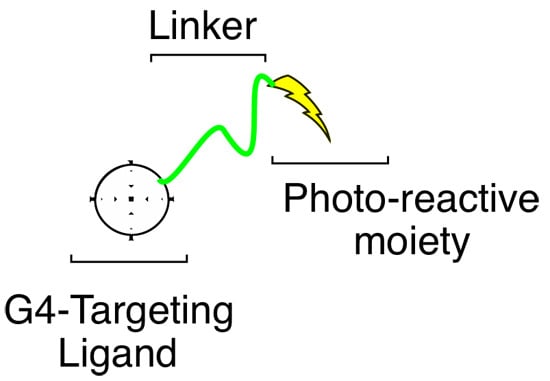
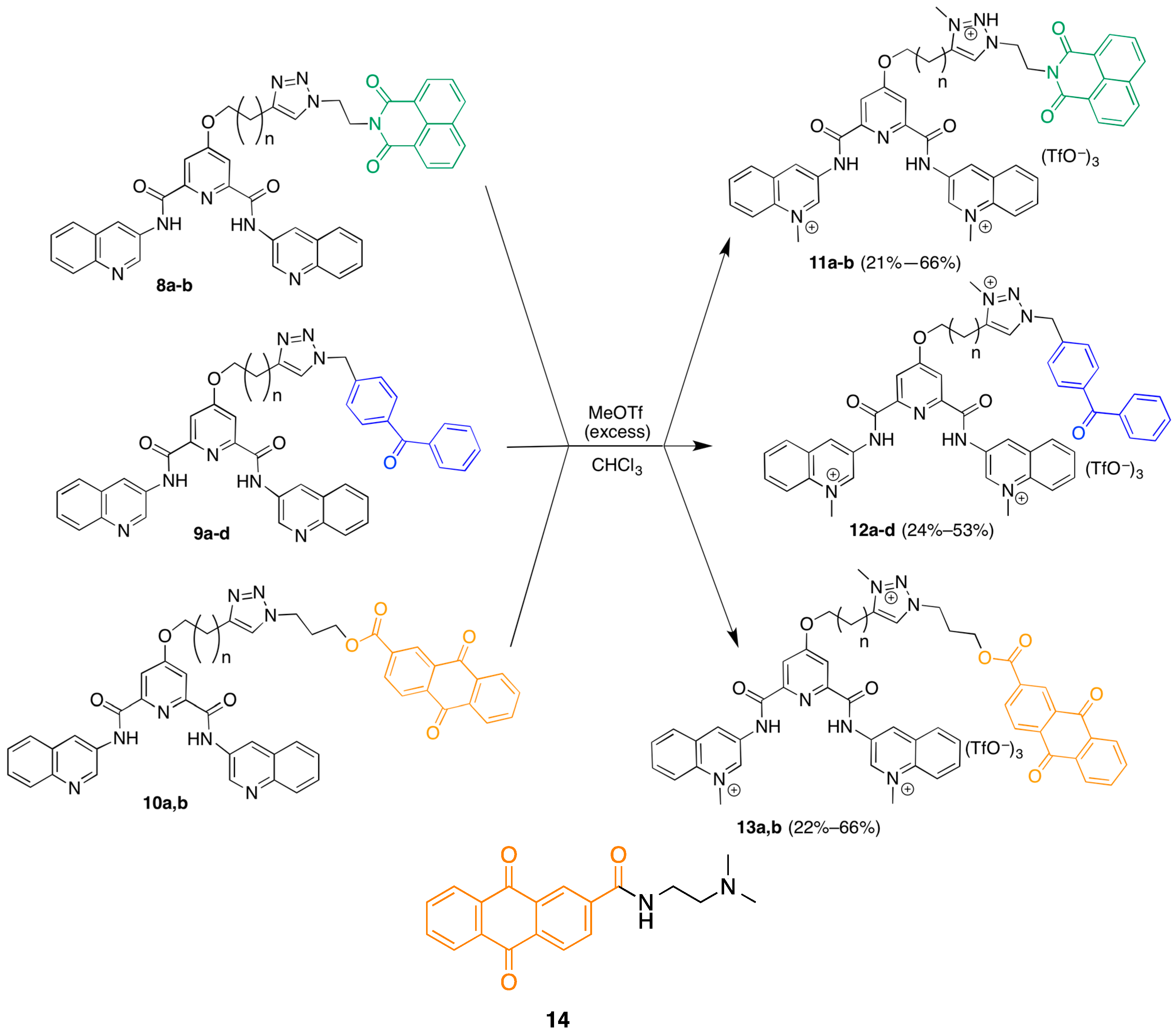
3.2. Library Synthesis
3-Azidopropyl 9,10-dioxo-9,10-dihydroanthracene-2-carboxylate (
7). 100 mg (1 eq, 0.37 mmol) of 9,10-dioxo-9,10-dihydroanthracene-2-carbonyl chloride was dissolved in 3 mL freshly distilled dichloromethane in an oven-dried flask under argon. This flask was then cooled to 0 °C. In a separate oven-dried flask, 150 mg (2.55 eq, 0.92 mmol) of 62 wt % 3-azidopropan-1-ol [
27] in DMF was dissolved in 1 mL of distilled dichloromethane under argon. 52 µL (1 eq, 0.37 mmol) of freshly distilled triethylamine was then added. The solution containing the alcohol and the amine was then added slowly under argon to the flask containing the acyl chloride at 0 °C. The reaction was then allowed to return to room temperature and was stirred for 19 h when it appeared complete by TLC (75% ethyl acetate in hexanes). After flash chromatography (25% ethyl acetate gradually up to 50% ethyl acetate in hexanes), 74 mg of the light yellow solid was obtained (approximately 80% purity, 45% yield). mp < 125 °C; IR (KBr) 3133.59, 2105.89, 1727.91, 1679.70, 1590.99, 1402.00, 1332.57, 1276.65, 1249.65, 1164.79, 1037.52, 939.16, 802.24, 703.89 cm
−1;
1H-NMR (400 MHz, CDCl
3) δ 8.9 (1H, s), 8.3–8.4 (4H, m), 7.80–7.83 (2H, m), 4.5 (2H, t,
J = 6.0 Hz), 3.5 (2H, t,
J = 6.6 Hz), 2.1 (2H, p,
J = 6.4 Hz); ESIMS
m/z 358.1 ([M + Na
+]
+, 100%); HRESIMS calc for C
18H
13N
3NaO
4+ 358.07983, found 358.07973.
Dimethyl 4-oxo-1,4-dihydropyridine-2,6-dicarboxylate (
2) [
