
Evolution of Macromolecular Docking Techniques: The Case Study of Nickel and Iron Metabolism in Pathogenic Bacteria



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
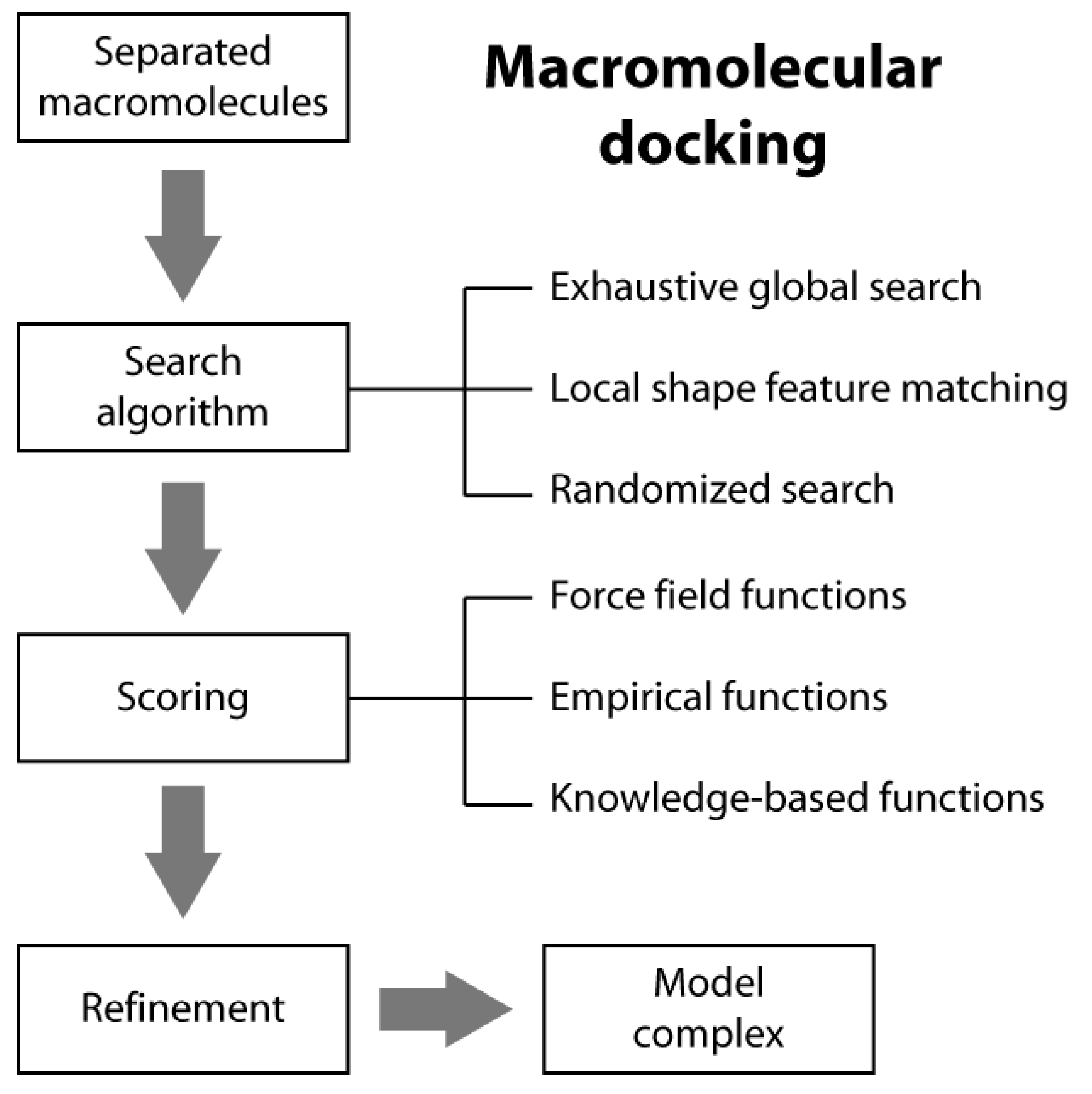
2. Macromolecular Docking Overview
2.1. Search Algorithms

2.2. Scoring Functions
2.3. A Peculiar Case: Protein-DNA Docking
3. Urease Activation


3.1. Calculation of the Structure of the UreEG Complex

3.2. Calculation of the Structure of the UreDFGE Complex
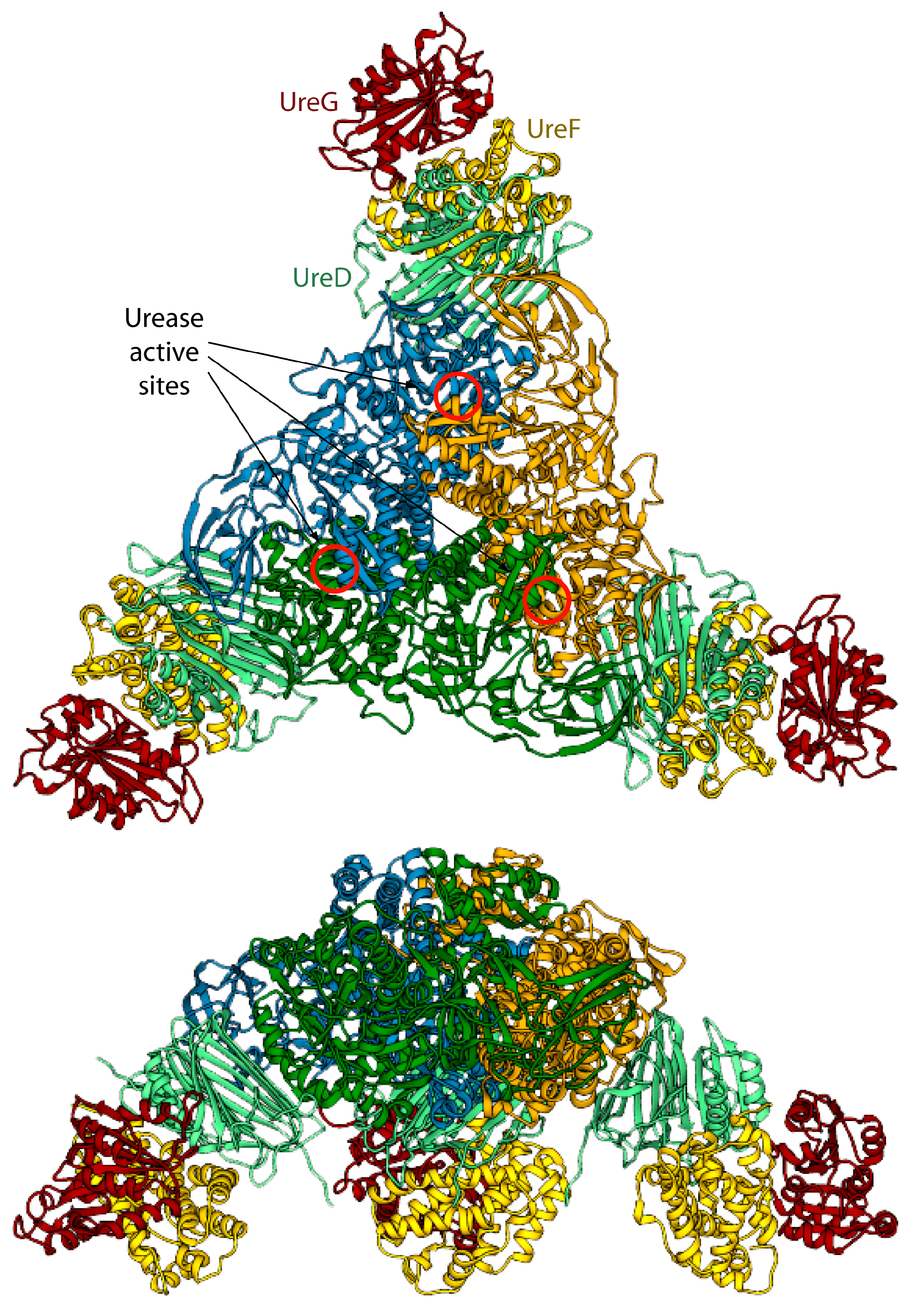
3.3. Calculation of the Structure of the Urease-UreDFG Complex

4. HpFur-DNA and HpNikR-DNA Complexes
4.1. HpFur-DNA Complexes

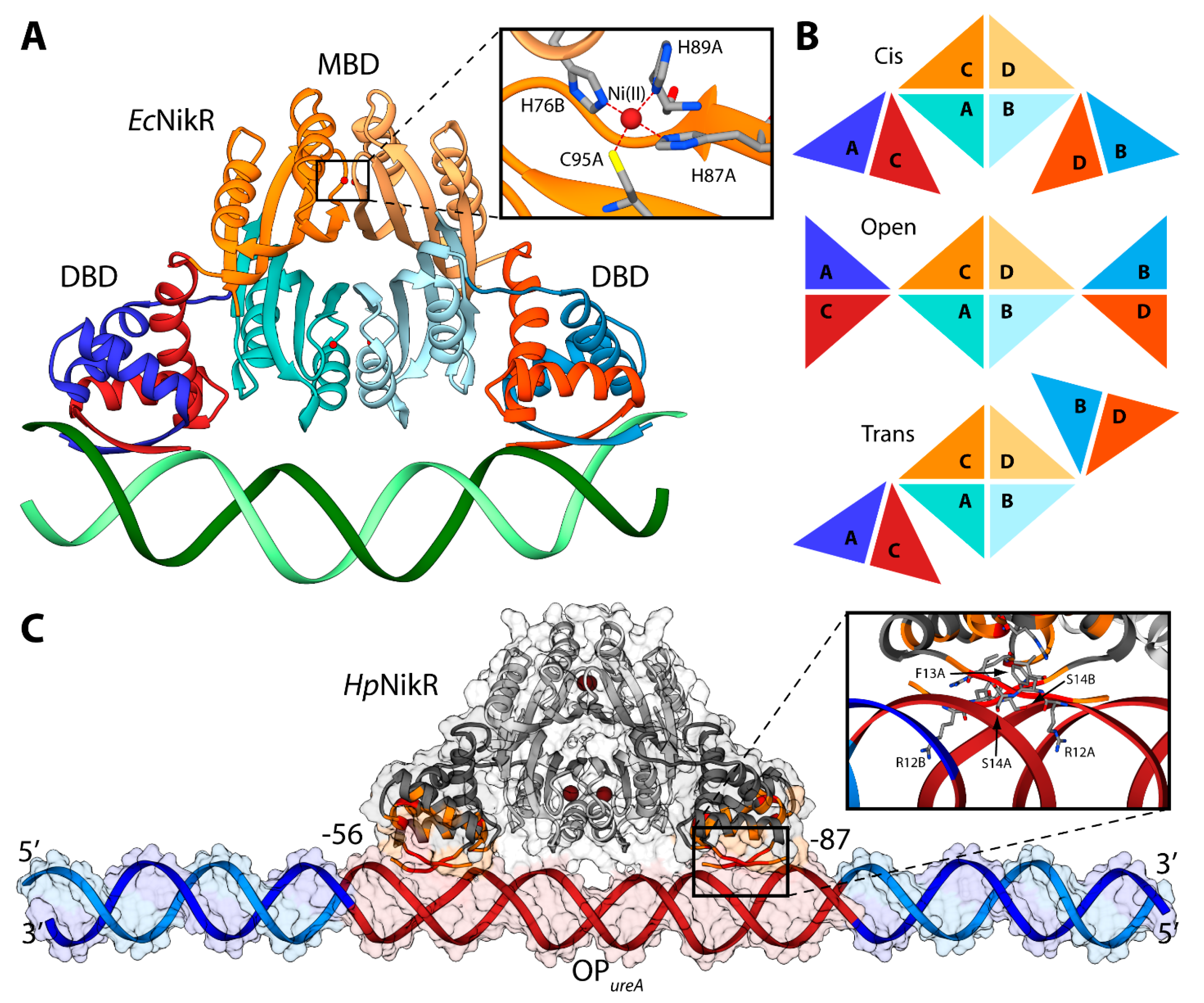
4.2. HpNikR-DNA Complex

5. Perspectives
Acknowledgements
Conflicts of Interest
References
- Cozzarelli, N.R.; Cost, G.J.; Nollmann, M.; Viard, T.; Stray, J.E. Giant proteins that move DNA: Bullies of the genomic playground. Nat. Rev. Mol. Cell Biol. 2006, 7, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Wodak, S.J.; Vlasblom, J.; Turinsky, A.L.; Pu, S. Protein-protein interaction networks: The puzzling riches. Curr. Opin. Struct. Biol. 2013, 23, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Lage, K. Protein-protein interactions and genetic diseases: The interactome. Biochim. Biophys. Acta 2014, 1842, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Re, A.; Joshi, T.; Kulberkyte, E.; Morris, Q.; Workman, C.T. RNA-protein interactions: An overview. Methods Mol. Biol. 2014, 1097, 491–521. [Google Scholar] [PubMed]
- Su, C.; Peregrin-Alvarez, J.M.; Butland, G.; Phanse, S.; Fong, V.; Emili, A.; Parkinson, J. Bacteriome.org—An integrated protein interaction database for E. coli. Nucleic Acids Res. 2008, 36, D632–D636. [Google Scholar] [CrossRef] [PubMed]
- Braun, P.; Aubourg, S.; van Leene, J.; de Jaeger, G.; Lurin, C. Plant protein interactomes. Annu. Rev. Plant Biol. 2013, 64, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Cukuroglu, E.; Gursoy, A.; Nussinov, R.; Keskin, O. Non-redundant unique interface structures as templates for modeling protein interactions. PLoS ONE 2014, 9, e86738. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.; Bonvin, A.M. Integrative computational modeling of protein interactions. FEBS J. 2014, 281, 1988–2003. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.B.; Alber, F.; Aloy, P.; Davis, F.P.; Korkin, D.; Pichaud, M.; Topf, M.; Sali, A. A structural perspective on protein-protein interactions. Curr. Opin. Struct. Biol. 2004, 14, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Protein-protein docking dealing with the unknown. J. Comput. Chem. 2010, 31, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, S.; Fernandez-Recio, J. Protein-protein docking and hot-spot prediction for drug discovery. Curr. Pharm. Des. 2012, 18, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y. Search strategies and evaluation in protein-protein docking: Principles, advances and challenges. Drug Discov. Today 2014, 19, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y. Exploring the potential of global protein-protein docking: An overview and critical assessment of current programs for automatic ab initio docking. Drug Discov. Today 2015. