
A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds

 , ,
, , 
Abstract
:1. Introduction
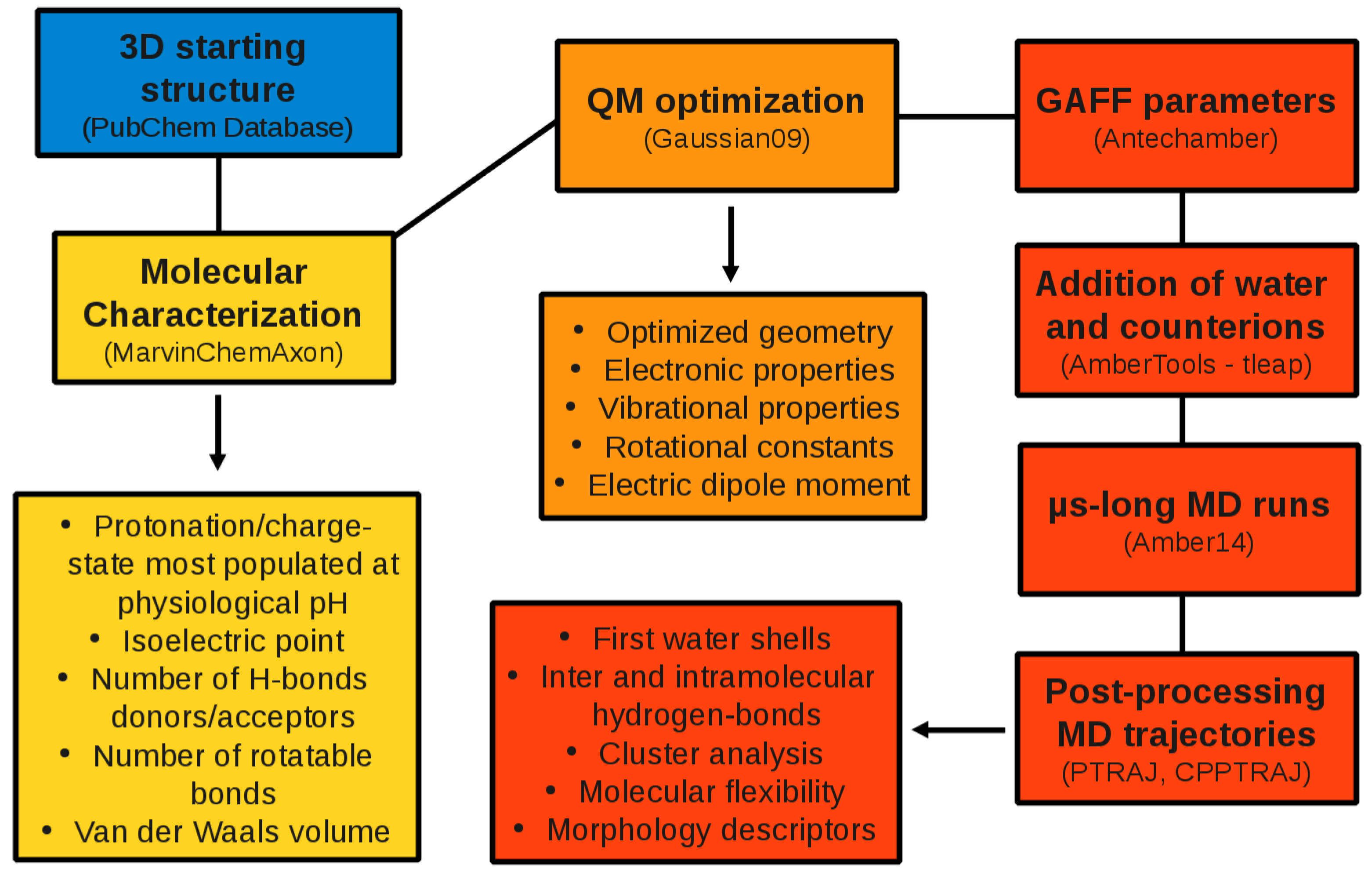
2. Computational Methods
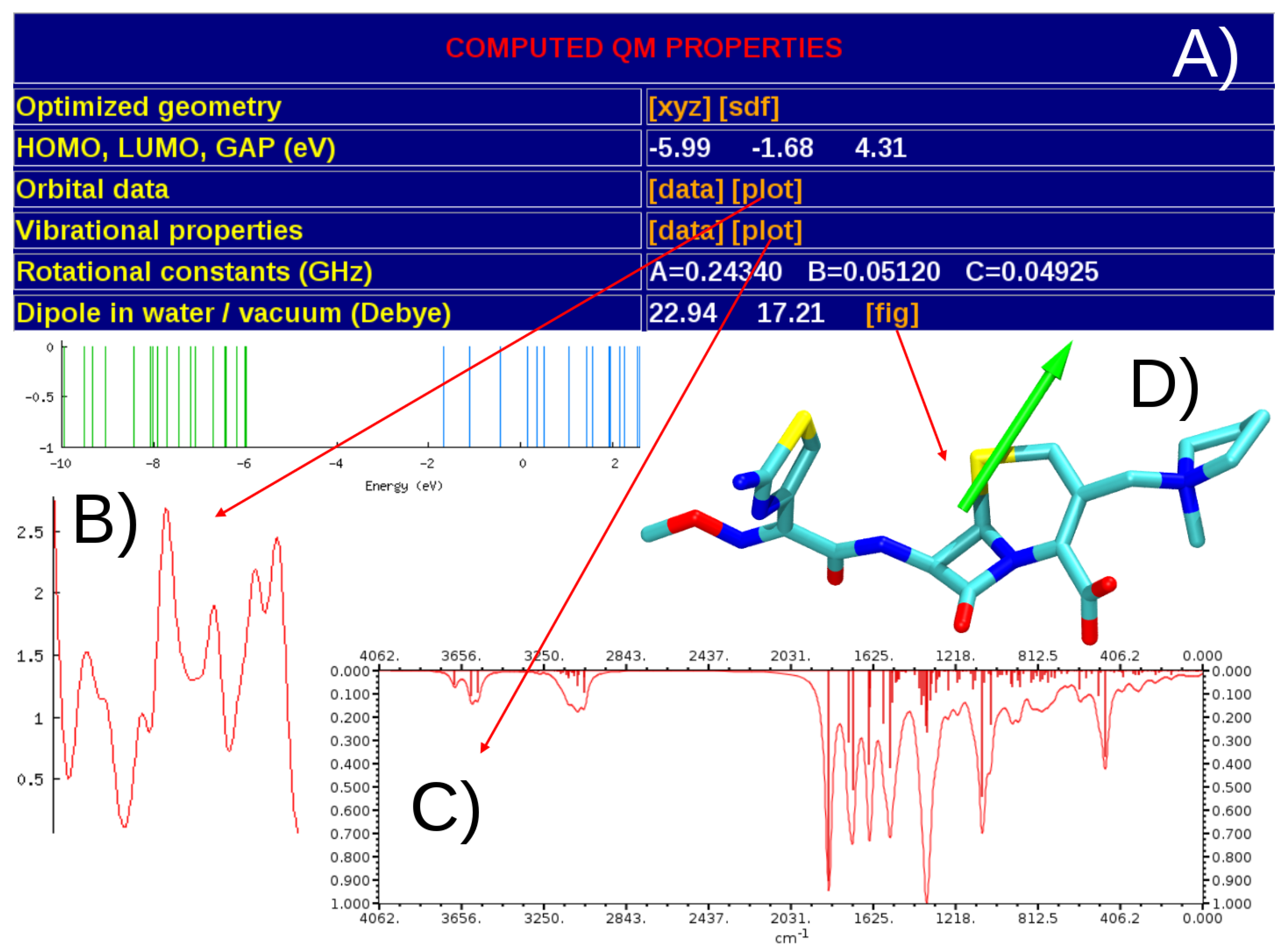
2.1. QM Calculations
2.2. MD Simulations
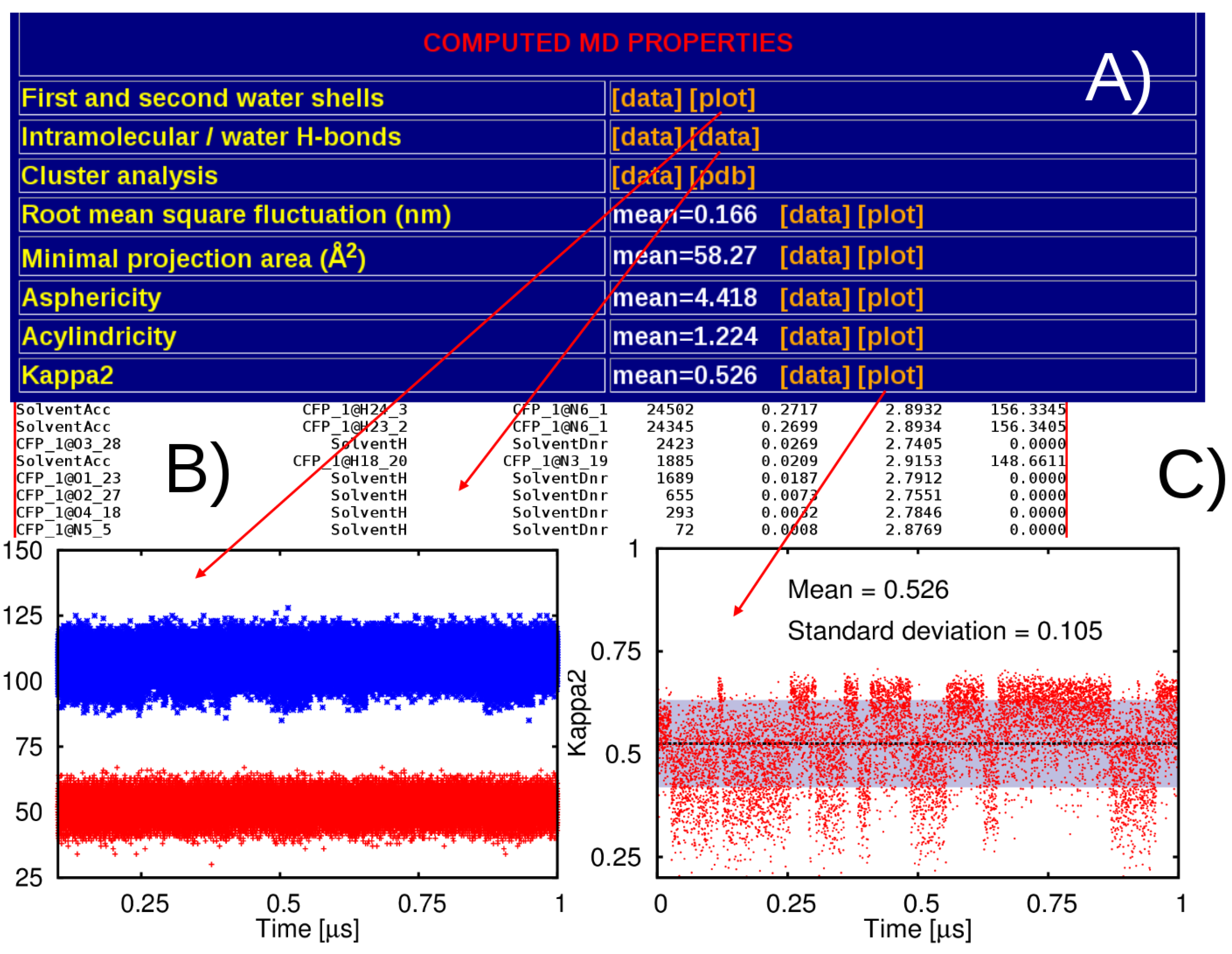
2.3. Post-Processing of the MD Trajectories

3. Results
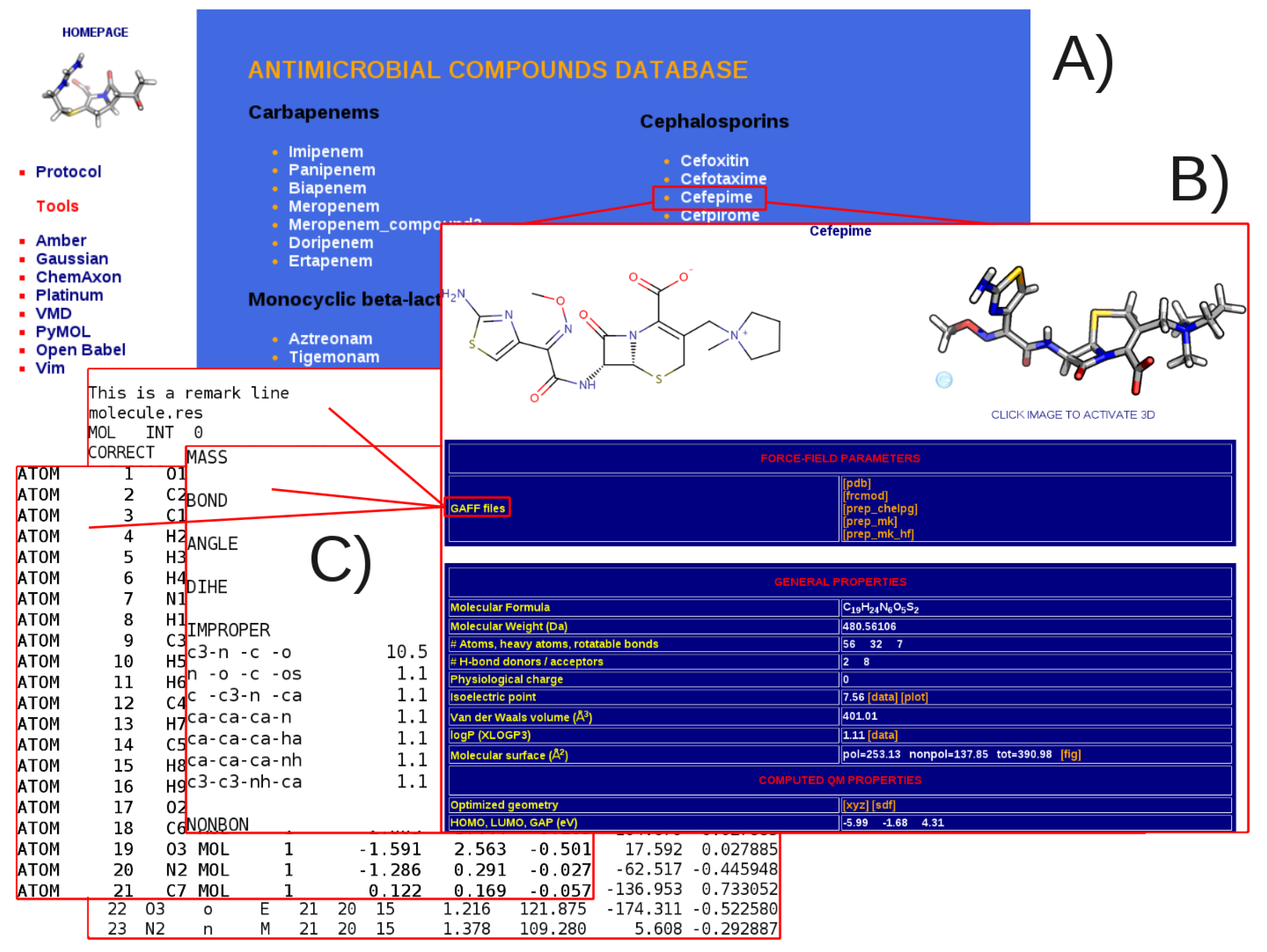
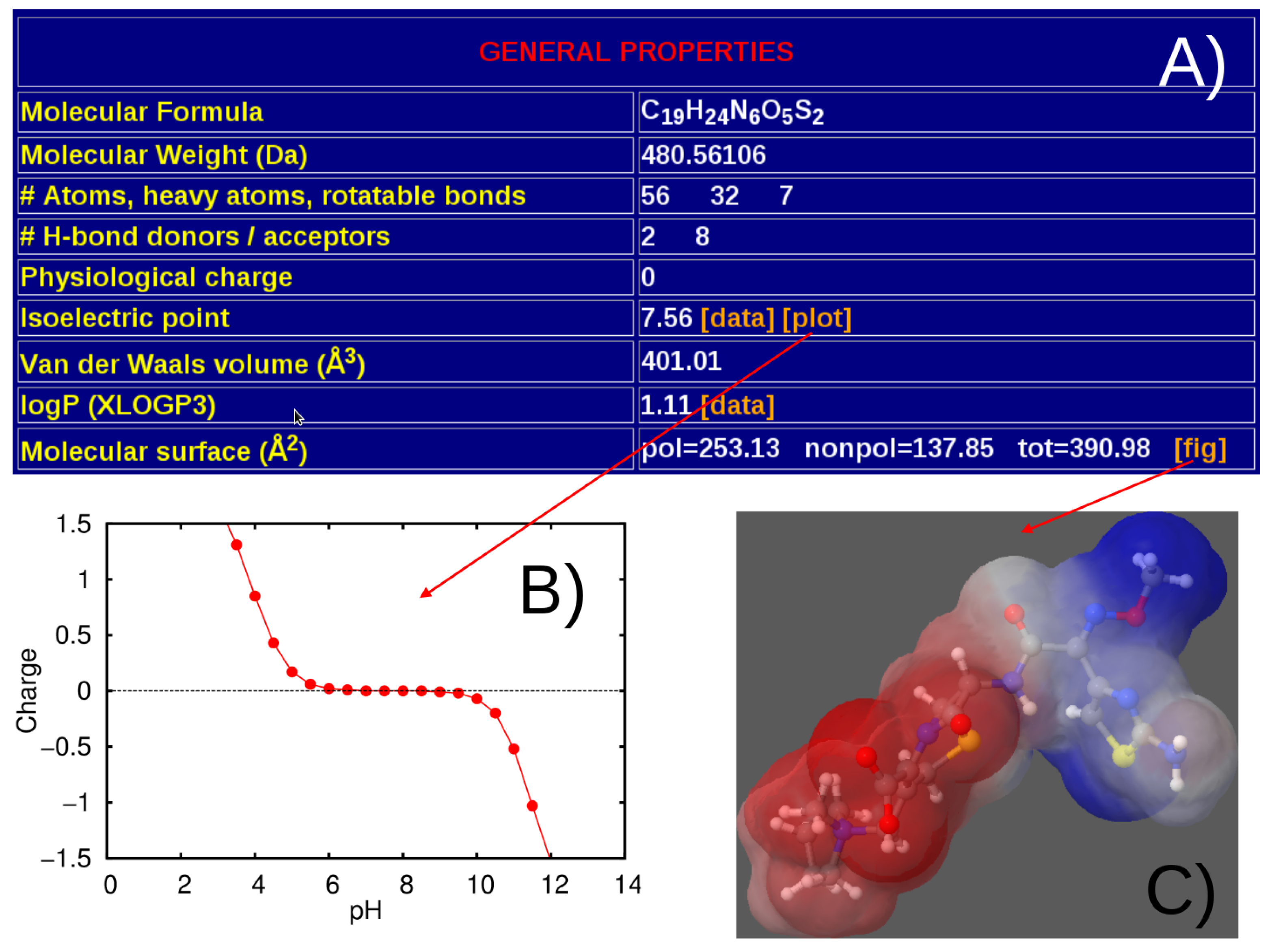
3.1. General Structure of the Database




4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Minimal Projection Area (Å) | ||
|---|---|---|---|
| Chemicalize | Mean ± Stdev | Min, Max (Delta) | |
| Carbapenems | |||
| Imipenem | n.a. | 44.7 ± 3.2 | 35.4, 56.6 (21.2) |
| Ertapenem | 72.6 | 57.4 ± 6.3 | 43.3, 82.5 (39.2) |
| Cephalosporins | |||
| Cefoxitin | 59.8 | 57.8 ± 5.2 | 41.9, 70.1 (28.2) |
| Ceftazidime | 66.0 | 68.3 ± 5.7 | 52.8, 88.1 (35.3) |
| Monocyclic beta-lactams | |||
| Aztreonam | 59.6 | 58.4 ± 2.7 | 48.3, 69.3 (21.0) |
| BAL30072 | n.a. | 64.7 ± 4.7 | 50.2, 81.4 (31.2) |
| Oxazolidinones | |||
| Linezolid | 47.6 | 43.4 ± 4.0 | 32.5, 53.5 (21.0) |
| Sutezolid | n.a. | 44.0 ± 3.9 | 32.6, 56.8 (24.2) |
| Penicillines | |||
| Aminopenicillanic acid | 39.9 | 38.1 ± 0.7 | 35.0, 41.5 (6.5) |
| Piperacillin | 86.6 | 77.5 ± 3.6 | 56.1, 90.0 (33.9) |
| Quinolones | |||
| Nalidixic acid | 34.3 | 36.5 ± 1.4 | 32.6, 42.7 (10.1) |
| Fleroxacin | 46.6 | 46.1 ± 1.9 | 39.8, 55.5 (15.7) |
| Tetracyclines | |||
| Minocycline | 66.2 | 67.5 ± 1.4 | 61.4, 72.9 (11.5) |
| Tigecycline | 76.2 | 76.6 ± 3.5 | 65.5, 86.7 (21.2) |
| Beta-lactamase inhibitors | |||
| Clavulanic acid | 38.6 | 37.5 ± 1.1 | 32.1, 40.4 (8.3) |
