
Stable Hemiaminals: 2-Aminopyrimidine Derivatives

Abstract
:
1. Introduction


2. Results and Discussion
General Remarks


{kind=link}
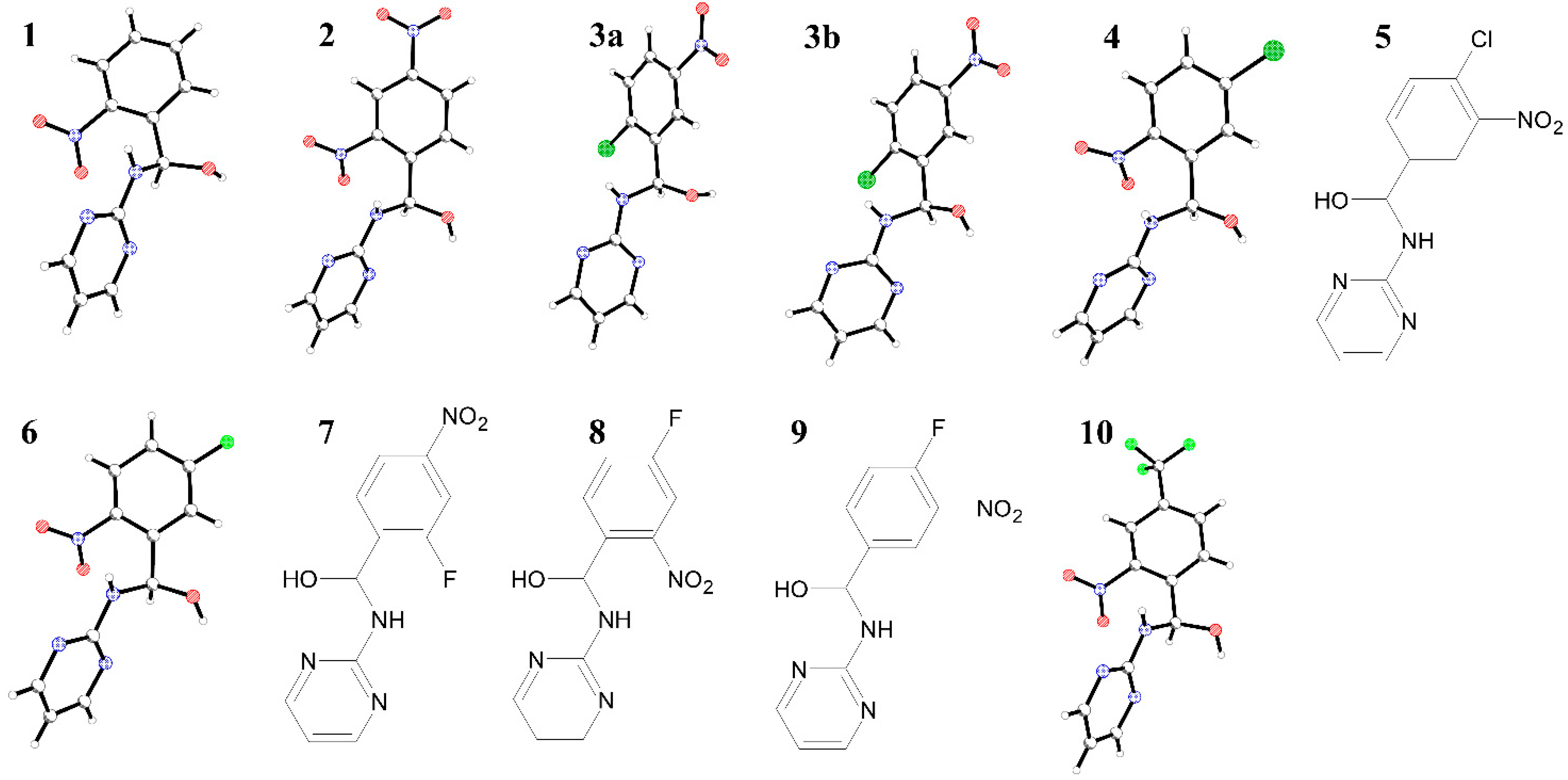{kind=link}
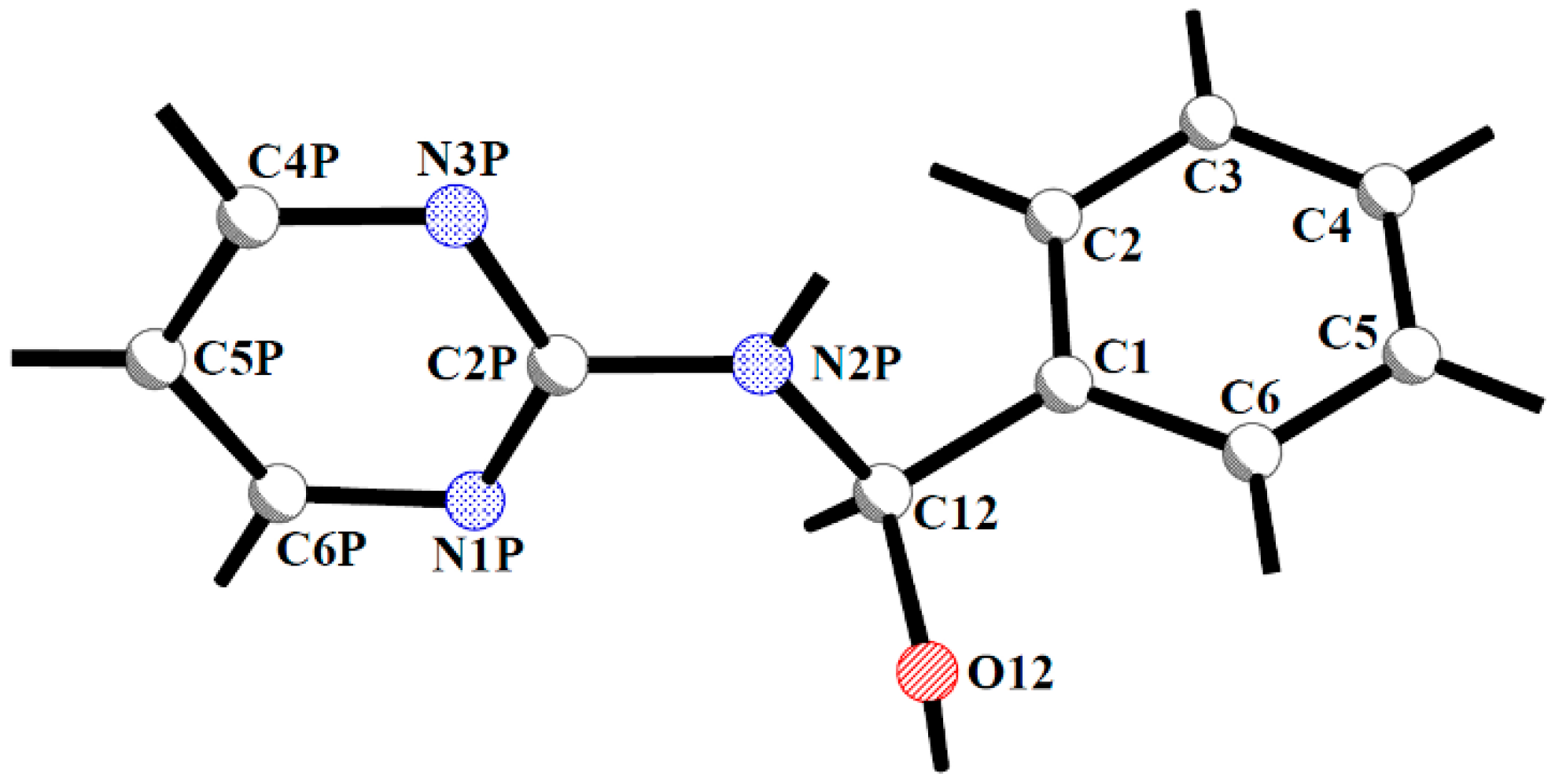{kind=link}
{kind=link}
{kind=link}
| Compound | Geometrical Parameter | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bond Lengths [Å] | Valence Angles [°] | Torsion Angles [°] | Phenyl-Pyrimidine Dihedral Angle [°] | ||||||
| C1-C12 | C12-N2P | N2P-C2P | C12-O12 | C1-C12-N2P | C12-N2P-C2P | C2P-N2P-C12-C1 | C2P-N2P-C12-O12 | ||
| 1 | - | ||||||||
| molecule 1 | 1.537(2) | 1.448(2) | 1.358(2) | 1.424(2) | 108.7(2) | 124.3(2) | −155.4(2) | 83.1(2) | 71.7(1 ) |
| molecule 2 | 1.527(2) | 1.430(2) | 1.356(2) | 1.412(2) | 108.4(2) | 123.8(2) | 145.8(2) | −91.5(2) | 68.9(1) |
| 2 | 1.525(2) | 1.444(2) | 1.369(2) | 1.409(2) | 106.8(1) | 121.8(1) | 157.2(1) | −82.8(1) | 67.4(1) |
| 3a | |||||||||
| molecule 1 | 1.534(3) | 1.442(2) | 1.362(2) | 1.413(2) | 108.9(2) | 121.5(2) | 173.1(2) | −66.3(2) | 88.5(1) |
| molecule 2 | 1.523(3) | 1.444(2) | 1.362(2) | 1.414(2) | 108.4(1) | 122.7(2) | 158.6(2) | −81.7(2) | 85.2(1) |
| 3b | 1.521(3) | 1.440(3) | 1.347(3) | 1.406(3) | 109.4(2) | 125.0(1) | 152.9(2) | −87.7(3) | 68.2(1) |
| 4 | 1.523(2) | 1.445(2) | 1.368(2) | 1.414(2) | 108.5(1) | 121.9(1) | 150.7(1) | −88.4(1) | 74.3(1) |
| 6 | 1.524(3) | 1.440(3) | 1.375(3) | 1.426(3) | 108.5(2) | 122.6(2) | 154.6(2) | −85.0(3) | 69.5(1) |
| 10 | 1.520(1) | 1.448(1) | 1.370(1) | 1.409(1) | 107.9(1) | 123.2(2) | 151.7(1) | −87.7(1) | 73.2(1) |

3. Experimental Section
3.1. General Information
3.2. Crystallography
3.3. Synthetic Procedures
General Procedure for the Synthesis of Hemiaminals
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Smith, J.M.B.; March, J. March’s Advanced Organic Chemistry, 6th ed.; Wiley Interscience: Hoboken, NJ, USA, 2007; pp. 1281–1284. [Google Scholar]
