
Ascorbic Acid-Initiated Tandem Radical Cyclization of N-Arylacrylamides to Give 3,3-Disubstituted Oxindoles

Abstract
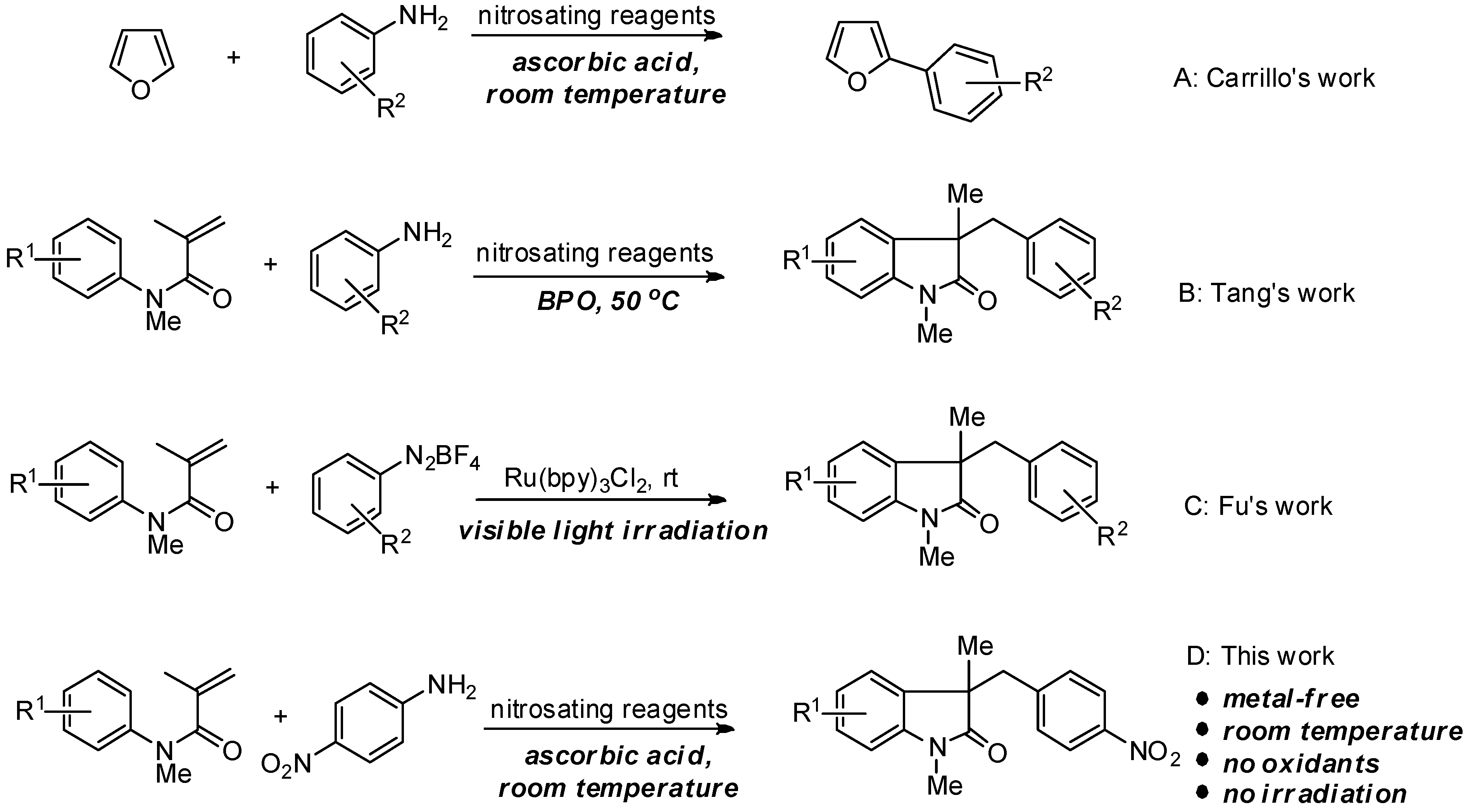
:1. Introduction

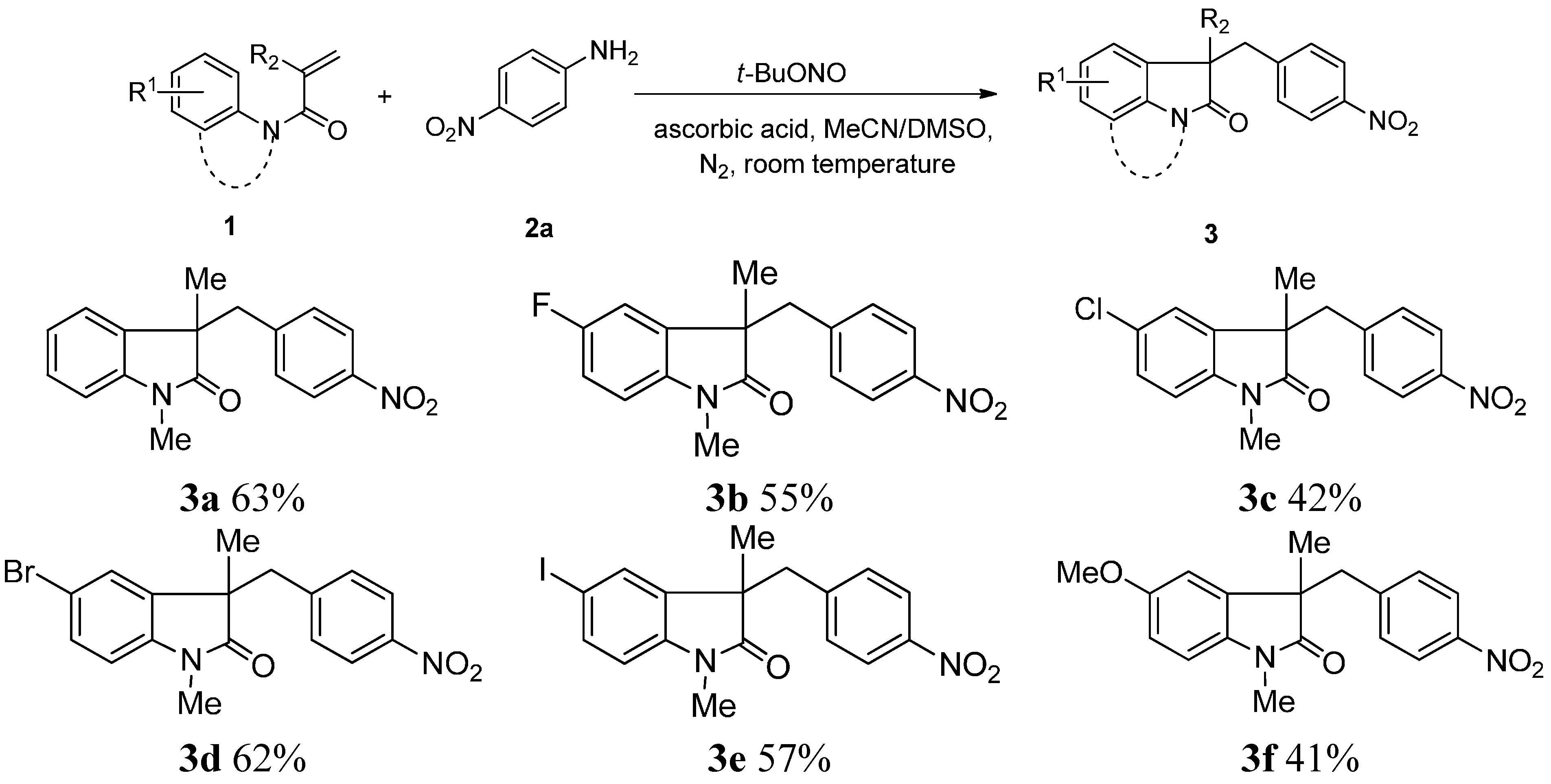
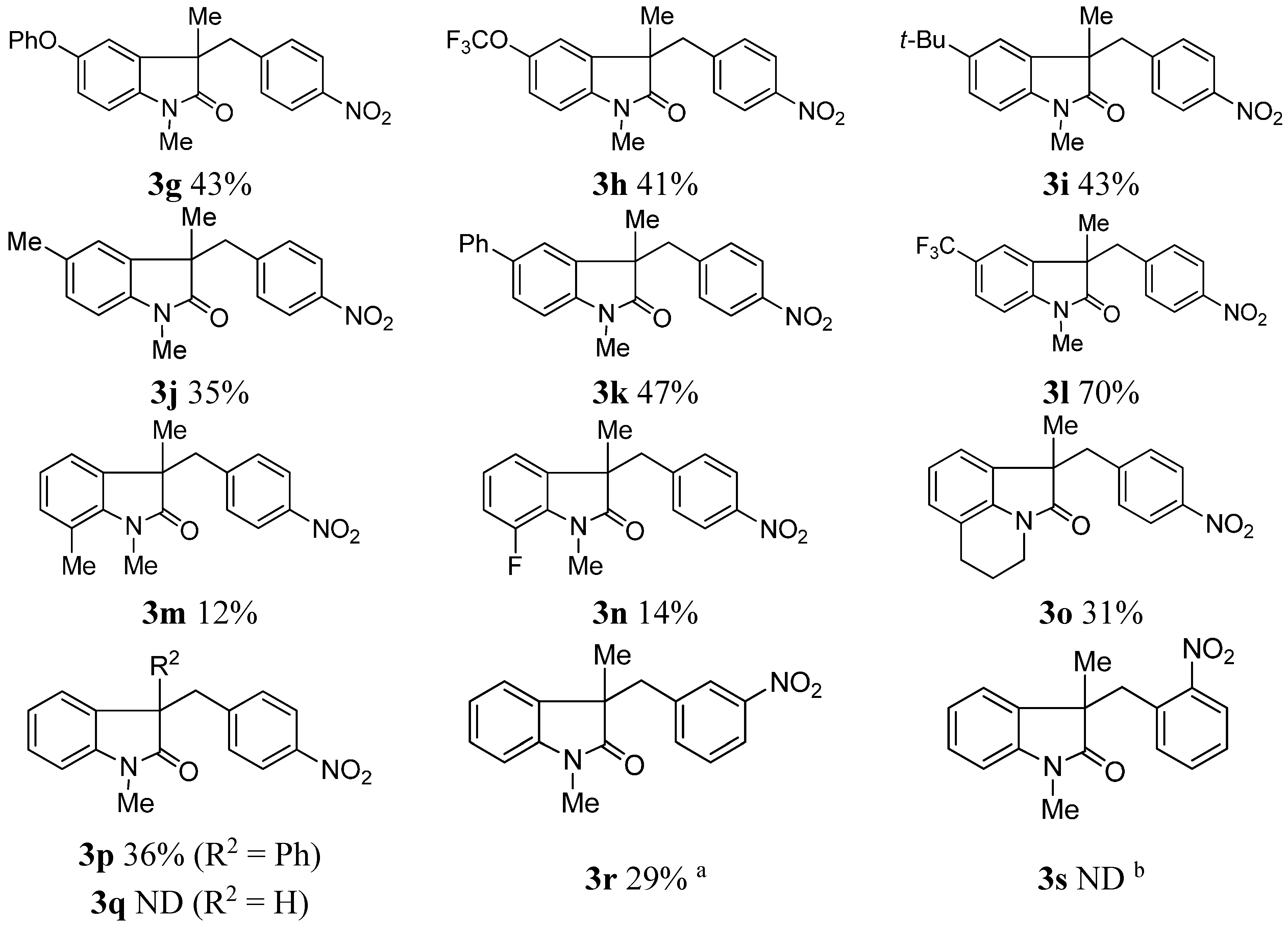
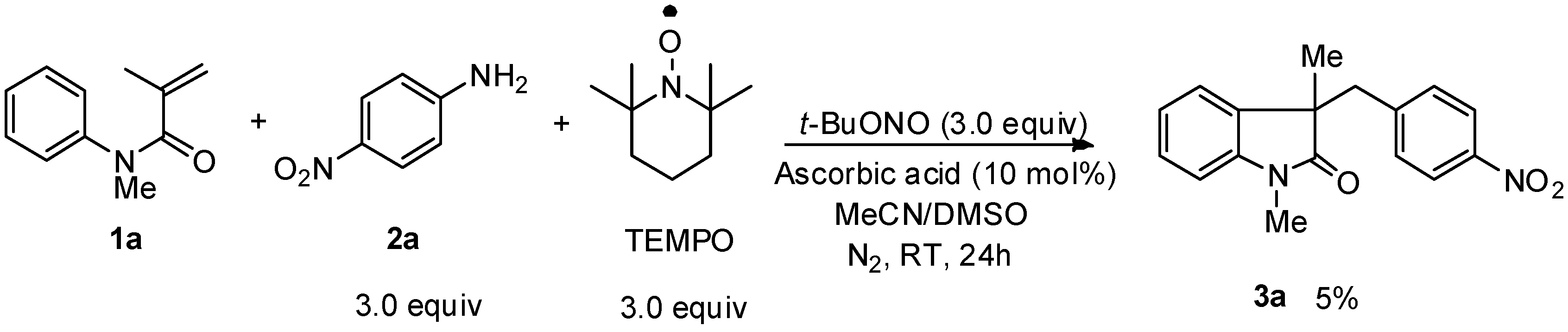
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
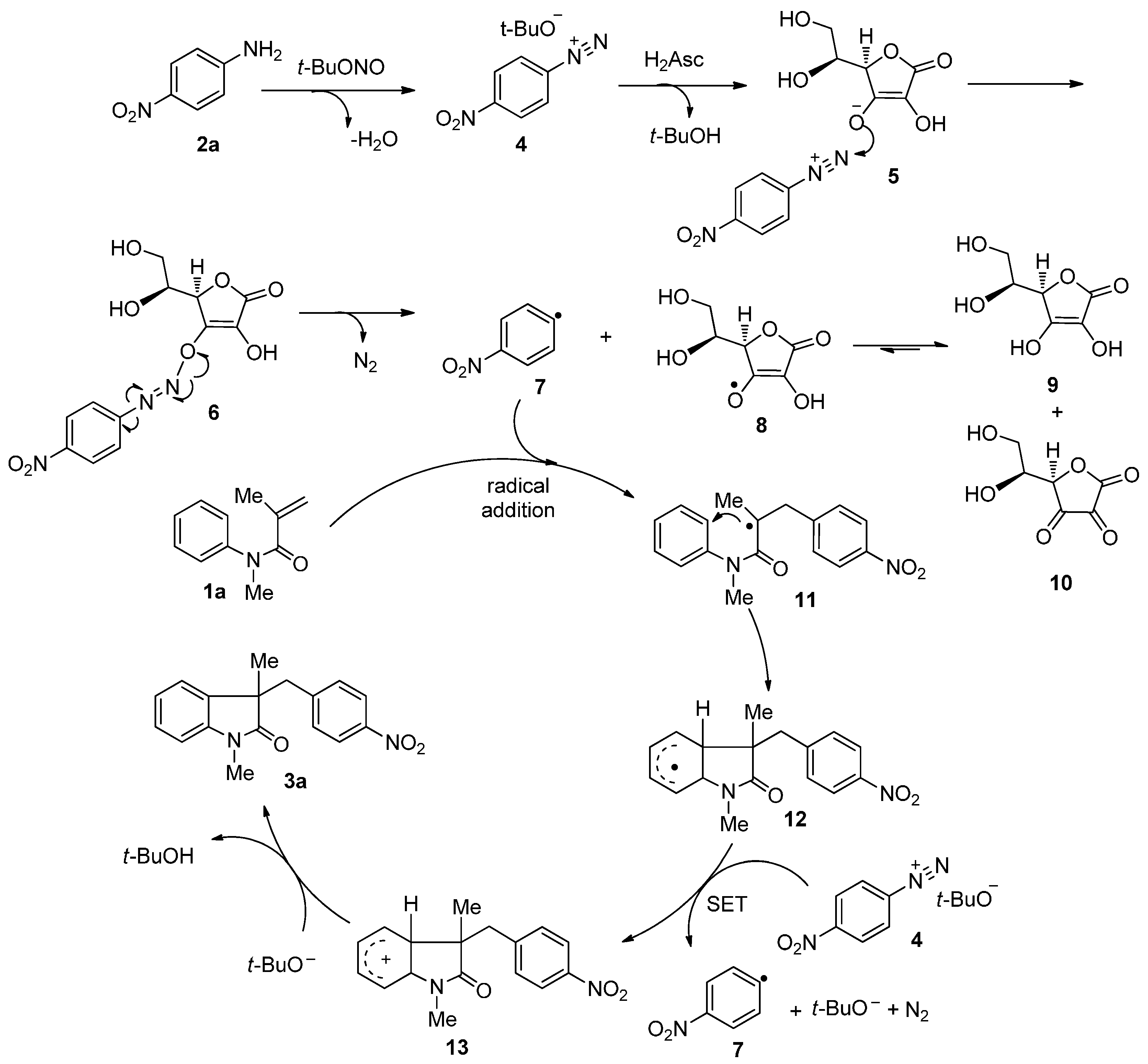{kind=link}
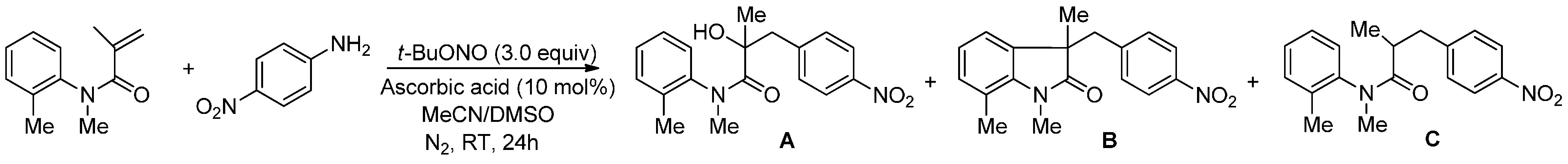{kind=link}
| Entry | Initiator 10 mol % | Nitrosating Reagent | 1a/2a/Nitrosating Reagent | Solvent | Yields% b |
|---|---|---|---|---|---|
| 1 | - | t-BuONO | 1:1.5:1.5 | MeCN | 21 |
| 2 | - | i-AmONO | 1:1.5:1.5 | MeCN | 13 |
| 3 | - | NaNO2 | 1:1.5:1.5 | DMSO | 9 |
| 4 | - | t-BuONO | 1:1.5:1.5 | DMSO | 19 |
| 5 | - | t-BuONO | 1:1.5:1.5 | DMF | 10 |
| 6 | - | t-BuONO | 1:1.5:1.5 | DCE | 12 |
| 7 | - | t-BuONO | 1:3:3 | MeCN | 33 |
| 8 | Ascorbic acid | t-BuONO | 1:3:3 | MeCN/DMSO c | 63 |
| 9 | - | t-BuONO | 1:3:3 | MeCN/DMSO c | 36 |
| 10 d | Ascorbic acid | t-BuONO | 1:3:3 | MeCN/DMSO c | 60 |