| Tazobactam | 47.0 | 44.2 ± 3.0 | 39.3, 52.5 (13.2) |
5. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
| Compound | Molecular | Number | Molecular | Physiological | |
|---|---|---|---|---|---|
| Formula | of Atoms | Weight | Charge | ||
| Carbapenems | |||||
| Imipenem |  | C12H17N3O4S | 37 | 299.3461 | 0 |
| Panipenem |  | C15H21N3O4S | 44 | 339.4099 | 0 |
| Biapenem |  | C15H18N4O4S | 42 | 350.3928 | 0 |
| Meropenem |  | C17H25N3O5S | 51 | 383.4625 | 0 |
| Meropenem 2 |  | C17H25N3O5S | 51 | 383.4625 | 0 |
| Doripenem |  | C15H24N4O6S | 51 | 420.5043 | 0 |
| Ertapenem |  | C22H24N3O7S | 57 | 474.5069 | –1 |
| Cephalosporins | |||||
| Cefoxitin |  | C16H16N3O7S2 | 44 | 426.4441 | –1 |
| Cefotaxime |  | C16H16N5O7S2 | 46 | 454.4575 | –1 |
| Cefepime |  | C19H24N6O5S2 | 56 | 480.5611 | 0 |
| Cefpirome |  | C22H22N6O5S2 | 57 | 515.5773 | 0 |
| Ceftobiprole |  | C20H22N8O6S2 | 58 | 534.5687 | 0 |
| Ceftazidime |  | C22H21N6O7S2 | 58 | 545.5681 | –1 |
| Monocyclic beta-lactams | |||||
| Aztreonam |  | C13H15N5O8S2 | 43 | 433.41690 | –2 |
| Tigemonam |  | C12H13N5O9S2 | 41 | 435.3897 | –2 |
| Carumonam |  | C12H12N6O10S2 | 42 | 464.3879 | –2 |
| BAL19764 |  | C14H13N6O9S2 | 44 | 473.4178 | –1 |
| Nocardicin |  | C23H22N4O9 | 58 | 498.4422 | –2 |
| BAL300072 |  | C16H17N6O10S2 | 51 | 517.47038 | –1 |
| Oxazolidinones | |||||
| Linezolid |  | C16H20FN3O4 | 44 | 337.3461 | 0 |
| Sutezolid |  | C16H20FN3O3S | 44 | 353.41170 | 0 |
| Penicillines | |||||
| Aminipenicillanic acid |  | C8H11N2O3S | 25 | 215.2495 | –1 |
| Benzylpenicillin |  | C16H17N2O4S | 40 | 333.3822 | –1 |