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Bonvin, A.M. A protein-DNA docking benchmark. Nucleic Acids Res. 2008, 36, e88. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Visscher, K.M.; Kastritis, P.L.; Bonvin, A.M. Solvated protein-DNA docking using HADDOCK. J. Biomol. NMR 2013, 56, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Tuszynska, I.; Matelska, D.; Magnus, M.; Chojnowski, G.; Kasprzak, J.M.; Kozlowski, L.P.; Dunin-Horkawicz, S.; Bujnicki, J.M. Computational modeling of protein-RNA complex structures. Methods 2014, 65, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I. PMF scoring revisited. J. Med. Chem. 2006, 49, 5895–5902. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; van Dijk, A.D.; Hsu, V.; Boelens, R.; Bonvin, A.M. Information-driven protein-DNA docking using HADDOCK: It is a matter of flexibility. Nucleic Acids Res. 2006, 34, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; van Dijk, A.D.; Krzeminski, M.; van Dijk, M.; Thureau, A.; Hsu, V.; Wassenaar, T.; Bonvin, A.M. HADDOCK versus HADDOCK: New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 2007, 69, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Bonvin, A.M. Pushing the limits of what is achievable in protein-DNA docking: Benchmarking HADDOCK’s performance. Nucleic Acids Res. 2010, 38, 5634–5647. [Google Scholar] [CrossRef] [PubMed]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [PubMed]
- Linge, J.P.; O’Donoghue, S.I.; Nilges, M. Automated assignment of ambiguous nuclear overhauser effects with ARIA. Methods Enzymol. 2001, 339, 71–90. [Google Scholar] [PubMed]
- Van Dijk, M.; Bonvin, A.M. 3D-DART: A DNA structure modelling server. Nucleic Acids Res. 2009, 37, W235–W239. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Musiani, F.; Benini, S.; Ciurli, S. Chemistry of Ni2+ in urease: Sensing, trafficking, and catalysis. Acc. Chem. Res. 2011, 44, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Maroney, M.J.; Ciurli, S. Nonredox nickel enzymes. Chem. Rev. 2014, 114, 4206–4228. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, M.A.; Macomber, L.; Hausinger, R.P. Biosynthesis of the urease metallocenter. J. Biol. Chem. 2013, 288, 13178–13185. [Google Scholar] [CrossRef] [PubMed]
- Steyert, S.R.; Rasko, D.A.; Kaper, J.B. Functional and phylogenetic analysis of ureD in Shiga toxin-producing Escherichia coli. J. Bacteriol. 2011, 193, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Kuchar, J.; Hausinger, R.P. Chemical cross-linking and mass spectrometric identification of sites of interaction for UreD, UreF, and urease. J. Biol. Chem. 2004, 279, 15305–15313. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.; Hausinger, R.P. GTP-dependent activation of urease apoprotein in complex with the UreD, UreF, and UreG accessory proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11140–11144. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Stola, M.; Musiani, F.; de Vriendt, K.; Samyn, B.; Devreese, B.; van Beeumen, J.; Turano, P.; Dikiy, A.; Bryant, D.A.; et al. UreG, a chaperone in the urease assembly process, is an intrinsically unstructured GTPase that specifically binds Zn2+. J. Biol. Chem. 2005, 280, 4684–4695. [Google Scholar] [CrossRef] [PubMed]