| 11 e | Ascorbic acid | t-BuONO | 1:3:3 | MeCN/DMSO c | 42 |





3. Experimental Section
3.1. General Information
3.2. Typical Procedure for Ascorbic Initiated Tandem Cyclization of N-Arylacrylamines 1 with 4-Nitroniline
3.3. Physical, Analytical and Spectral Data
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Widengren, J.; Chmyrov, A.; Eggeling, C.; Löfdahl, P.Å.; Seidel, C.A. Strategies to Improve Photostabilities in Ultrasensitive Fluorescence Spectroscopy. J. Phys. Chem. 2007, 111, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Lee, K.M.; Wang, F.; Yoon, J. Visual detection of copper ions based on azide- and alkyne-functionalized polydiacetylene vesicles. J. Mater. Chem. 2011, 21, 15214–15217. [Google Scholar] [CrossRef]
- Browne, D.L.; Baxendale, I.R.; Ley, S.V. Piecing together the puzzle: Understanding a mild, metal free reduction method for the large scale synthesis of hydrazines. Tetrahedron 2011, 67, 10296–10303. [Google Scholar] [CrossRef]
- Roginsky, V.; Barsukova, T.; Stegmann, H.B. Kinetics of redox interaction between substituted quinones and ascorbate under aerobic conditions. Chem. Biol. Interact. 1999, 121, 177–197. [Google Scholar]
- Pethig, R.; Gascoyne, P.R.C.; Mclaughlin, J.A.; Szent-Gyorgyi, A. Enzyme-controlled scavenging of ascorbyl and 2,6-dimethoxy-semiquinone free radicals in Ehrlich ascites tumor cells. Proc. Natl. Acad. Sci. USA 1983, 80, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Cadrobbi, J.; Delvaux, M.; Jamison, J.M.; Gilloteaux, J.; Summers, J.L.; Taper, H.S.; Calderon, P.B. The association of vitamins C and K 3 kills cancer cells mainly by autoschizis, a novel form of cell death. Basis for their potential use as coadjuvants in anticancer therapy. Eur. J. Med. Chem. 2003, 38, 451–457. [Google Scholar] [CrossRef]
- Silveira-Dorta, G.; Monzόn, D.M.; Crisόstomo, F.P.; Martín, T.; Martín, V.S.; Carrillo, R. Oxidation with air by ascorbate-driven quinone redox cycling. Chem. Commun. 2015, 51, 7027–7030. [Google Scholar]