28]. Distilled methanol that had been stored under argon over 4 Å molecular sieves (3.1 mL, 31 eq, 77.5 mmol) was added to a oven-dried flask under argon and the flask was then placed in an ice bath. Slowly, 1.1 mL (6.2 eq, 15.5 mmol) of thionyl chloride was added. The solution was allowed to stir for 5–10 min before 500 mg (1 eq, 2.5 mmol) of chelidamic acid was added under increased argon flow. The flask was outfitted with an oven-dried condenser. The mixture was then stirred for 72 h, under argon, allowing the ice bath to slowly warm to room temperature. The faintly yellow solution was then diluted approximately 2× with methanol, transferred to a larger flask, and the solvent removed under reduced pressure to give a white solid residue. The flask was then placed in an ice bath for 15 min before 3 mL of chilled distilled water was added with swirling, followed by 0.75 mL of chilled 10% sodium carbonate solution and 0.75 mL of chilled 50% aqueous methanol. After swirling, the mixture was allowed to stand in the ice bath for 20 min before being filtered under reduced pressure and washed with 3 mL, 3 mL, and 1 mL portions of chilled 50% aqueous methanol, giving 500 mg of crude white product. The crude product was adsorbed to 1 g of silica and purified through chromatography on a silica plug (about 5–6 g SiO
2, EtOAc as eluent), giving 417 mg of purified product as a white solid (79% yield).
1H-NMR (400 MHz, CDCl
3) δ 10.0 (1H, s), 7.5 (2H, s), 4.0 (6H, s) (matches lit. [
21]);
13C-NMR (100 MHZ, CDCl
3) δ 172.8, 163.3, 144.1, 117.7, 53.3 (matches lit. [
21]).