| Ampicillin |  | C16H19N3O4S | 43 | 349.4048 | 0 |
| Carbenicillin |  | C17H16N2O6S | 42 | 376.3837 | –2 |
| Ticarcillin |  | C15H14N2O6S2 | 39 | 382.4115 | –2 |
| Oxacillin |  | C19H18N3O5S | 46 | 400.4283 | –1 |
| Piperacillin |  | C23H26N5O7S | 62 | 516.5468 | –1 |
| Quinolones | |||||
| Nalidixic acid |  | C12H11N2O3 | 28 | 231.2273 | –1 |
| Norfloxacin |  | C16H18FN3O3 | 41 | 319.3308 | 0 |
| Ciprofloxacin |  | C17H18FN3N3O3 | 42 | 331.3415 | 0 |
| Enrofloxacin |  | C19H21FN3N3O3 | 47 | 358.3867 | –1 |
| Levofloxacin |  | C18H19FN3N3O4 | 45 | 360.3596 | –1 |
| Fleroxacin |  | C17H17F3N3O3 | 43 | 368.3304 | –1 |
| Tetracyclines | |||||
| Minocycline |  | C23H27N3O7 | 60 | 457.4764 | 0 |
| Tigecycline |  | C29H40N5O8 | 82 | 586.6566 | 1 |
| Beta-lactamase inhibitors | |||||
| Clavulanic acid |  | C8H8NO5 | 22 | 198.1528 | –1 |
| Sulbactam |  | C8H10NO5S | 25 | 232.2337 | –1 |
| Avibactam |  | C7H10N3O6S | 27 | 264.2358 | –1 |
| Tazobactam |  | C10H11N4O5S | 31 | 299.2831 | –1 |
| Class | Compound | RMSD (Å) |
|---|---|---|
| Carbapenems | Imipenem | 1.59 |
| Panipenem | 1.70 | |
| Biapenem | 1.69 | |
| Meropenem | 1.66 | |
| Meropenem-2 | 1.65 | |
| Doripenem | 1.66 | |
| Ertapenem | 1.71 | |
| Cephalosporins | Cefoxitin | 1.63 |
| Cefotaxime | 1.63 | |
| Cefepime | 1.62 | |
| Ceftobiprole | 1.64 | |
| Ceftazidime | 1.60 | |
| Monocyclic beta-lactams | Aztreonam | 1.60 |
| Tigemonam | 1.69 | |
| Carumonam | 1.64 | |
| BAL19764 | 1.65 | |
| Nocardicin | 1.66 | |
| BAL30072 | 1.76 | |
| Oxazolidinones | Linezolid | 1.68 |
| Sutezolid | 1.67 | |
| Penicillines | Aminopenicillanic Acid | 1.73 |
| Benzylpenicillin | 1.64 | |
