical modification, SH-groups can be introduced into proteins also by genetic modifications. Badelt-Lichtblau [
22] successfully constructed S-layer proteins with the fusion of a cysteine without influencing the self-assembling properties, thus offering free sulfhydryl groups for the covalent attachment of differently sized molecules as well as for binding of gold nanoparticles. However, generally the introduction of cysteines into proteins, especially into cysteine-lacking proteins, can potentially induce massive structural changes. SH-groups are able to form disulfide bonds and can change the crystallization structure, or even disturb the crystallization process. For this, additional chemicals would be necessary to inhibit the formation of disulfide bonds. Conversely, genetic engineering allows a directed modification of the protein at defined sites, whereas the localization of chemically introduced molecules cannot be controlled. In conclusion, chemical modifications, as performed in this study, are easier and faster to perform than genetic modifications, which presume a deeper knowledge of the protein structure, although the exact location of the modifications within the protein cannot be determined.
In further experiments the SATA modified SlfB proteins were incubated with EDC in order to verify if the NH
2-groups of SlfB had been successfully blocked by SATA. If so, blocking should prohibit internal crosslinking of S-layer proteins. The internal crosslinking was checked by means of SDS-PAGE as described in
Section 3.3.
In these experiments, using a SATA-excess of up to four times, a crosslinking can still be detected (see arrows
Figure 2), which indicates that NH
2-groups are still available for modification. In contrast, when using a SATA-excess of twenty times, no internal crosslinking could be monitored indicating that all NH
2-groups are blocked by SATA. Furthermore, a deacetylation with hydroxylamine does not destroy the protein as observed for other proteins [
23]. These results show that SATA is a suitable candidate not only for introduction of SH-groups into the protein, but also as a blocking reagent for free NH
2-groups. The thus SH-modified SlfB proteins are not only attractive for crosslinking with functional molecules, but also for its immobilization on gold surfaces. Currently, we use in our lab polyelectrolytes as an intermediate layer for the attachment of S-layer proteins on surfaces [
20]. However, especially for the construction of biosensors that use SPR technologies for detection, a direct attachment to gold surfaces might be more suitable. Another attractive approach for the SH-modified S-layers is the attachment of gold nanoparticles [
24]. Such materials can be used for the design of sensory devices or catalytic surfaces.
Figure 2.
SDS-PAGE of SlfB with different modifications: lane 1—native SlfB; lane 3—native SlfB crosslinked with EDC; lane 4—SlfB modified with an 4-fold excess of SATA and modified with EDC and; lane 5—same protein as in lane 4 and deacetylated; lane 8—SlfB modified with an 20-fold excess of SATA and modified with EDC; lane 9—same protein as in lane 8 and deacetylated; lane 2, 6, and 7—molecular weight standard with 200, 150, 120, 100, 85, 70, 50, 40, 30, 25, and 20 kDa.
Figure 2.
SDS-PAGE of SlfB with different modifications: lane 1—native SlfB; lane 3—native SlfB crosslinked with EDC; lane 4—SlfB modified with an 4-fold excess of SATA and modified with EDC and; lane 5—same protein as in lane 4 and deacetylated; lane 8—SlfB modified with an 20-fold excess of SATA and modified with EDC; lane 9—same protein as in lane 8 and deacetylated; lane 2, 6, and 7—molecular weight standard with 200, 150, 120, 100, 85, 70, 50, 40, 30, 25, and 20 kDa.
2.4. Biofunctionalization of Surfaces
Due to their self-assembling properties, their arrangement in two-dimensional arrays, and high potential in nanotechnologies, S-layer proteins are attractive as matrices for bioconjugations, thus linking functional molecules to technical surfaces in a highly ordered manner. The use for these purposes implies that the chemical modification does not alter the protein structure, the polymer composition, or the self-assembling properties. In a previous study [
15] it was shown that modification of SlfB monomers alter the self-assembling properties seriously. These labeled monomers were not able to form typical S-layer arrays demonstrating a severe change of the protein properties. In contrast, the same study demonstrated that the modification of protein polymers in suspension did not alter the polymeric structure. Modification of these polymers with fluorescent dyes resulted in a FRET, indicating a close proximity of FRET pairs and intact self-assemblies.