Mitsunobu Coupling General Procedure: Dimethyl 4-(pent-4-yn-1-yloxy)pyridine-2,6-dicarboxylate (3a). 300 mg (1 eq, 1.42 mmol) of 7 was suspended in 11 mL of freshly distilled THF under argon. 745 mg (2 eq, 2.84 mmol) of triphenylphosphine was then added, followed by 198 µL (1.5 eq, 2.13 mmol) of 4-pentyn-1-ol. The flask was then placed in an ice bath and stirred for 10 min. 391 µL (1.4 eq, 1.99 mmol) of diisopropylazodicarboxylate (DIAD) was then added drop-wise. The reaction was stirred, allowing it to return to room temperature, for 72 h at which point the reaction appeared done by TLC (EtOAc, KMnO4 stain). The solvent was removed under reduced pressure, giving a viscous oil that was then subjected to high vacuum for 1 h before being dissolved in the minimum amount of ethyl acetate. After purification by flash chromatography (50% EtOAc in hexanes), 350 mg (89% yield) of the product was obtained as a white powder. mp 107.6–108.6 °C; IR (KBr) 3270.68, 2964.05, 1727.91, 1604.48, 1444.42, 1371.14, 1270.86, 1112.73, 1045.23, 1008.59, 883.24, 788.74, 705.82, 592.04 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.8 (2H, s), 4.2 (2H, t, J = 6.1 Hz), 4.0 (6H, s), 2.4 (2H, t of d, J = 6.8 Hz), 2.02 (2H, p, J = 6.4 Hz), 1.96 (1H, t, J = 2.7 Hz); 13C-NMR (100 MHz, CDCl3) δ 166.8, 165.1, 149.7, 114.5, 82.5, 69.5, 67.1, 53.2, 27.5, 14.9; ESIMS m/z 300.1 ([M + Na]+, 100%), 278.1 ([M + H]+, 25%); HRESIMS calc for C14H15NO5Na+ 300.08424, found 300.08404.
Dimethyl 4-(hex-5-yn-1-yloxy)pyridine-2,6-dicarboxylate (3b). Following the general procedure above but employing 5-hexyn-1-ol afforded 180 mg (87% yield) of 3b after flash chromatography as a white solid. 1H-NMR (400 MHz, CDCl3) δ 7.8 (s, 2H), 4.1 (2H, t, J = 6.3 Hz), 4.0 (6H, s), 2.3 (2H, t of d, J = 7.0 Hz), 1.9–2.0 (m, 3H), 1.7 (2H, p, J = 7.2 Hz); 13C-NMR (100 MHz, CDCl3) δ 166.9, 165.1, 149.7, 114.5, 83.5, 69.0, 68.4, 53.2, 27.6, 24.6, 18.0; ESIMS m/z 605.2 ([2M + Na]+, 100%), 314.1 ([M + Na]+, 25%), 292.1 ([M + H]+,10%); HRESIMS calc for C15H18NO5+ 292.11790, found 292.11800.
Dimethyl 4-(hept-6-yn-1-yloxy)pyridine-2,6-dicarboxylate (3c). Following the general procedure above but employing 6-heptyn-1-ol afforded 164 mg (87% yield) of 3c after flash chromatography as a white solid. 1H-NMR (400 MHz, CDCl3) δ 7.8 (2H, s), 4.11 (2H, t, J = 6.4 Hz), 3.96 (6H, s), 2.20 (2H, p, J = 2.7 Hz), 1.9 (1H, t, J = 2.7 Hz), 1.8 (2H, p, J = 6.8 Hz), 1.6 (4H, m). 13C-NMR (100 MHz, CDCl3) δ 167.0,165.1, 149.6, 114.4, 84.0, 68.8, 68.6, 53.2, 28.2, 27.9, 24.9, 18.2; ESIMS m/z 633.2 ([2M + Na]+, 100%), 328.1 ([M + Na]+, 33%), 306.1 ([M + H]+, 15%); HRESIMS calc for C16H19NO5Na+ 328.11550, found 328.11580.
Dimethyl 4-(non-8-yn-1-yloxy)pyridine-2,6-dicarboxylate (3d). Following the general procedure above but employing 7-octyn-1-ol afforded 221 mg (quantitative yield) of 3d after flash chromatography as a white powder. 1H-NMR (400 MHz, CDCl3) δ 7.7 (2H, s), 4.1 (2H, t, J = 6.3 Hz), 4.0 (6H, s), 2.1 (2H, t, J = 6.9 Hz), 1.9 (1H, t, J = 2.6Hz), 1.8 (2H, p, J = 7.0 Hz), 1.3–1.5 (8H, m); 13C-NMR (100 MHz, CDCl3) δ 167.0, 165.1, 149.6, 114.4, 84.4, 68.9, 68.2, 53.1, 28.60, 28.55, 28.4, 28.2, 25.6, 18.3; CIMS m/z 334 ([M + H]+,100%); HRCIMS for C18H25NO5+ 334.1654, found 134.1655.