| Ampicillin | 1.65 | |
| Carbenicillin | 1.68 | |
| Ticarcillin | 1.68 | |
| Oxacillin | 1.64 | |
| Piperacillin | 1.74 | |
| Quinolones | Nalidixic Acid | 1.62 |
| Norfloxacin | 1.58 | |
| Ciprofloxacin | 1.58 | |
| Enrofloxacin | 1.58 | |
| Levofloxacin | 1.56 | |
| Fleroxacin | 1.59 | |
| Tetracyclines | Minocycline | 1.62 |
| Tigecycline | 1.65 | |
| Beta-lactamase inhibitors | Clavulanic Acid | 1.66 |
| Sulbactam | 1.63 | |
| Avibactam | 1.62 | |
| Tazobactam | 1.66 |
References
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; McCammon, J.A. Molecular Recognition and Ligand Association. Ann. Rev. Phys. Chem. 2013, 64, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.X. Calculation of Protein-Ligand Binding Affinities. Ann. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; McCammon, J.A. Molecular Recognition and Ligand Association. Ann. Rev. Phys. Chem. 2013, 64, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Magistrato, A. Atomistic-Level Portrayal of Drug-DNA Interplay: A History of Courtships and Meetings Revealed by Molecular Simulations. ChemMedChem 2014, 9, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glaettli, A.; Huenenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. 2006, 45, 4064–4092. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, P.; Brooks, B.R.; Sansom, M.S.P. Multiscale methods for macromolecular simulations. Curr. Opin. Struc. Biol. 2008, 18, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, T.E., III; Case, D.A. Twenty-Five Years of Nucleic Acid Simulations. Biopolymers 2013, 99, 969–977. [Google Scholar] [PubMed]