- Crisόstomo, F.P.; Martín, T.; Carrillo, R. Ascorbic acid as an initiator for the dirct C-H arylation of (hetero)arenes with anilines nitrosated in situ. Angew. Chem. Int. Ed. 2014, 53, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.R.; Yu, X.Y.; Xiao, W.J. Tandem radical cyclization of N-arylacrylamides: An emerging platform for the construction of 3,3-disubstituted oxindoles. Synthesis 2015, 47, 604–629. [Google Scholar] [CrossRef]
- Xu, Z.; Yan, C.; Liu, Z.Q. A free-radical cascade methylation/cyclization of N-arylacrylamides and isocyanides with dicumyl peroxide. Org. Lett. 2014, 16, 5670–5673. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, L.N.; Duan, X.H. Silver-catalyzed oxidative coupling/cyclization of acrylamides with 1,3-dicarbonyl compounds. Chem. Commun. 2013, 49, 10370–10372. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.B.; Song, R.J.; Ouyang, X.H.; Liu, Y.; Wei, W.T.; Deng, G.B.; Li, J.H. Metal-free oxidative tandem coupling of activated alkenes with carbonyl C(sp2)-H bonds and aryl C(sp2)-H bonds using TBHP. Chem. Sci. 2013, 4, 2690–2694. [Google Scholar] [CrossRef]