- Musiani, F.; Ippoliti, E.; Micheletti, C.; Carloni, P.; Ciurli, S. Conformational fluctuations of UreG, an intrinsically disordered enzyme. Biochemistry 2013, 52, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Salomone-Stagni, M.; Zambelli, B.; Musiani, F.; Ciurli, S. A model-based proposal for the role of UreF as a GTPase-activating protein in the urease active site biosynthesis. Proteins 2007, 68, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.L.; Flugga, N.; Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Interplay of metal ions and urease. Metallomics 2009, 1, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.; Colpas, G.J.; Hausinger, R.P. UreE stimulation of GTP-dependent urease activation in the UreD-UreF-UreG-urease apoprotein complex. Biochemistry 2000, 39, 12435–12440. [Google Scholar] [CrossRef] [PubMed]
- Rain, J.C.; Selig, L.; de Reuse, H.; Battaglia, V.; Reverdy, C.; Simon, S.; Lenzen, G.; Petel, F.; Wojcik, J.; Schachter, V.; et al. The protein-protein interaction map of Helicobacter pylori. Nature 2001, 409, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Voland, P.; Weeks, D.L.; Marcus, E.A.; Prinz, C.; Sachs, G.; Scott, D. Interactions among the seven Helicobacter pylori proteins encoded by the urease gene cluster. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G96–G106. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, M.; Zambelli, B.; Musiani, F.; Turano, P.; Ciurli, S. Helicobacter pylori UreE, a urease accessory protein: Specific Ni(2+)- and Zn(2+)-binding properties and interaction with its cognate UreG. Biochem. J. 2009, 422, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Lam, R.; Romanov, V.; Johns, K.; Battaile, K.P.; Wu-Brown, J.; Guthrie, J.L.; Hausinger, R.P.; Pai, E.F.; Chirgadze, N.Y. Crystal structure of a truncated urease accessory protein UreF from Helicobacter pylori. Proteins 2010, 78, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.H.; Wong, H.C.; Chuck, C.P.; Chen, Y.W.; Sun, H.; Wong, K.B. Assembly of preactivation complex for urease maturation in Helicobacter pylori: Crystal structure of UreF-UreH protein complex. J. Biol. Chem. 2011, 286, 43241–43249. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.H.; Wong, H.C.; Yuen, M.H.; Lau, P.H.; Chen, Y.W.; Wong, K.B. Structure of UreG/UreF/UreH complex reveals how urease accessory proteins facilitate maturation of Helicobacter pylori urease. PLoS Biol. 2013, 11, e1001678. [Google Scholar] [CrossRef] [PubMed]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Quiroz-Valenzuela, S.; Anderson, K.L.; Hausinger, R.P. Mutagenesis of Klebsiella aerogenes UreG to probe nickel binding and interactions with other urease-related proteins. Biochemistry 2010, 49, 5859–5869. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Turano, P.; Musiani, F.; Neyroz, P.; Ciurli, S. Zn2+-linked dimerization of UreG from Helicobacter pylori, a chaperone involved in nickel trafficking and urease activation. Proteins 2009, 74, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Real-Guerra, R.; Staniscuaski, F.; Zambelli, B.; Musiani, F.; Ciurli, S.; Carlini, C.R. Biochemical and structural studies on native and recombinant Glycine max UreG: A detailed characterization of a plant urease accessory protein. Plant. Mol. Biol. 2012, 78, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Musiani, F.; Savini, M.; Tucker, P.; Ciurli, S. Biochemical studies on Mycobacterium tuberculosis UreG and comparative modeling reveal structural and functional conservation among the bacterial UreG family. Biochemistry 2007, 46, 3171–3182. [Google Scholar] [CrossRef] [PubMed]
- Remaut, H.; Safarov, N.; Ciurli, S.; van Beeumen, J. Structural basis for Ni2+ transport and assembly of the urease active site by the metallochaperone UreE from Bacillus pasteurii. J. Biol. Chem. 2001, 276, 49365–49370. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Banaszak, K.; Merloni, A.; Kiliszek, A.; Rypniewski, W.; Ciurli, S. Selectivity of Ni(II) and Zn(II) binding to Sporosarcina pasteurii UreE, a metallochaperone in the urease assembly: A calorimetric and crystallographic study. J. Biol. Inorg. Chem. 2013, 18, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Song, H.K.; Mulrooney, S.B.; Huber, R.; Hausinger, R.P. Crystal structure of Klebsiella aerogenes UreE, a nickel-binding metallochaperone for urease activation. J. Biol. Chem. 2001, 276, 49359–49364. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Munger, C.; Asinas, A.; Benoit, S.L.; Miller, E.; Matte, A.; Maier, R.J.; Cygler, M. Crystal structures of apo and metal-bound forms of the UreE protein from Helicobacter pylori: Role of multiple metal binding sites. Biochemistry 2010, 49, 7080–7088. [Google Scholar] [CrossRef] [PubMed]
- Banaszak, K.; Martin-Diaconescu, V.; Bellucci, M.; Zambelli, B.; Rypniewski, W.; Maroney, M.J.; Ciurli, S. Crystallographic and X-ray absorption spectroscopic characterization of Helicobacter pylori UreE bound to Ni2+ and Zn2+ reveals a role for the disordered C-terminal arm in metal trafficking. Biochem. J. 2012, 441, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Musiani, F.; Zambelli, B.; Stola, M.; Ciurli, S. Nickel trafficking: Insights into the fold and function of UreE, a urease metallochaperone. J. Inorg. Biochem. 2004, 98, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Lai, T.P.; Sun, H. UreE-UreG complex facilitates nickel transfer and preactivates GTPase of UreG in Helicobacter pylori. J. Biol. Chem. 2015, 290, 12474–12485. [Google Scholar] [CrossRef] [PubMed]
- Grossoehme, N.E.; Mulrooney, S.B.; Hausinger, R.P.; Wilcox, D.E. Thermodynamics of Ni2+, Cu2+, and Zn2+ binding to the urease metallochaperone UreE. Biochemistry 2007, 46, 10506–10516. [Google Scholar] [CrossRef] [PubMed]
- Gasper, R.; Scrima, A.; Wittinghofer, A. Structural insights into HypB, a GTP-binding protein that regulates metal binding. J. Biol. Chem. 2006, 281, 27492–27502. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Merloni, A.; Dobrovolska, O.; Zambelli, B.; Agostini, F.; Bazzani, M.; Musiani, F.; Ciurli, S. Molecular landscape of the interaction between the urease accessory proteins UreE and UreG. Biochim. Biophys. Acta 2014, 1844, 1662–1674. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Sanejouand, Y.H. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, W610–W614. [Google Scholar] [CrossRef] [PubMed]