- Lee, E.H.; Hsin, J.; Sotomayor, M.; Comellas, G.; Schulten, K. Discovery through the Computational Microscope. Structure 2009, 17, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, G.F.; Scorciapino, M.A.; Messina, A.; De Pinto, V.; Ceccarelli, M. Charged residues distribution modulates selectivity of the open state of human isoforms of the voltage dependent anion-selective channel. PLoS ONE 2014, 9, e103879. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.G.; Lane, D.P.; Verma, C.S. Molecular simulations of protein dynamics: New windows on mechanisms in biology. EMBO Rep. 2008, 9, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Ann. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Lavery, R. Significance of Molecular Dynamics Simulations for Life Sciences. Isr. J. Chem. 2014, 54, 1042–1051. [Google Scholar] [CrossRef]
- Mortier, J.; Rakers, C.; Bermudez, M.; Murgueitio, M.S.; Riniker, S.; Wolber, G. The impact of molecular dynamics on drug design: Applications for the characterization of ligand-macromolecule complexes. Drug Discov. Today 2015, 20, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Schlick, T. Molecular Modeling and Simulation: An Interdisciplinary Guide; Interdisciplinary applied mathematics; Springer: New York, NY, USA, 2010. [Google Scholar]
- Gelin, B.R. Testing and comparison of empirical force fields: Techniques and problems. In Computer Simulation of Biomolecular Systems: Theoretical and Experimental Applications; Gunsteren, W.F.V., Weiner, P., Wilkinson, A.J., Eds.; ESCOM: Leiden, The Netherlands, 1993; Volume 2, pp. 127–146. [Google Scholar]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.G.; Merz, K.M.J.