- Hooley, R.J.; Iwasawa, T.; Rebek, J., Jr. Detection of reactive tetrahedral intermediates in a deep cavitand with an introverted functionality. J. Am. Chem. Soc. 2007, 129, 15330–15339. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, T.; Hooley, R.J.; Rebek, J., Jr. Stabilization of Labile Carbonyl Addition Intermediates by a Synthetic Receptor. Science 2007, 317, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hua, S.; Li, S. Insight into the reaction between a primary amine and a cavitand with an introverted aldehyde group: an enzyme-like mechanism. Chem. Commun. 2013, 49, 1542–1544. [Google Scholar] [CrossRef] [PubMed]
- Kawamichi, T.; Haneda, T.; Kawano, M.; Fujita, M. X-ray observation of a transient hemiaminal trapped in a porous network. Nature 2009, 461, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Morris, W.; Doonan, C.J.; Yaghi, O.M. Postsynthetic modification of a metal–organic framework for stabilization of a hemiaminal and ammonia uptake. Inorg. Chem. 2011, 50, 6853–6855. [Google Scholar] [CrossRef] [PubMed]
- Dolotko, O.; Wiench, J.W.; Dennis, K.W.; Pecharsky, V.K.; Balema, V.P. Mechanically induced reactions in organic solids: Liquid eutectics or solid-state processes? New J. Chem. 2010, 34, 25–28. [Google Scholar] [CrossRef]
- García, J.M.; Jones, G.O.; Virwani, K.; McCloskey, B.D.; Boday, D.J.; ter Huurne, G.M.; Horn, H.W.; Coady, D.J.; Bintaleb, A.M.; Alabdulrahman, A.M.S.; et al. Recyclable, Strong Thermosets and Organogels via Paraformaldehyde Condensation with Diamines. Science 2014, 344, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.H.; ter Hurrne, G.M.; Wojtecki, R.J.; Jones, G.O.; Horn, H.W.; Meijer, E.W.; Frank, C.W.; Hedrick, J.L.; García, J.M. Supramolecular motifs in dynamic covalent PEG-hemiaminal organogels. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.O.; García, J.M.; Horn, H.W.; Hedrick, J.L. Computational and Experimental Studies on the Mechanism of Formation of Poly(hexahydrotriazine)s and Poly(hemiaminal)s from the Reactions of Amines with Formaldehyde. Org. Lett. 2014, 16, 5502–5505. [Google Scholar] [CrossRef] [PubMed]
- Baymak, M.S.; Zuman, P. Equilibria of formation and dehydration of the carbinolamine intermediate in the reaction of benzaldehyde with hydrazine. Tetrahedron 2007, 63, 5450–5454. [Google Scholar] [CrossRef]
