
Influence of Solid Drug Delivery System Formulation on Poorly Water-Soluble Drug Dissolution and Permeability

Abstract
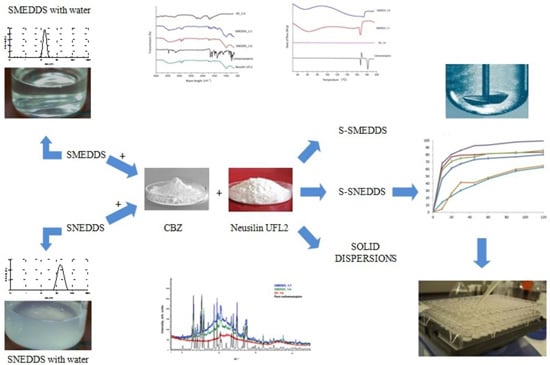
:
1. Introduction
2. Results and Discussion
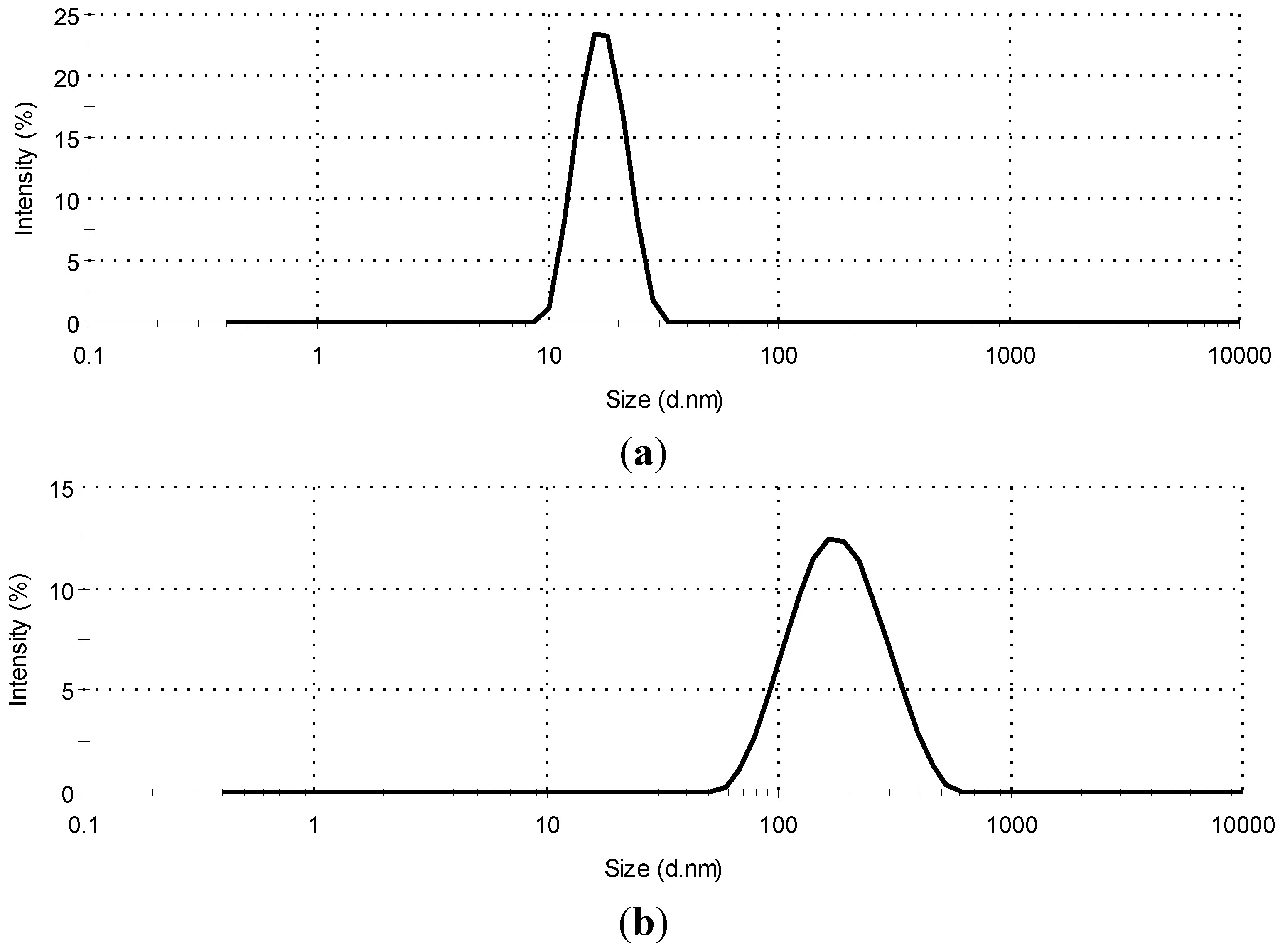
2.1. Droplet Size Analysis

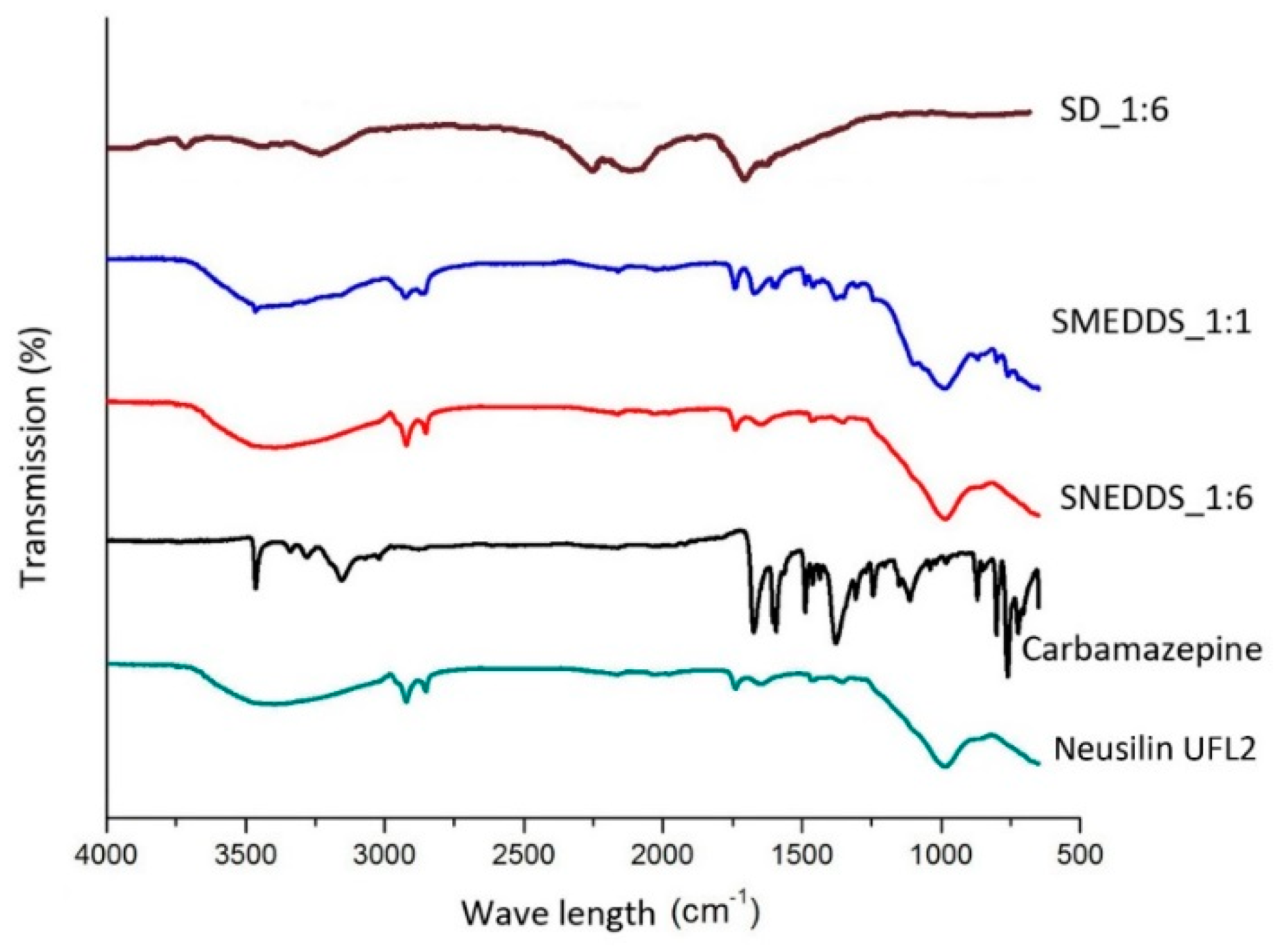
2.2. Fourier-Transform Infrared Spectroscopy (FT-IR)

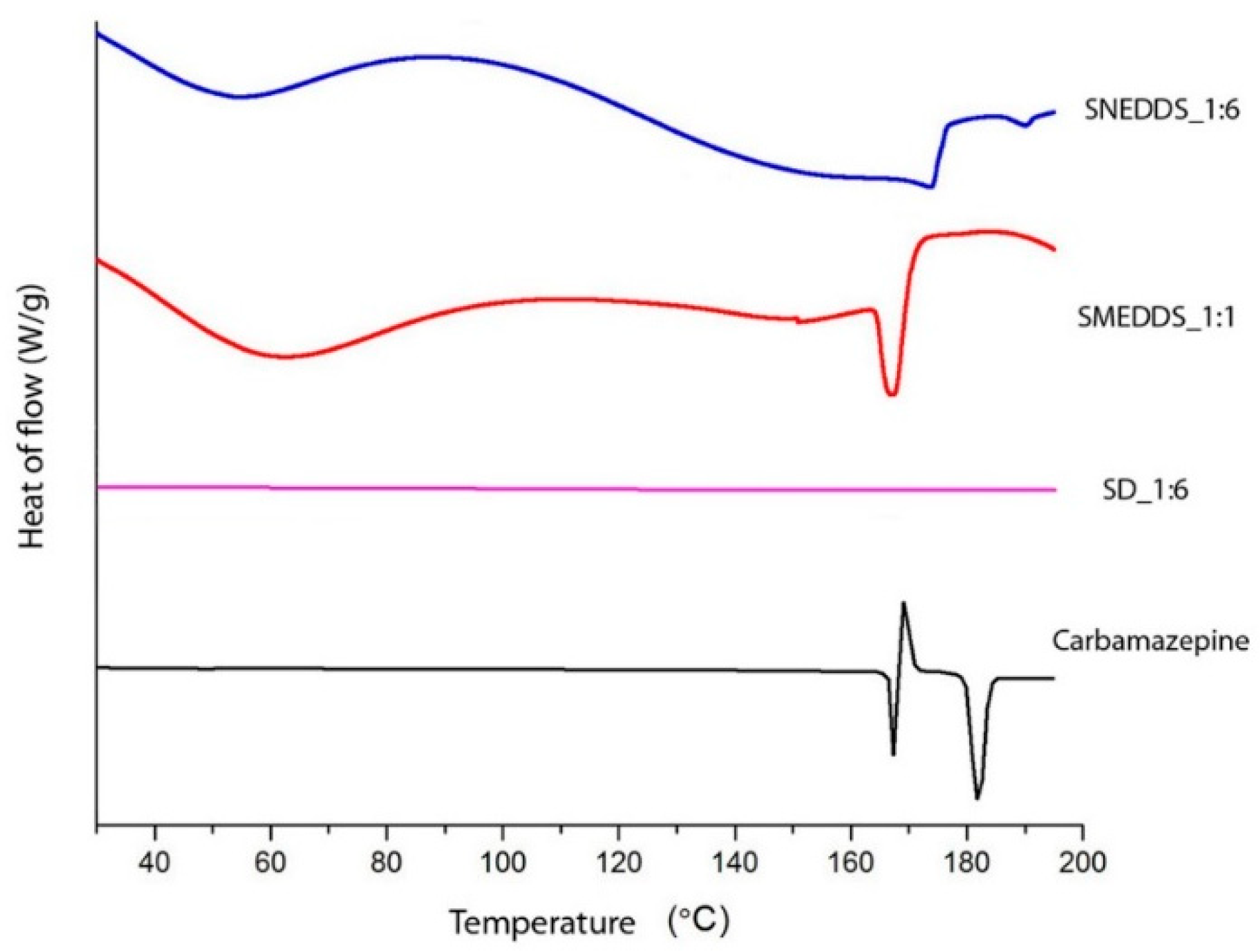
2.3. Differential Scanning Calorimetry (DSC)

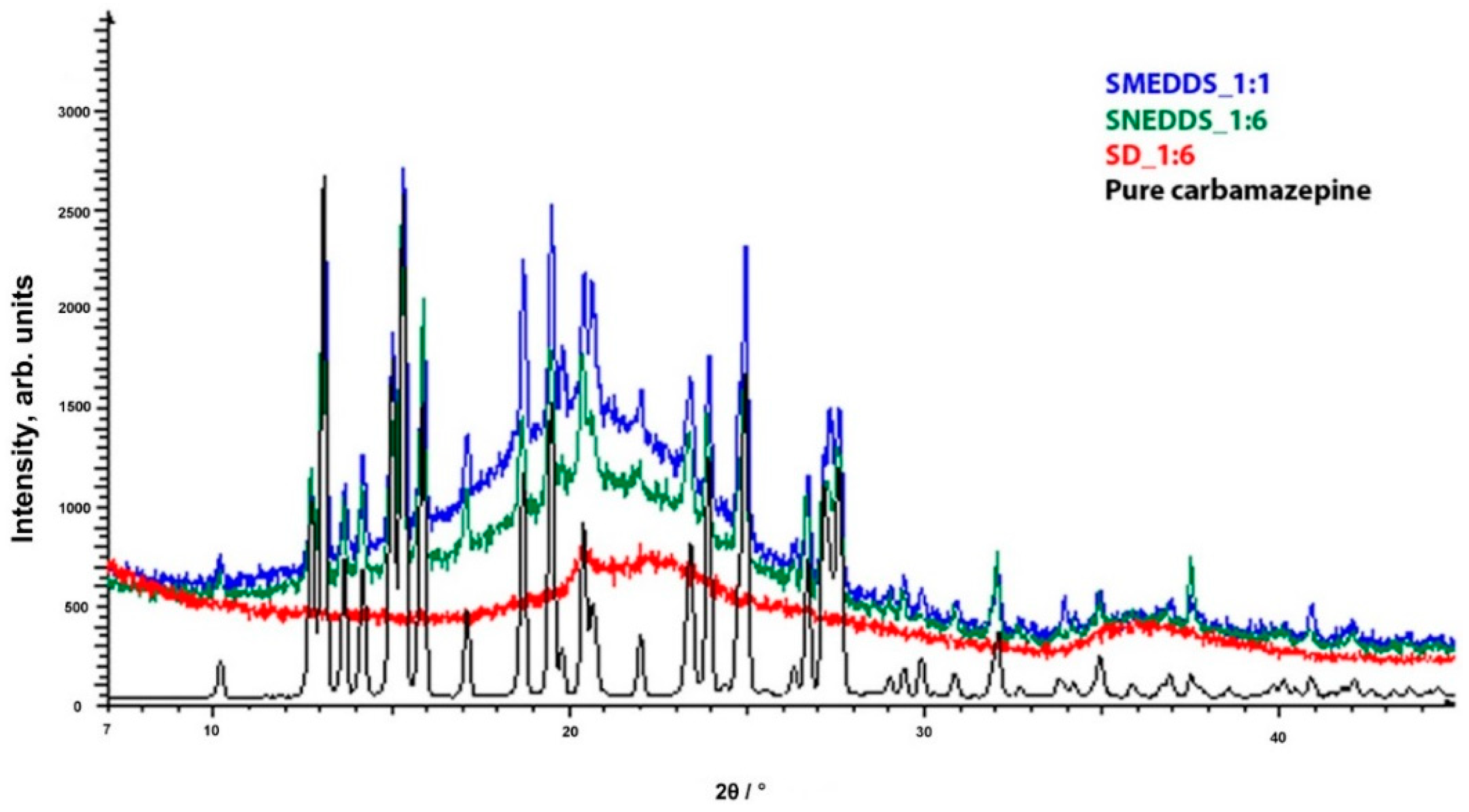
2.4. Powder X-ray Diffraction Analysis (PXRD)

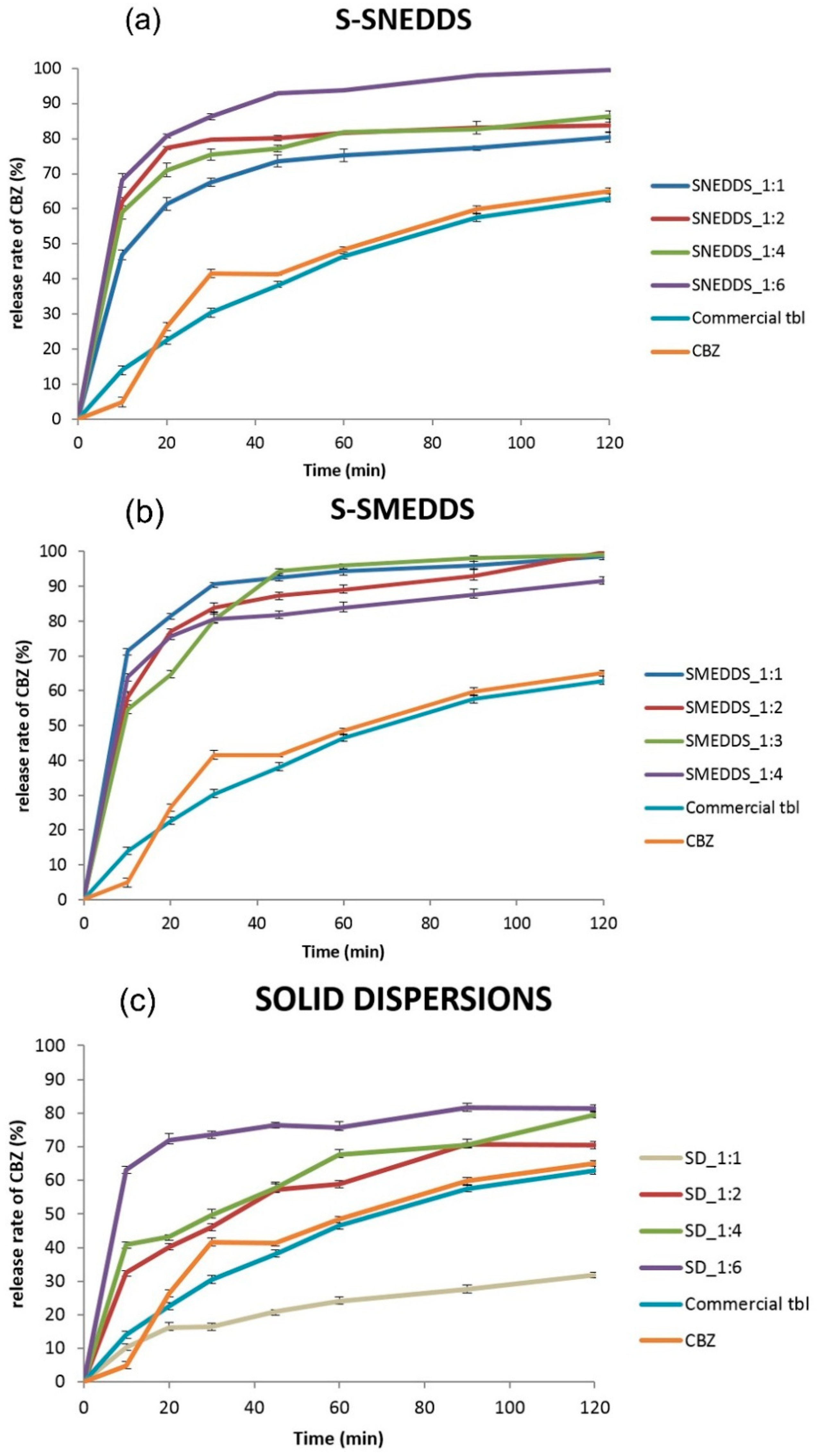
2.5. In Vitro Drug Release Profile


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pure Carbamazepine | Commercial Tablets | ||
|---|---|---|---|
| SMEDDS_1:1 | f1 | 62.17 | 56.50 |
| f2 | 18.49 | 15.95 | |
| SNEDDS_1:6 | f1 | 55.33 | 49.05 |
| f2 | 24.40 | 22.17 | |
| SD_1:6 | f1 | 54.86 | 44.02 |
| f2 | 24.41 | 25.62 | |
2.6. Parallel Artificial Membrane Permeability Assay (PAMPA)
| Formulations | Papp × 10−6 (cm/s) |
|---|---|
| Carbamazepine | 11.77 ± 0.34 |
| SD_1:6 | 16.12 ± 2.27 |
| SNEDDS_1:6 | 19.12 ± 0.87 |
| SMEDDS_1:1 | 21.42 ± 1.67 |
3. Experimental Section
3.1. Materials
3.2. Methods
3.2.1. Solid Dispersions
3.2.2. Solid Self-Microemulsifying Drug Delivery Systems (S-SMEDDS)