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Lutz, M.; Marchi, M. A density functional normal mode calculation of a bacteriochlorophyll a derivative. J. Am. Chem. Soc. 2000, 122, 3532–3533. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Marchi, M. Simulation and Modeling of the Rhodobacter sphaeroides Bacterial Reaction Center II: Primary Charge Separation. J. Phys. Chem. B 2003, 107, 5630–5641. [Google Scholar] [CrossRef]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Banáš, P.; Jurečka, P.; Zgarbová, M.; Kührová, P.; Havrila, M.; Krepl, M.; Stadlbauer, P.; Otyepka, M. Molecular Dynamics Simulations of Nucleic Acids. From Tetranucleotides to the Ribosome. J. Phys. Chem. Lett. 2014, 5, 1771–1782. [Google Scholar] [CrossRef]
- Schmidt, T.H.; Kandt, C. LAMBADA and InflateGRO2: Efficient Membrane Alignment and Insertion of Membrane Proteins for Molecular Dynamics Simulations. J. Chem. Inf. Mod. 2012, 52, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Graen, T.; Hoefling, M.; Grubmueller, H. AMBER-DYES: Characterization of Charge Fluctuations and Force Field Parameterization of Fluorescent Dyes for Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 5505–5512. [Google Scholar] [CrossRef]
- Dupradeau, F.Y.; Cézard, C.; Lelong, R.; Stanislawiak, E.; Pêcher, J.; Delepine, J.C.; Cieplak, P. R.E.DD.B.: A database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 2008, 36, D360–D367. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [PubMed]
- AMBER Parameter Database. Available online: http://www.pharmacy.manchester.ac.uk/bryce/amber/ (accessed on 20 July 2014).