- Namli, H.; Turhan, O. Simultaneous observation of reagent consumption and product formation with the kinetics of benzaldehyde and aniline reaction in FTIR liquid cell. Vib. Spectrosc. 2007, 43, 274–283. [Google Scholar] [CrossRef]
- Evans, D.A.; Borg, G.; Scheidt, K.A. Remarkably stable tetrahedral intermediates: Carbinols from nucleophilic additions to N-Acylpyrroles. Angew. Chem. Int. Ed. 2002, 41, 3188–3191. [Google Scholar] [CrossRef]
- Yufit, D.S.; Howard, J.A.K. Structure of hemiaminal intermediate of the reaction of diethylamine with cyclobutanone. J. Mol. Struct. 2010, 984, 182–185. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Pellarin, K.R.; Puddephatt, R.J. Oxidation of a Dimethylplatinum(II) Complex with Oxaziridines: A Hemiaminal Intermediate but No Oxo Complex. Organometallics 2013, 32, 3604–3610. [Google Scholar] [CrossRef]
- Suni, V.; Kurup, M.R.P.; Nethaji, M. Unusual isolation of a hemiaminal product from 4-cyclohexyl-3-thiosemicarbazide and di-2-pyridyl ketone: Structural and spectral investigations. J. Mol. Struct. 2005, 749, 177–182. [Google Scholar] [CrossRef]
- Lee, S.K.; Park, J.K. Room-Temperature Transition-Metal-Free One-Pot Synthesis of 3-Aryl Imidazo[1,2-a]pyridines via Iodo-hemiaminal Intermediate. J. Org. Chem. 2015, 80, 3723–3729. [Google Scholar] [CrossRef] [PubMed]
- Barys, M.; Ciunik, Z.; Drabent, K.; Kwiecień, A. Stable hemiaminals containing a triazole ring. New J. Chem. 2010, 34, 2605–2611. [Google Scholar] [CrossRef]
- Kwiecień, A.; Barys, M.; Ciunik, Z. Stable Hemiaminals with a Cyano Group and a Triazole Ring. Molecules 2014, 19, 11160–11177. [Google Scholar] [CrossRef] [PubMed]
- Ciunik, Z.; Drabent, K.; Szterenberg, L. Molecular conformation versus C–H···Ph weak hydrogen bonds in 4-(4-H-1,2,4-triazol-4-yl)-2-X-phenylmethanimine (X = CH3, Cl, Br) crystals. J. Mol. Struct. 2002, 641, 175–182. [Google Scholar] [CrossRef]
- Ciunik, Z.; Drabent, K. The C-H··· [FBF3]− hydrogen bonds as origin of the linear polytetrameric self-organisation of Schiff base containing substituted 1,2,4-triazole. Pol. J. Chem. 2001, 75, 1475–1482. [Google Scholar]