- Agriesti, F.; Roncarati, D.; Musiani, F.; del Campo, C.; Iurlaro, M.; Sparla, F.; Ciurli, S.; Danielli, A.; Scarlato, V. FeON-FeOFF: The Helicobacter pylori Fur regulator commutates iron-responsive transcription by discriminative readout of opposed DNA grooves. Nucleic Acids Res. 2014, 42, 3138–3151. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janezic, D. ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics 2010, 26, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janezic, D. ProBiS: A web server for detection of structurally similar protein binding sites. Nucleic Acids Res. 2010, 38, W436–W440. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janezic, D. ProBiS-2012: Web server and web services for detection of structurally similar binding sites in proteins. Nucleic Acids Res. 2012, 40, W214–W221. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Hausinger, R.P. Klebsiella aerogenes UreF: Identification of the UreG binding site and role in enhancing the fidelity of urease activation. Biochemistry 2012, 51, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Biagi, F.; Musiani, F.; Ciurli, S. Structure of the UreD-UreF-UreG-UreE complex in Helicobacter pylori: A model study. J. Biol. Inorg. Chem. 2013, 18, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Szasz, C.; Buday, L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005, 30, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Ligabue-Braun, R.; Real-Guerra, R.; Carlini, C.R.; Verli, H. Evidence-based docking of the urease activation complex. J. Biomol. Struct. Dyn. 2013, 31, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Macindoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.D.; Ritchie, D.W. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010, 38, W445–W449. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.W.; Kourinov, I.V.; Andrews, L.C. The Fourier-Green’s function and the rapid evaluation of molecular potentials. Protein. Eng. 1994, 7, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Kemp, G.J.L. Protein docking using spherical polar Fourier correlations. Proteins 2000, 39, 178–194. [Google Scholar] [CrossRef]
- Feig, M.; Karanicolas, J.; Brooks, C.L., 3rd. MMTSB Tool Set: Enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 2004, 22, 377–395. [Google Scholar] [CrossRef] [PubMed]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Sali, A. FoXS: A web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010, 38, W540–W544. [Google Scholar] [CrossRef] [PubMed]
- Quiroz-Valenzuela, S.; Sukuru, S.C.; Hausinger, R.P.; Kuhn, L.A.; Heller, W.T. The structure of urease activation complexes examined by flexibility analysis, mutagenesis, and small-angle X-ray scattering. Arch. Biochem. Biophys. 2008, 480, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.L.; Tronrud, D.E.; Taber, S.R.; Karplus, P.A.; Hausinger, R.P. Iron-containing urease in a pathogenic bacterium. Proc. Natl. Acad. Sci. USA 2011, 108, 13095–13099. [Google Scholar] [CrossRef] [PubMed]