- Marvin 14.8.25.0. ChemAxon 2014. Available online: http://www.chemaxon.com (accessed on 30 November 2014).
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09 Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stavenger, R.A.; Winterhalter, M. TRANSLOCATION Project: How to Get Good Drugs into Bad Bugs. Sci. Transl. Med. 2014, 6, 228ed7. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Prevention of drug access to bacterial targets: Permeability barriers and active efflux. Science 1994, 264, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Ruggerone, P. Physical insights into permeation of and resistance to antibiotics in bacteria. Curr. Drug Targ. 2008, 9, 779–788. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Bolton, E.E.; Wangand, Y.; Thiessenand, P.A.; Bryant, S.H. PubChem: Integrated Platform of Small Molecules and Biological Activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Kohn, W. Nobel Lecture: Electronic structure of matter-wave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253–1266. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Pople, J.A. Quantum Chemical Models (Nobel Lecture). Angew. Chem. Int. Ed. 1999, 38, 1894–1902. [Google Scholar] [CrossRef]
- Malloci, G.; Joblin, C.; Mulas, G. On-line database of the spectral properties of polycyclic aromatic hydrocarbons. Chem. Phys. 2007, 332, 353–359. [Google Scholar] [CrossRef]
- Malloci, G.; Cappellini, G.; Mulas, G.; Mattoni, A. Electronic and optical properties of families of polycyclic aromatic hydrocarbons: A systematic (time-dependent) density functional theory study. Chem. Phys. 2011, 384, 19–27. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Allouche, A.R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol-Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Pyrkov, T.V.; Chugunov, A.O.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PLATINUM: A web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics 2009, 25, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Nag, A.; Chakraborty, D.; Chandra, A. Effects of ion concentration on the hydrogen bonded structure of water in the vicinity of ions in aqueous NaCl solutions. J. Chem. Sci. 2008, 120, 71–77. [Google Scholar] [CrossRef]
- Luzar, A.; Chandler, D. Effect of Environment on Hydrogen Bond Dynamics in Liquid Water. Phys. Rev. Lett. 1996, 76, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Luzar, A.; Chandler, D. Hydrogen-bond kinetics in liquid Water. Nature 1996, 379, 55–57. [Google Scholar] [CrossRef]
- Sutmann, G.; Vallauri, R. Hydrogen bonded clusters in the liquid phase: I. Analysis of the velocity correlation function of water triplets. J. Phys. Condens. Matter 1998, 10, 9231–9240. [Google Scholar]
- Chandra, A. Effects of Ion Atmosphere on Hydrogen-Bond Dynamics in Aqueous Electrolyte Solutions. Phys. Rev. Lett. 2000, 85, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar]
- van der Spoel, D.; van Buuren, A.R.; Apol, E.; Meulenhoff, P.J.; Tieleman, D.P.; Sijbers, A.L.T.M.; Hess, B.; Feenstra, K.A.; Lindahl, E.; van Drunen, R.; Berendsen, H.J.C. Gromacs User Manual version 3.0; Nijenborgh 4, 9747 AG Groningen: The Netherlands, 2001; Available online: http://www.gromacs.org (accessed on 29 June 2014).