- Berski, S.; Ciunik, Z.; Drabent, K.; Latajka, Z.; Panek, J. Dominant role of C-Br···N halogen bond in molecular self-organization. Crystallographic and quantum-chemical study of Schiff-base-containing triazoles. J. Phys. Chem. B 2004, 108, 12327–12332. [Google Scholar] [CrossRef]
- Kitaev, Y.P.; Savin, V.I.; Zverev, V.V.; Popova, G.V. Structures and reactivities of nitrogen-containing derivatives of carbonyl compounds. Chem. Heterocycl. Compd. 1971, 7, 522–526, (translated from Khim. Geterotsikl. Soedin. 1971, 7, 559–564. [Google Scholar] [CrossRef]
- Barys, M.; Ciunik, Z. N-[(E)-(2,4-Dinitrophenyl)methylene]aniline. Acta Crystallogr., Sect. E: Struct. Rep. Online 2005, 61, o1967–o1969. [Google Scholar] [CrossRef]
- Wilson, A.J.C. (Ed.) International Tables for Crystallography Volume C: Mathematical, Physical and Chemical Tables; Kluwer Academic: Dordrecht, The Netherlands; Boston, MA, USA, London, UK, 1992; pp. 707–791.
- Cosier, J.; Glazer, A.M. A nitrogen-gas-stream cryostat for general X-ray diffraction studies. J. Appl. Cryst. 1986, 19, 105–107. [Google Scholar] [CrossRef]
- CrysAlis CCD, CrysAlis RED, CrysAlisPRO; Oxford Diffraction/Agilent Technologies UK Ltd: Yarnton, UK, 2008/2011/2013.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- XP—INTERACTIVE MOLECULAR GRAPHICS, v. 5.1, Bruker Analytical X-ray System. 1998.
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 28 July 2015).
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwiecień, A.; Ciunik, Z. Stable Hemiaminals: 2-Aminopyrimidine Derivatives. Molecules 2015, 20, 14365-14376. https://doi.org/10.3390/molecules200814365
Kwiecień A, Ciunik Z. Stable Hemiaminals: 2-Aminopyrimidine Derivatives. Molecules. 2015; 20(8):14365-14376. https://doi.org/10.3390/molecules200814365
Chicago/Turabian StyleKwiecień, Anna, and Zbigniew Ciunik. 2015. "Stable Hemiaminals: 2-Aminopyrimidine Derivatives" Molecules 20, no. 8: 14365-14376. https://doi.org/10.3390/molecules200814365
APA StyleKwiecień, A., & Ciunik, Z. (2015). Stable Hemiaminals: 2-Aminopyrimidine Derivatives. Molecules, 20(8), 14365-14376. https://doi.org/10.3390/molecules200814365