3.2.3. Solid Self-Nanoemulsifying Drug Delivery Systems (S-SNEDDS)
3.3. Photon Correlation Spectroscopy (PCS)
3.4. In Vitro Drug Release Studies
3.5. Characterization of Selected Formulations
3.5.1. Fourier-Transform Infrared Spectroscopy (FT-IR)
3.5.2. Differential Scanning Calorimetry (DSC)
3.5.3. Powder X-ray Diffraction Analysis (PXRD)
3.6. PAMPA Test
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neduri, K.; Bontha, V.K.; Vemula, S.K. Different techniques to enhance the dissolution rate of lovastatin: Formulation and evaluation. Asian J. Pharm. Clin. Res. 2013, 6, 56–60. [Google Scholar]
- Nesamony, J.; Karla, A.; Majrad, M.S.; Boddu, S.H.S.; Jung, R.; Williams, F.E.; Schnapp, A.M.; Nauli, S.M.; Kalinoski, A.L. Development and characterization of nanostructured mists with potential for actively targeting poorly water-soluble compounds into the lungs. Pharm. Res. 2013, 30, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, B.; Quan, G.; Li, F.; Wu, Q.; Dian, L.; Dong, Y.; Li, G.; Wu, C. Increasing the oral bioavailability of poorly water-soluble carbamazepine using immediate-release pellets supported on SBA-15 mesoporous silica. Int. J. Nanomed. 2012, 7, 5807–5818. [Google Scholar]
- Löbenberg, R.; Amidon, G.L. Modern bioavailability, bioequivalence and biopharmaceutics classifcation system. New scientifc approaches to international regulatory standards. Eur. J. Pharm. Biopharm. 2000, 50, 3–12. [Google Scholar] [CrossRef]