- Delany, I.; Ieva, R.; Soragni, A.; Hilleringmann, M.; Rappuoli, R.; Scarlato, V. In vitro analysis of protein-operator interactions of the NikR and fur metal-responsive regulators of coregulated genes in Helicobacter pylori. J. Bacteriol. 2005, 187, 7703–7715. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Schroll, A.; Sonnweber, T.; Weiss, G. The struggle for iron—A metal at the host-pathogen interface. Cell. Microbiol. 2010, 12, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Coale, K.H.; Johnson, K.S.; Fitzwater, S.E.; Gordon, R.M.; Tanner, S.; Chavez, F.P.; Ferioli, L.; Sakamoto, C.; Rogers, P.; Millero, F.; et al. A massive phytoplankton bloom induced by an ecosystem-scale iron fertilization experiment in the equatorial Pacific Ocean. Nature 1996, 383, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, P.; Wei, Q.; Andrews, S.C.; Vinckx, T. Iron homeostasis and management of oxidative stress response in bacteria. Met. Integr. Biomet. Sci. 2011, 3, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Bagg, A.; Neilands, J.B. Ferric uptake regulation protein acts as a repressor, employing iron (II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 1987, 26, 5471–5477. [Google Scholar] [CrossRef] [PubMed]
- Delany, I.; Spohn, G.; Rappuoli, R.; Scarlato, V. The Fur repressor controls transcription of iron-activated and -repressed genes in Helicobacter pylori. Mol. Microbiol. 2001, 42, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Bereswill, S.; Greiner, S.; van Vliet, A.H.; Waidner, B.; Fassbinder, F.; Schiltz, E.; Kusters, J.G.; Kist, M. Regulation of ferritin-mediated cytoplasmic iron storage by the ferric uptake regulator homolog (Fur) of Helicobacter pylori. J. Bacteriol. 2000, 182, 5948–5953. [Google Scholar] [CrossRef] [PubMed]
- Fillat, M.F. The FUR (ferric uptake regulator) superfamily: Diversity and versatility of key transcriptional regulators. Arch. Biochem. Biophys. 2014, 546, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Dian, C.; Vitale, S.; Leonard, G.A.; Bahlawane, C.; Fauquant, C.; Leduc, D.; Muller, C.; de Reuse, H.; Michaud-Soret, I.; Terradot, L. The structure of the Helicobacter pylori ferric uptake regulator Fur reveals three functional metal binding sites. Mol. Microbiol. 2011, 79, 1260–1275. [Google Scholar] [CrossRef] [PubMed]
- Pohl, E.; Haller, J.C.; Mijovilovich, A.; Meyer-Klaucke, W.; Garman, E.; Vasil, M.L. Architecture of a protein central to iron homeostasis: Crystal structure and spectroscopic analysis of the ferric uptake regulator. Mol. Microbiol. 2003, 47, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Tiss, A.; Barre, O.; Michaud-Soret, I.; Forest, E. Characterization of the DNA-binding site in the ferric uptake regulator protein from Escherichia coli by UV crosslinking and mass spectrometry. FEBS Lett. 2005, 579, 5454–5460. [Google Scholar] [CrossRef] [PubMed]
- Iwig, J.S.; Chivers, P.T. Coordinating intracellular nickel-metal-site structure-function relationships and the NikR and RcnR repressors. Nat. Prod. Rep. 2010, 27, 658–667. [Google Scholar] [CrossRef] [PubMed]
- De Pina, K.; Desjardin, V.; Mandrand-Berthelot, M.A.; Giordano, G.; Wu, L.F. Isolation and characterization of the nikR gene encoding a nickel-responsive regulator in Escherichia coli. J. Bacteriol. 1999, 181, 670–674. [Google Scholar] [PubMed]