Trimethyl Aluminum Amidation General Procedure: 4-(pent-4-yn-1-yloxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (4a). 320 mg (1 eq, 1.04 mmol) of 3a and 357 mg (2.4 eq, 2.5 mmol) of 3-aminoquinoline were dissolved in 15 mL distilled 1,2-dichloroethane under argon. 4.5 mL (4.2 eq, 4.5 mmol) of 1.0 M trimethylaluminium in heptane was then added under increased argon flow resulting in a yellow solution. The flask was then fitted with an oven-dried condenser and placed in an oil bath at 94 °C and stirred at reflux for 90 min at which point the reaction appeared complete by TLC (EtOAc). The dark red solution was cooled to room temperature and quenched with 3 mL of methanol, resulting in the formation of a yellow gel. The gel was diluted and partially dissolved with additional methanol and chloroform, silica gel added, and the solvent removed under reduced pressure. The silica-adsorbed residue was partially purified through a silica column plug (EtOAc). Combining the fractions from the column gave 564 mg of the crude product after removal of the solvent under reduced pressure. The crude product was then suspended in 20 mL of methanol and stirred for 2 h before being filtered under reduced pressure and washed three times with methanol giving 487 mg of the product as a yellow solid. The filtrate was concentrated under reduced pressure, suspended in 3–5 mL of methanol and filtered again after 30 min, giving an additional 8 mg of the product as a yellow solid. The combined crops gave 495 mg (89% yield) of the product. mp = 235 °C; IR (KBr) 3126.18, 1675.34, 1606.65, 1543.35, 1492.28, 1376.56, 1340.15, 1225.99, 1045.45, 902.46, 750.69 cm−1; 1H-NMR (400 MHz, CDCl3, 4% MeOD) δ 11.1 (0.5H, s), 9.02–9.04 (4H, m), 7.971 (2H, s), 7.969 (2H, d, J = 8.0 Hz), 7.8 (2H, d, J = 8.1 Hz), 7.6 (2H, t, J = 7.6 Hz), 7.5 (2H, t, J = 7.2 Hz), 4.3 (2H, t, J = 6.1 Hz), 2.4 (2H, t of d, J = 6.9 Hz), 2.1 (2H, p, J = 6.5 Hz), 2.0 (1H, t, J = 2.6 Hz); 13C (100 MHz, CDCl3, 4% MeOD) δ 167.8, 162.8, 150.7, 144.6, 144.5, 131.6, 128.7, 128.2, 127.9, 127.3, 125.6, 125.5, 112.1, 82.6, 69.5, 67.2, 27.5, 14.9; ESIMS m/z 502.3 ([M + H]+, 100%); HRESIMS calc for C30H24N5O3+ 502.18737, found 502.1873.
4-(Hex-5-yn-1-yloxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (4b). Following the general procedure above but employing 3b afforded 146 mg (64% yield) of 4b as a yellow solid. mp = 204 °C; IR (KBr) 3293.32, 2941.37, 1667.96, 1604.95, 1537.16, 1491.22, 1468.93, 1423.99, 1374.2, 1337.97, 1278.61, 1226.4, 1176.96, 1144.2, 1098.44, 1034.38, 899.15, 780.04, 748.42, 661.48 cm−1; 1H-NMR (400 MHz, CDCl3 (2% MeOD)) δ 10.9 (1H, s), 9.0 (2H, s), 8.9 (2H, s), 7.92 (2H, d, J = 8.4 Hz), 7.88 (2H, s), 7.7 (2H, d, J = 8.1 Hz), 7.6 (2H, t, J = 7.5 Hz), 7.5 (2H, t, J = 7.4 Hz), 4.2 (2H, t, J = 6.3 Hz), 2.3 (2H, t, J = 6.9 Hz), 2.00 (1H, t, J = 2.6 Hz), 1.99 (2H, p, J = 6.3 Hz), 1.7 (2H, p, J = 7.1 Hz); 13C-NMR (100 MHz, CDCl3 (2% MeOD)) δ 167.8, 162.7, 162.6, 150.6, 144.64, 144.59, 131.5, 131.4, 128.6, 128.3, 128.1, 127.8, 127.3, 125.4, 125.3, 112.0, 83.6, 69.0, 68.5, 27.7, 24.7, 18.0 ; ESIMS m/z 516.2 ([M + H]+, 100%), 252.2 (M2+, 70%); HRESIMS calc for C31H26N5O3+ 516.20302, found 516.20327.