- Mohanachandran, P.S.; Sindhumol, P.G.; Kiran, T.S. Enhancement of solubility and dissolution rate: An overview. Pharm. Glob. IJCP 2010, 4, 1–10. [Google Scholar]
- Raghavendra Rao, N.G.; Upendra, K. Development of carbamazepine fast dissolving tablets: Effect of functionality of hydrophilic carriers on solid dispersion technique. Asian J. Pharm. Clin. Res. 2010, 3, 114–117. [Google Scholar]
- Rane, Y.; Mashru, R.; Sankalia, M.; Sankalia, J. Effect of hydrophilic swellable polymers on dissolution enhancement of carbamazepine solid dispersions studied using response surface methodology. AAPS PharmSciTech 2007, 8, E1–E11. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, N.; Okamoto, H.; Danjo, K. Lactose as a low molecular weight carrier of solid dispersions for carbamazepine and ethenzamide. Chem. Pharm. Bull. 1999, 47, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Agarwal, S.P.; Iqbal, Z. Effect of fulvic acid on oral delivery of carbamazepine. Sci. Adv. Mater. 2011, 3, 1–10. [Google Scholar] [CrossRef]
- Nisha, G.S.; Vaishali, P.; Geeta, R.; Prabhakar, P.; Harish, N.M.; Marina, K.; Charayulu, R.N. Formulation and evaluation of self microemulsifying drug delivery system of carbamazepine. Int. J. Res. Pharm. Sci. 2011, 2, 162–169. [Google Scholar]