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; Parrinello, M. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Comm. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Theodorou, D.N.; Suter, U.W. Shape of unperturbed linear polymers: Polypropylene. Macromolecules 1985, 18, 1206–1214. [Google Scholar] [CrossRef]
- O’Boyle, N.; Banck, M.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 1.3r1. The PyMOL Molecular Graphics System, Version 1.3, Schrödinger. LLC: Mannheim, Germany.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumar, A.; Hajjar, E.; Ruggerone, P.; Ceccarelli, M. Molecular simulations reveal the mechanism and the determinants for ampicillin translocation through OmpF. J. Phys. Chem. B 2010, 114, 9608–9616. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, E.; Bessonov, A.; Molitor, A.; Kumar, A.; Mahendran, K.R.; Winterhalter, M.; Pagès, J.M.; Ruggerone, P.; Ceccarelli, M. Toward screening for antibiotics with enhanced permeation properties through bacterial porins. Biochemistry 2010, 49, 6928–6935. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Chen, M.; Black, S.S.; Bushell, S.R.; Ceccarelli, M.; Mach, T.; Beis, K.; Low, A.S.; Bamford, V.A.; Booth, I.R.; Bayley, H.; Naismith, J.H. Altered Antibiotic Transport in OmpC Mutants Isolated from a Series of Clinical Strains of Multi-Drug Resistant E. coli. PLoS ONE 2011, 6, e25825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collu, F.; Vargiu, A.V.; Dreier, J.; Cascella, M.; Ruggerone, P. Recognition of Imipenem and Meropenem by the RND-Transporter MexB Studied by Computer Simulations. J. Am. Chem. Soc. 2012, 134, 19146–19158. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Nikaido, H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 20637–20642. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Ruggerone, P.; Opperman, T.J.; Nguyen, S.T.; Nikaido, H. Molecular Mechanism of MBX2319 Inhibition of Escherichia coli AcrB Multidrug Efflux Pump and Comparison with Other Inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6224–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JSmol: An Open-Source HTML5 Viewer for Chemical Structures in 3D. Available online: http://wiki.jmol.org/index.php/JSmol#JSmol (accessed on 27 March 2015).
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, M. Protein-protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003, 12, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, M. Accounting for conformational changes during protein-protein docking. Curr. Opin. Struct. Biol. 2010, 20, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, A. Flexible protein-protein docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Mashiach, E.; Nussinov, R.; Wolfson, H.J. Principles of flexible protein-protein docking. Proteins 2008, 73, 271–289. [Google Scholar] [CrossRef] [PubMed]
- B-Rao, C.; Subramanian, J.; Sharma, S.D. Managing protein flexibility in docking and its applications. Drug Discov. Today 2009, 14, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Agostino, M.; Ramsland, P.A. Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 2011, 24, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Sinko, W.; Lindert, S.; McCammon, J.A. Accounting for Receptor Flexibility and Enhanced Sampling Methods in Computer-Aided Drug Design. Chem. Biol. Drug Des. 2013, 81, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Orry, A.J.W. Ligand docking and structure-based virtual screening in drug discovery. Curr. Opin. Struct. Biol. 2007, 7, 1006–1014. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. -Aided Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Collu, F.; Schulz, R.; Pos, K.M.; Zacharias, M.; Kleinekathöfer, U.; Ruggerone, P. Effect of the F610A Mutation on Substrate Extrusion in the AcrB Transporter: Explanation and Rationale by Molecular Dynamics Simulations. J. Am. Chem. Soc. 2011, 133, 10704–10707. [Google Scholar] [CrossRef] [PubMed]
- Asthana, S.; Shukla, S.; Ruggerone, P.; Vargiu, A.V. Molecular Mechanism of Viral Resistance to a Potent Non-nucleoside Inhibitor Unveiled by Molecular Simulations. Biochemistry 2014, 53, 6941–6953. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.J.; Müller, R.T.; Pos, K.M. Switch-Loop Flexibility Affects Transport of Large Drugs by the Promiscuous AcrB Multidrug Efflux Transporter. Antimicrob. Agents Chemother. 2014, 58, 4767–4772. [Google Scholar] [CrossRef] [PubMed]
- Ruggerone, P.; Murakami, S.; Pos, K.M.; Vargiu, A.V. RND efflux pumps: Structural information translated into function and inhibition mechanisms. Curr. Top. Med. Chem. 2013, 13, 3079–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Nikaido, H. Efflux-Mediated Drug Resistance in Bacteria An Update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.; Richmond, G.; Piddock, L. Multidrug efflux pumps in Gram-negative bacteria and their role in antibiotic resistance. Future Microbiol. 2014, 9, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Zgurskaya, H.I.; Totrov, M.; Watkins, W.J. Waltzing transporters and ‘the dance macabre’ between humans and bacteria. Nat. Rev. Drug Discov. 2007, 6, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 2005, 56, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Pagés, J.M. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol. Rev. 2012, 36, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Chemicalize. ChemAxon 2014. Available online: http://www.chemicalize.org (accessed on 29 January 2014).
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Fukunishi, H.; Watanabe, O.; Takada, S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J. Chem. Phys. 2002, 116, 9058–9067. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malloci, G.; Vargiu, A.V.; Serra, G.; Bosin, A.; Ruggerone, P.; Ceccarelli, M. A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds. Molecules 2015, 20, 13997-14021. https://doi.org/10.3390/molecules200813997
Malloci G, Vargiu AV, Serra G, Bosin A, Ruggerone P, Ceccarelli M. A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds. Molecules. 2015; 20(8):13997-14021. https://doi.org/10.3390/molecules200813997
Chicago/Turabian StyleMalloci, Giuliano, Attilio Vittorio Vargiu, Giovanni Serra, Andrea Bosin, Paolo Ruggerone, and Matteo Ceccarelli. 2015. "A Database of Force-Field Parameters, Dynamics, and Properties of Antimicrobial Compounds" Molecules 20, no. 8: 13997-14021. https://doi.org/10.3390/molecules200813997