4-(Hept-6-yn-1-yloxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (4c). Following the general procedure above but employing 3c afforded 104.6 mg (37% yield) of 4c as a pale yellow solid. mp 199.8–202.1 °C (decomp); IR (KBr) = 3354.65, 3199.37, 1686.04, 1637.74, 1609.93, 1539.93, 1492.12, 1399.91, 1375.81, 1337.48, 1207.02, 1175.14, 1145.52, 1028.99, 901.20, 782.60, 752.33, 736.65, 656.88 cm−1; 1H-NMR (400 MHz, DMF) δ 11.4 (2H, s), 9.5 (2H, d, J = 2.5 Hz), 9.1 (2H, d, J = 2.4 Hz), 8.1 (4H, t, J = 9.3 Hz), 7.8 (2H, t, J = 7.6 Hz), 7.7 (2H, t, J = 7.5 Hz), 4.4 (2H, t, J = 6.5 Hz), 2.8 (1H, t, J = 2.6 Hz), 2.3 (2H, p, J = 4.0 Hz), 1.9 (2H, p, J = 7.0 Hz), 1.7 (4H, m); 13C-NMR (100 MHz, DMF) δ 168.8, 163.0, 151.6, 146.4, 145.9, 133.0, 129.6, 128.9, 128.8, 128.6, 127.8, 124.4, 112.1, 84.9, 70.9, 69.7, 28.8, 25.6, 18.5. ESIMS m/z 530.22([M + H]+, 100%); HRESIMS calc for C32H28N5O3+ 530.21867, found 530.21987.
4-(Non-8-yn-1-yloxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (4d). Following the general procedure above but employing 3d afforded 134 mg (36% yield) of 4d as pale yellow crystals. mp 160.7–162.4 °C (decomp); IR (KBr) 3299.15, 2933.39, 2373.65, 2344.64, 1870.59, 1846.21, 1793.81, 1773.91, 1751.61, 1735.65, 1718.92, 1700.86, 1685.47, 1664.83, 1637.67, 1607.89, 1560.25, 1542.40, 1490.85, 1458.79, 1421.72, 1375.18, 1399.93, 1208.00, 1143.95, 1097.50, 1015.73, 897.82, 778.95, 746.47, 669.71, 473.38 cm−1; 1H-NMR (400 MHz, CDCl3) δ 10.2 (2H, s), 8.9 (2H, d, J = 2.5 Hz), 8.5 (2H, d, J = 2.3 Hz), 7.9 (2H, d, J = 8.3 Hz), 7.7 (2H, s), 7.53 (2H, t, J = 8.1 Hz), 7.50 (2H, t, J = 7.7 Hz), 7.4 (2H, t, J = 7.5 Hz), 4.0 (2H, t, J = 6.6 Hz), 2.2 (2H, t of d, J = 7.0 Hz), 2.0 (1H, t, J = 2.6 Hz), 1.8 (2H, p, J = 6.7 Hz), 1.3–1.6 (8H, m); 13C-NMR (100 MHz, CDCl3) δ 167.9, 162.3, 150.2, 145.2, 144.9, 130.8, 128.71, 128.67, 127.8, 127.6, 127.2, 125.2, 111.8, 84.6, 69.2, 68.3, 28.8, 28.7, 28.6, 28.3, 25.7, 18.44, 18.38. ESIMS m/z 558.25 ([M + H]+, 100%); HRESIMS calc for C34H32N5O3+ 558.24997, found 558.25046.
Copper-catylized coupling General Procedure: 4-(3-(1-(2-(1,3-dioxo-1
H-benzo[de]isoquinolin-2(3
H)-yl)ethyl)-1
H-1,2,3-triazol-4-yl)propoxy)-
N2,
N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (
8a). The alkyne
3a 20 mg (1 eq, 0.04 mmol) and 12.7 mg (1.2 eq, 0.048 mmol) of azide 5 [
29] were dissolved in 3 mL DMF and placed under argon. A solution of 0.1 M copper triflate in water (440 µL, 0.044 mmol) was added, resulting in a dark green solution. A freshly prepared solution of 0.1 M sodium ascorbate in water (160 µL, 0.016 mmol) was added, and the flask was placed in an oil bath at 25 °C and stirred for 68 h until the reaction appeared complete by TLC (EtOAc). The DMF was removed under high vacuum and the residue partitioned between about 40 mL of chloroform and 40 mL of water. This mixture was stirred vigorously for 24 h before being transferred to a separation funnel. The aqueous layer was extracted twice more with smaller volumes of chloroform (about 10–20 mL) and the organic layers were combined, and the solvent removed. The residue was dissolved in a mixture of methanol and chloroform and absorbed to silica by removal of the solvent under reduced pressure. A mini column was then run (EtOAc up to 10% MeOH in EtOAc), giving 23 mg (76% yield) of 8a as a pale yellow solid.
1H-NMR (400 MHz, CDCl
3 (4% MeOD)) δ 11.1 (exchanged, s), 9.1 (4H, s), 8.5 (2H, d,
J = 7.6 Hz), 8.2 (2H, d,
J = 7.8 Hz), 8.0 (4H, s), 7.9 (2H, d,
J = 7.7 Hz), 7.6–7.7 (4H, m), 7.5–7.6 (3H, m), 4.72 (2H, s), 4.62 (2H, s), 4.2 (2H, s), 2.9 (2H, t,
J = 7.3 Hz), 2.2 (2H, t,
J = 6.1 Hz). ESIMS
m/
z 790.26 ([M + Na]
+,100%), 768.28 ([M + H]
+, 25%); HRESIMS calc for (C
44H
33N
9O
5Na)
+ 790.24970, found 790.25100.