- Kokare, C.R.; Kumbhar, S.A.; Patil, A. Formulation and evaluation of self-emulsifying drug delivery system of carbamazepine. Ind. J. Pharm. Educ. Res. 2013, 47, 172–177. [Google Scholar]
- Nan, Z.; Lijun, G.; Tao, W.; Dongqin, Q. Evaluation of carbamazepine (CBZ) supersaturable self-microemulsifying (S-SMEDDS) formulation in vitro and in vivo. Iran. J. Pharm. Res. 2012, 11, 257–264. [Google Scholar] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Basalious, E.B.; Shawky, N.; Badr-Eldin, S.M. SNEDDS containing bioenchancers for improvement of dissolution and oral absorption of lacidipine. I: Development and optimization. Int. J. Pharm. 2010, 391, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Katteboina, S.; Chandrasekhar, P.; Balaji, S. Approaches for the development of solid self-emulsifying drug delivery systems and dosage forms. Asian J. Pharm. Sci. 2009, 4, 240–253. [Google Scholar]
- Ingle, L.M.; Wankhade, V.P.; Udasi, T.A.; Tapar, K.K. New approaches for development and characterization of SMEDDS. Int. J. Pharm. Pharm. Sci. Res. 2013, 3, 7–14. [Google Scholar]
- Krstic, M.; Razic, S.; Djekic, L.; Dobricic, V.; Momcilovic, M.; Vasiljevic, D.; Ibric, S. Application of a mixture experimental design in the optimization of the formulation of solid self-emulsifying drug delivery systems containing carbamazepine. Lat. Am. J. Pharm. 2015, 34, 885–894. [Google Scholar]
- Dahan, A.; Miller, J.M. The solubility-permeability interplay and its implications in formulation design and development for poorly soluble drugs. AAPS J. 2012, 14, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Gibaud, S.; Attivi, D. Microemulsions for oral administration and their therapeutic applications. Expert Opin. Drug Deliv. 2012, 9, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.; Santos, H.A. Improving oral absorption via drug-loaded nanocarriers: Absorption mechanisms, intestinal models and rational fabrication. Curr. Drug Metab. 2013, 14, 28–56. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Jiang, L.; Chen, T.M.; Hwang, K.K. A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur. J. Med. Chem. 2002, 37, 399–407. [Google Scholar] [CrossRef]
- Shakeel, F.; Baboota, S.; Ahuja, A.; Ali, J.; Aqil, M.; Shafiq, S. Nanoemulsions as vehicles for transdermal delivery of aceclofenac. AAPS PharmSciTech 2007, 8, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Shivakumar, H.N.; Kumar, K. Effect of β cyclodextrin complexation on the solubility and dissolution rate of carbamazepine from tablets. Indian J. Pharm. Sci. 2006, 68, 301–307. [Google Scholar]