- Chivers, P.T.; Sauer, R.T. Regulation of high affinity nickel uptake in bacteria. Ni2+-Dependent interaction of NikR with wild-type and mutant operator sites. J. Biol. Chem. 2000, 275, 19735–19741. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.H.; Ernst, F.D.; Kusters, J.G. NikR-mediated regulation of Helicobacter pylori acid adaptation. Trends Microbiol. 2004, 12, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Musiani, F.; Bertoša, B.; Magistrato, A.; Zambelli, B.; Turano, P.; Losasso, V.; Micheletti, C.; Ciurli, S.; Carloni, P. Computational study of the DNA-binding protein Helicobacter pylori NikR: The role of Ni2+. J. Chem. Theory Comput. 2010, 6, 3503–3515. [Google Scholar] [CrossRef]
- West, A.L.; St. John, F.; Lopes, P.E.M.; MacKerell, A.D.; Pozharski, E.; Michel, S.L.J. Holo-Ni(II) HpNikR Is an asymmetric tetramer containing two different nickel-binding sites. J. Am. Chem. Soc. 2010, 132, 14447–14456. [Google Scholar] [CrossRef] [PubMed]
- Benini, S.; Cianci, M.; Ciurli, S. Holo-Ni2+ Helicobacter pylori NikR contains four square-planar nickel-binding sites at physiological pH. Dalton Trans. 2011, 40, 7831–7833. [Google Scholar] [CrossRef] [PubMed]
- Dian, C.; Schauer, K.; Kapp, U.; McSweeney, S.M.; Labigne, A.; Terradot, L. Structural basis of the nickel response in Helicobacter pylori: Crystal structures of HpNikR in apo and nickel-bound states. J. Mol. Biol. 2006, 361, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Chivers, P.T.; Sauer, R.T. NikR is a ribbon-helix-helix DNA-binding protein. Protein Sci. 1999, 8, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Schreiter, E.R.; Wang, S.C.; Zamble, D.B.; Drennan, C.L. NikR-operator complex structure and the mechanism of repressor activation by metal ions. Proc. Natl. Acad. Sci. USA 2006, 103, 13676–13681. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Danielli, A.; Romagnoli, S.; Neyroz, P.; Ciurli, S.; Scarlato, V. High-affinity Ni2+ binding selectively promotes binding of Helicobacter pylori NikR to its target urease promoter. J. Mol. Biol. 2008, 383, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Bahlawane, C.; Dian, C.; Muller, C.; Round, A.; Fauquant, C.; Schauer, K.; de Reuse, H.; Terradot, L.; Michaud-Soret, I. Structural and mechanistic insights into Helicobacter pylori NikR activation. Nucleic Acids Res. 2010, 38, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- West, A.L.; Evans, S.E.; Gonzalez, J.M.; Carter, L.G.; Tsuruta, H.; Pozharski, E.; Michel, S.L. Ni(II) coordination to mixed sites modulates DNA binding of HpNikR via a long-range effect. Proc. Natl. Acad. Sci. USA 2012, 109, 5633–5638. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.J.; Chivers, P.T.; Baker, N.A. Molecular dynamics simulation of the Escherichia coli NikR protein: Equilibrium conformational fluctuations reveal interdomain allosteric communication pathways. J. Mol. Biol. 2008, 378, 1155–1173. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Merz, K.M., Jr. The intrinsic dynamics and function of nickel-binding regulatory protein: Insights from elastic network analysis. Biophys. J. 2008, 94, 3769–3778. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, L.; Dobrovolska, O.; Musiani, F.; Zambelli, B.; Ciurli, S. On the interaction of Helicobacter pylori NikR, a Ni(II)-responsive transcription factor, with the urease operator: In solution and in silico studies. J. Biol. Inorg. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Jubb, H.; Higueruelo, A.P.; Winter, A.; Blundell, T.L. Structural biology and drug discovery for protein-protein interactions. Trends Pharmacol. Sci. 2012, 33, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Bonvin, A.M. Protein-protein interactions. Curr. Opin. Struct. Biol. 2013, 23, 859–861. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Schueler-Furman, O. Druggable protein-protein interactions—From hot spots to hot segments. Curr. Opin. Chem. Biol. 2013, 17, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [PubMed]
- Aeluri, M.; Chamakuri, S.; Dasari, B.; Guduru, S.K.; Jimmidi, R.; Jogula, S.; Arya, P. Small molecule modulators of protein-protein interactions: Selected case studies. Chem. Rev. 2014, 114, 4640–4694. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Blundell, T.L.; Burke, D.F.; Chirgadze, D.; Dhanaraj, V.; Hyvonen, M.; Innis, C.A.; Parisini, E.; Pellegrini, L.; Sayed, M.; Sibanda, B.L. Protein-protein interactions in receptor activation and intracellular signalling. Biol. Chem. 2000, 381, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Koehler, A.N. A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol. 2010, 14, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- Berg, T. Inhibition of transcription factors with small organic molecules. Curr. Opin. Chem. Biol. 2008, 12, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Blundell, T.L.; Sibanda, B.L.; Montalvao, R.W.; Brewerton, S.; Chelliah, V.; Worth, C.L.; Harmer, N.J.; Davies, O.; Burke, D. Structural biology and bioinformatics in drug design: Opportunities and challenges for target identification and lead discovery. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Hamilton, A.D. Protein surface recognition and proteomimetics: Mimics of protein surface structure and function. Curr. Opin. Chem. Biol. 2005, 9, 632–638. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musiani, F.; Ciurli, S. Evolution of Macromolecular Docking Techniques: The Case Study of Nickel and Iron Metabolism in Pathogenic Bacteria. Molecules 2015, 20, 14265-14292. https://doi.org/10.3390/molecules200814265
Musiani F, Ciurli S. Evolution of Macromolecular Docking Techniques: The Case Study of Nickel and Iron Metabolism in Pathogenic Bacteria. Molecules. 2015; 20(8):14265-14292. https://doi.org/10.3390/molecules200814265
Chicago/Turabian StyleMusiani, Francesco, and Stefano Ciurli. 2015. "Evolution of Macromolecular Docking Techniques: The Case Study of Nickel and Iron Metabolism in Pathogenic Bacteria" Molecules 20, no. 8: 14265-14292. https://doi.org/10.3390/molecules200814265
APA StyleMusiani, F., & Ciurli, S. (2015). Evolution of Macromolecular Docking Techniques: The Case Study of Nickel and Iron Metabolism in Pathogenic Bacteria. Molecules, 20(8), 14265-14292. https://doi.org/10.3390/molecules200814265