Despite this success, experiments in our lab demonstrated that the thusly-modified proteins were not applicable for a two-dimensional arrangement on surfaces. Therefore, these constructs are not suitable for a direct application as an agent for the functionalization of surfaces. Instead, another approach was chosen. In a first step, PEI (polyethyleneimine) activated silicon surfaces were coated with SlfB as described in
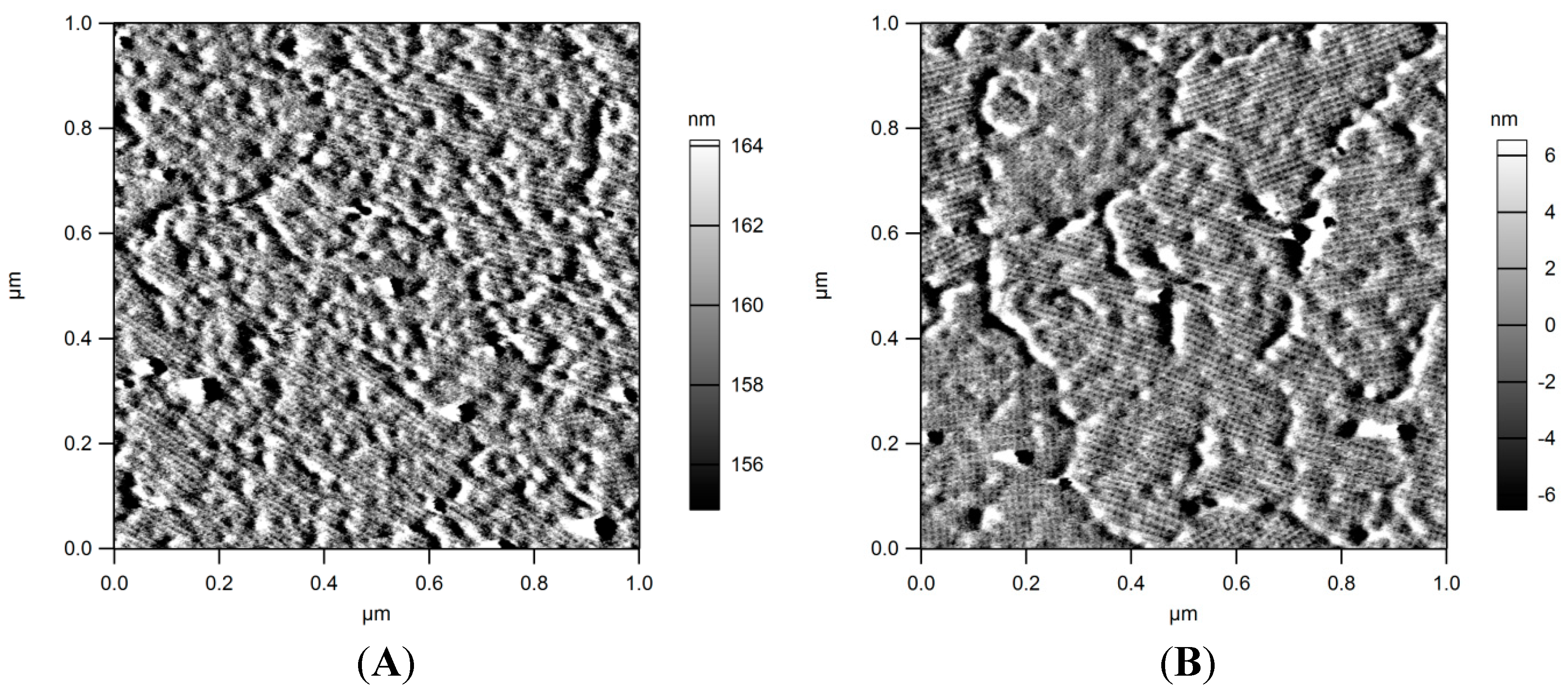
Section 3.5. AFM (Atomic Force Microscopy) analyses proved the formation of the typical S-layer arrays exhibiting p4 symmetry (
Figure 3A). In a second step the protein layers were EDC activated and conjugated with tryptophan. AFM analyses proved the intactness of the arrays after this treatment (
Figure 3B). These results confirm that the chemical modification of the protein layers do not alter their structure. In a recent work S-layer proteins of
L. sphaericus JG-B53 were recrystallized on PEI modified silicon surfaces and layer thickness was determined by AFM and QCM-D. The formed layers exhibited a thickness of about 12.9 nm indicating the formation of monolayers [
12].
Figure 3.
AFM images of SlfB recrystallized on silicon surfaces. (A) amplitude image of SlfB prior modification with tryptophan; (B) amplitude image of SlfB after crosslinking with tryprophan; S-layer coatings as well as p4-symmetries stay intact.
Figure 3.
AFM images of SlfB recrystallized on silicon surfaces. (A) amplitude image of SlfB prior modification with tryptophan; (B) amplitude image of SlfB after crosslinking with tryprophan; S-layer coatings as well as p4-symmetries stay intact.
It has to be emphasized that assembly of SlfB on surfaces almost certainly decreases the number of modifiable COOH- and NH2-groups. Properties and accessibility of the proteins in suspension naturally differ from those of the proteins placed on surfaces. On the other hand, a reliable determination of available modifiable groups on protein coatings is in view of low protein amounts (about 5 µg∙cm−2) not possible with current methods. However, the previous experiments showed that in suspensions numerous COOH- and NH2-groups of the proteins could be modified. These data allow a rough estimation of the modifiability of the proteins. It can be expected that a reasonable amount of these groups are still accessible after coating.
There are several studies that describe the immobilization of S-layer proteins followed by chemical modification with enzymes, antibodies, or antigens, thus producing functionalized surfaces. These approaches used cell wall preparations, peptidoglycan, liposomes, and ultrafiltration membranes [
13,
17,
18,
25,
26] as immobilization matrices. Further, there are numerous examples of immobilized genetically engineered fusion proteins on different surfaces (listed in [
1]). In contrast to these works, the present work describes a method that allows a fast and reliable patterning of various kinds of technical surfaces and an easy immobilization of organic molecules on the these nanostructured surfaces. In a current study [
27] S-layer proteins were modified with thrombin-binding or ofloxacin-binding aptamers (short oligonucleotides). The thus immobilized aptamers kept their functionality, thus proving the suitability of the presented bio-functionalization method.



{kind=link}
{kind=link}
{kind=link}