- Grzesiak, A.L.; Lang, M.; Kim, K.; Matzeger, A.J. Comparison of the Four Anhydrous Polymorphs of Carbamazepine and the Crystal Structure of Form. Int. J. Pharm. Sci. 2003, 92, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Rustichelli, C.; Gamberini, G.; Ferioli, V.; Gamberini, M.C.; Ficarra, R.; Tommasini, S. Solid-state study of polymorphic drugs: Carbamazepine. J. Pharm. Biomed. Anal. 2000, 23, 41–54. [Google Scholar] [CrossRef]
- Emami, J. In vitro-in vivo correlation: From theory to applications. J. Pharm. Pharm. Sci. 2006, 9, 169–189. [Google Scholar] [PubMed]
- Milović, M.; Djuriš, J.; Djekić, L.; Vasiljević, D.; Ibrić, S. Characterization and evaluation of solid self-microemulsifying drug delivery systems with porous carriers as systems for improved carbamazepine release. Int. J. Pharm. 2012, 436, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Siddiqui, A.; Ali, A.; Nazzal, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEEDS). Int. J. Pharm. 2009, 366, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Beig, A.; Carr, R.A.; Spence, J.K.; Dahan, A. A win-win solution in oral delivery of lipophilic drugs: Supersaturation via amorphous solid dispersions increases apparent solubility without sacrifice of intestinal membrane permeability. Mol. Pharm. 