4-(3-(1-(4-Benzoylbenzyl)-1H-1,2,3-triazol-4-yl)propoxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (9a). Following the general procedure above but employing azide 6 afforded 5.5 mg (52% yield) of the 9a as a pale yellow solid. 1H-NMR (400 MHz, CDCl3 (1.5% MeOD)) δ 10.8 (exchanged amide, s), 9.1 (2H, s), 9.0 (2H, s), 7.99 (2H, d, J = 7.2 Hz), 7.91 (2H, s), 7.8 (2H, d, J = 8.6 Hz), 7.68–7.72 (4H, m), 7.6 (2H, t, J = 7.3 Hz), 7.52 (2H, t, J = 7.3 Hz), 7.49 (1H, t, J = 1.3 Hz), 7.4 (2H, t, J = 7.6 Hz), 7.3 (2H, t, J = 8.3 Hz), 5.6 (2H, s), 4.2 (2H, t, J = 6.2 Hz), 2.9 (2H, t, J = 7.3 Hz), 2.3 (2H, p, J = 6.6 Hz). ESIMS m/z 739.3 ([M + H]+, 50%), 761.3 ([M + Na]+, 40%); HRESIMS calc for C44H35N8O4+ = 739.27760, found 739.27870.
3-(4-(3-((2,6-Bis(quinolin-3-ylcarbamoyl)pyridin-4-yl)oxy)propyl)-1H-1,2,3-triazol-1-yl)propyl 9,10-dioxo-9,10-dihydroanthracene-2-carboxylate (10a). Following the general procedure above but employing azide 7 afforded 7 mg of 10a (21% yield). 1H-NMR (400 MHz, CDCl3) δ 10.3 (2H, s), 9.1 (2H, s), 8.8 (1H, s), 8.7 (2H, s), 8.2–8.3 (2H, m), 8.16–8.18 (1H, m), 7.93 (2H, d, J = 8.0 Hz), 7.8 (2H, s), 7.71–7.74 (2H, m), 7.69 (2H, d, J = 8.9 Hz), 7.6 (2H, t, J = 7.6 Hz), 7.45–7.49 (3H, m), 4.5 (2H, t, J = 6.7 Hz), 4.3 (2H, t, J = 6.7 Hz), 4.2 (2H, t, J = 5.8 Hz), 3.0 (2H, t, J = 7.2 Hz), 2.4 (2H, p, J = 6.4 Hz), 2.3 (2H, p, J = 6.7 Hz). ESIMS m/z 859.3 ([M + Na]+, 50%); HRESIMS calc for C48H36N8O7Na+ 859.25990, found 859.26200.
4-(4-(1-(2-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)ethyl)-1H-1,2,3-triazol-4-yl)butoxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (8b). Following the general procedure above but employing alkyne 4b afforded 13.4 mg (44% yield) of 8b. 1H-NMR (400 MHz, CDCl3 (20% MeOD)) δ 9.0 (2H, s), 8.9 (2H, s), 8.4 (2H, d, J = 7.0 Hz), 8.1 (2H, d, J = 8.0 Hz), 7.91 (2H, d, J = 8.5 Hz), 7.89 (2H, s), 7.8 (2H, d, J = 8.3 Hz), 7.64 (2H, t, J = 7.8 Hz), 7.58 (2H, t, J = 7.6 Hz), 7.48 (2H, t, J = 7.6 Hz), 7.45 (1H, s), 4.6 (2H, t, J = 6.3 Hz), 4.5 (2H, t, J = 6.0 Hz), 4.1 (2H, t, J = 6.3 Hz), 2.7 (2H, t, J = 7.5 Hz), 1.8 (4H, m). ESIMS m/z 804.3 ([M + Na]+, 100%); HRESIMS calc for C45H35N9O5Na+ 804.26530, found 804.26690.