- Tang, S.; Zhou, D.; Wang, Y.C. Metal-free Meerwein carboarylation of alkenes with anilines: A straightforward approach to 3-benzyl-3-alkyloxindole. Eur. J. Org. Chem. 2014, 3656–3661. [Google Scholar] [CrossRef]
- Fu, W.J.; Xu, F.J.; Fu, Y.Q.; Zhu, M.; Yu, J.Q.; Xu, C.; Zou, D.P.J. Synthesis of 3,3-disubstituted oxindoles by visible-light-mediated radical reactions of aryl diazonium salts with N-Arylacrylamides. J. Org. Chem. 2013, 78, 12202–12206. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.H.; Li, Y.M.; Zhou, A.X.; Yang, T.T.; Yang, S.D. Silver-catalyzed carboazidation of arylacrylamides. Org. Lett. 2013, 15, 4158–4161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, D.; Liu, Z.Q. A free radical cascade silylarylation of activated alkenes: Highly selective activation of the Si-H/C-H bonds. Org. Lett. 2015, 17, 2534–2537. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Sun, M.; Wang, H.L.; Tian, Q.P.; Yang, S.D. Direct annulations toward phosphorylated oxindoles: Silver-catalyzed carbon-phophorus functionalization of akenes. Angew. Chem. Int. Ed. 2013, 52, 3972–3976. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Klumphu, P.; Liang, Y.M.; Lipshuz, B.H. Copper-catalyzed trifluoromethylation of N-arylacrylamides “on water” at room temperature. Chem. Commun. 2014, 50, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.B.; Wang, C.Y.; Song, R.J.; Liu, Y.; Wei, W.T.; Li, J.H. Oxidative 1,2-difunctionalization of activated alkenes with benzylic C(sp3)-H bonds. Chem. Commun. 2013, 49, 10817–10819. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Nesloney, C.L.; Shanklin, M.S.; Marsh, C.A.; Brown, K.C. Formation and characterization of 3-O-arenediazoascorbic acids. New stable diazo ethers. J. Org. Chem. 1989, 54, 3785–3789. [Google Scholar] [CrossRef]
- Warren, J.J.; Mayer, J.M. Surprisingly long-lived ascorbyl radicals in acetonitrile: Concerted proton-electron transfer reactions and thermochemistry. J. Am. Chem. Soc. 2008, 130, 7546–7547. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 3a–3p and 3r are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Cheng, P.; Liu, W.; Zeng, J.-G. Ascorbic Acid-Initiated Tandem Radical Cyclization of N-Arylacrylamides to Give 3,3-Disubstituted Oxindoles. Molecules 2015, 20, 15631-15642. https://doi.org/10.3390/molecules200915631
Liu S, Cheng P, Liu W, Zeng J-G. Ascorbic Acid-Initiated Tandem Radical Cyclization of N-Arylacrylamides to Give 3,3-Disubstituted Oxindoles. Molecules. 2015; 20(9):15631-15642. https://doi.org/10.3390/molecules200915631
Chicago/Turabian StyleLiu, Sheng, Pi Cheng, Wei Liu, and Jian-Guo Zeng. 2015. "Ascorbic Acid-Initiated Tandem Radical Cyclization of N-Arylacrylamides to Give 3,3-Disubstituted Oxindoles" Molecules 20, no. 9: 15631-15642. https://doi.org/10.3390/molecules200915631
APA StyleLiu, S., Cheng, P., Liu, W., & Zeng, J.-G. (2015). Ascorbic Acid-Initiated Tandem Radical Cyclization of N-Arylacrylamides to Give 3,3-Disubstituted Oxindoles. Molecules, 20(9), 15631-15642. https://doi.org/10.3390/molecules200915631