2012, 9, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, S.; Alayoubi, A.; Nazzal, S.; Sylvester, P.W.; Kaddoumi, A. Enhanced solubility and oral bioavailability of γ-tocotrienol using a self-emulsifying drug delivery system (SEEDS). Lipids 2014, 49, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Djekic, L.; Primorac, M.; Jockovic, J. Phase behavior, microstructure and ibuprofen solubilization capacity of pseudo-ternary nonionic microemulsions. J. Mol. Liq. 2011, 160, 81–87. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krstić, M.; Popović, M.; Dobričić, V.; Ibrić, S. Influence of Solid Drug Delivery System Formulation on Poorly Water-Soluble Drug Dissolution and Permeability. Molecules 2015, 20, 14684-14698. https://doi.org/10.3390/molecules200814684
Krstić M, Popović M, Dobričić V, Ibrić S. Influence of Solid Drug Delivery System Formulation on Poorly Water-Soluble Drug Dissolution and Permeability. Molecules. 2015; 20(8):14684-14698. https://doi.org/10.3390/molecules200814684
Chicago/Turabian StyleKrstić, Marko, Miljana Popović, Vladimir Dobričić, and Svetlana Ibrić. 2015. "Influence of Solid Drug Delivery System Formulation on Poorly Water-Soluble Drug Dissolution and Permeability" Molecules 20, no. 8: 14684-14698. https://doi.org/10.3390/molecules200814684