4-(4-(1-(4-Benzoylbenzyl)-1H-1,2,3-triazol-4-yl)butoxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (9b). Following the general procedure above but employing alkyne 4b and azide 6 afforded 27.9 mg (95% yield) of 9b. 1H-NMR (400 MHZ, CDCl3) δ 10.8 (s, exchanged), 9.1 (2H, s), 9.0 (2H, s), 8.01 (2H, d, J = 8.8 Hz), 7.93 (2H, s), 7.8 (2H, d, J = 8.4 Hz), 7.71–7.73 (4H, m), 7.6 (2H, t, J = 7.5 Hz), 7.5–7.6 (3H, m), 7.42 (2H, t, J = 7.7 Hz), 7.35 (1H, s), 7.3 (2H, d, J = 8.0 Hz), 5.6 (2H, s), 4.2 (2H, t, J = 5.4 Hz), 2.8 (2H, t, J = 6.8 Hz), 1.90–1.91 (4H, m). ESIMS m/z 775.28 ([M + Na]+, 100%), 753.29 ([M + H]+, 33%); HRESIMS calc for (C45H36N8O4Na)+ 775.27520, found 775.27540.
4-((5-(1-(4-Benzoylbenzyl)-1H-1,2,3-triazol-4-yl)pentyl)oxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (9c). Following the general procedure above but employing alkyne 4c and azide 6 afforded 34.6 mg (88% yield) of 9c as an off-white solid mp 125 °C; IR (KBr) 3128.25, 2935.02, 2385.38, 1794.12, 1774.05, 1735.74, 1719.09, 1701.17, 1686.19, 1655.31, 1637.99, 1607.92, 1578.00, 1560.39, 1543.06, 1509.50, 1490.94, 1459.13, 1400.02, 1099.73, 750.87, 612.45 cm−1; 1H-NMR (400 MHZ, CDCl3) δ 10.2 (2H, s), 9.1 (2H, d, J = 2.5 Hz), 8.8 (2H, d, J = 2.3 Hz), 8.0 (2H, d, J = 9.3 Hz), 7.9 (2H, s), 7.8 (2H, d, J = 8.2 Hz), 7.69–7.72 (4H, m), 7.6 (2H, t, J = 7.7 Hz), 7.5–7.6 (3H, m), 7.4 (2H, t, J = 7.0 Hz), 7.26–7.28 (3H, m), 5.6 (2H, s), 4.1 (2H, t, J = 6.4 Hz), 2.8 (2H, t, J = 7.5 Hz), 1.9 (2H, p, J = 6.9 Hz), 1.8 (2H, p, J = 7.6 Hz), 1.5 (2H, p, J = 7.6 Hz); ESIMS m/z 789.29 ([M + Na]+, 100%), 767.31 ([M + H]+, 70%); HRESIMS calc for (C46H38N8O4Na)+ 789.29080, found 789.29120.
4-((7-(1-(4-Benzoylbenzyl)-1H-1,2,3-triazol-4-yl)heptyl)oxy)-N2,N6-di(quinolin-3-yl)pyridine-2,6-dicarboxamide (9d). Following the general procedure above but employing alkyne 4d and azide 6 afforded 20.9 mg (49% yield) of 9d as a white powder. 1H-NMR (400 MHz, CDCl3) δ 10.1 (2H, s), 9.1 (2H, d, J = 2.9 Hz), 8.8 (2H, d, J = 2.6 Hz), 8.0 (2H, d, J = 8.6 Hz), 7.9 (2H, s), 7.7–7.8 (6H, m), 7.62 (2H, t, J = 7.7 Hz), 7.5–7.56 (3H, m), 7.4 (2H, t, J = 7.2 Hz), 7.2–7.3 (3H, m), 5.5 (2H, s), 4.2 (2H, t, J = 6.4 Hz), 2.7 (2H, t, J = 7.7 Hz), 1.8 (2H, p, J = 7.0 Hz), 1.7 (2H, p, J = 7.5 Hz), 1.5 (2H, p, J = 6.4 Hz), 1.4–1.47 (4H, m). ESIMS m/z 795.3 ([M + H]+, 50%), 817.3 ([M + Na]+, 100%), 1612.7 ([2M + Na]+, 30%); HRESIMS calc for C48H42N8O4Na+ 817.32210, found 817.32210.
3-(4-(4-((2,6-Bis(quinolin-3-ylcarbamoyl)pyridin-4-yl)oxy)butyl)-1H-1,2,3-triazol-1-yl)propyl 9,10-dioxo-9,10-dihydroanthracene-2-carboxylate (10b). Following the general procedure above but employing alkyne 4b and azide 7 afforded 9.7 mg of 10b as a white solid (29% yield). 1H-NMR (400 MHz, CDCl3 (5% MeOD)) δ 9.0 (4H, s), 8.8 (1H, s), 8.2–8.4 (4H, m), 7.99 (2H, d, J = 8.2 Hz), 7.95 (2H, s), 7.8 (2H, d, J = 8.4 Hz), 7.75–7.77 (2H, m), 7.6 (2H, t, J = 7.3 Hz), 7.54 (2H, t, J = 7.5 Hz), 7.45 (1H, s), 4.5 (2H, t, J = 7.0 Hz), 4.4 (2H, t, J = 6.1 Hz), 4.2 (2H, t, J = 6.4 Hz), 2.8 (2H, t, J = 6.7 Hz), 2.4 (2H, p, J = 6.4 Hz), 1.9 (4H, m); ESIMS m/z 873.3 ([M + Na]+, 50%); HRESIMS calc for C49H38N8O7Na+ 873.27560, found 873.27450.
Methylation General Procedure: 3,3'-((4-(4-(1-(2-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)ethyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)butoxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) trifluoromethanesulfonate (11b). 13.4 mg (1 eq, 0.016 mmol) of 8b was dissolved in 3 mL of 3.8% methanol in chloroform and placed under argon. The flask was placed in an ice bath and 10 µL (5.6 eq, 0.09 mmol) of methyl triflate was added slowly. The solution was then stirred for 4 h, allowing the bath to return to room temperature. A significant amount of precipitate began to form after 2 h. This precipitate was isolated by filtration under reduced pressure and washed twice with small (1 mL) portions of chloroform, giving 13.4 mg (66% yield). 1H-NMR (400 MHz, d6-DMSO) δ 11.9 (2H, s), 10.0 (2H, s), 9.6 (2H, s), 8.9–9.0 (2H, m), 8.4–8.6 (5H, m), 8.3 (2H, t, J = 8.0 Hz), 8.05–8.11 (2H, m), 7.92 (2H, s), 7.86 (2H, t, J = 7.5 Hz), 7.7–7.8 (2H, m), 5.0 (2H, t, J = 5.5 Hz), 4.8 (6H, s), 4.6 (2H, t, J = 5.7 Hz), 4.3 (2H, s), 4.2 (3H, s), 3.0 (2H, t, J = 6.6 Hz), 1.8 (4H, m). MALDIMS m/z 1124 ([M − OTf], 100%); HRMALDIMS calc for C50H44N9O11F6S2+ 1124.25004, found 1124.24914.
3,3'-((4-(3-(1-(2-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)ethyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)propoxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) trifluoromethanesulfonate (11a). Following the general procedure above but starting with the triazole 8a afforded 3.5 mg (21% yield) of 11a 1H-NMR (400 MHz, d6-DMSO) δ 10.1 (2H, s), 9.6 (2H, s), 9.0 (2H, s), 8.6 (2H, t, J = 8.1 Hz), 8.4 (3H, m), 8.3 (2H, t, J = 8.0 Hz), 8.07–8.11 (3H, m), 7.93–7.97 (2H, m), 7.7–7.8 (4H, m), 5.0 (2H, s), 4.8 (6H, s), 4.6 (2H, s), 4.4 (2H, s), 4.2 (3H, s), 3.1 (2H, t, J = 7.2 Hz), 2.2 (2H , s). MALDIMS m/z 1110.1 ([M − OTf]+, 8%). HRMALDIMS calc for C49H42N9O11F6S2+ 1110.23439, found 1110.2335.
3,3'-((4-(3-(1-(4-Benzoylbenzyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)propoxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) trifluoromethanesulfonate (12a). Following the general procedure above but starting with the triazole 9a afforded 8.7 mg (50% yield) of 12a. 1H-NMR (600 MHz, d6-DMSO) δ 11.8 (2H, s), 10.0 (2H, s), 9.6 (2H, s), 9.0 (1H, s), 8.6 (2H, d, J = 9.0 Hz), 8.5 (2H, d, J = 7.8 Hz), 8.3 (2H, t, J = 7.9 Hz), 8.1 (2H, t, J = 7.8 Hz), 8.0 (2H, s), 7.8 (2H, d, J = 6.6 Hz), 7.6–7.7 (5H, m), 7.5 (2H, t, J = 6.8 Hz), 6.0 (2H, s), 4.8 (6H, s), 4.5 (2H, t, J = 6.0 Hz), 4.3 (3H, s), 3.1 (2H, t, J = 7.8 Hz), 2.3 (2H, p, J = 6.9 Hz ). ESIMS m/z 261 (M3+, 80%); HRESIMS calc for C47H43N8O43+ 261.11300, found 261.11310.
3,3'-((4-(4-(1-(4-Benzoylbenzyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)butoxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) trifluoromethanesulfonate (12b). Following the general procedure above but starting with the triazole 9b afforded 5.7 mg (35% yield) of 12b 1H-NMR (400 MHz, d6-DMSO) δ 10.0 (2H, s), 9.6 (2H, s), 8.9 (2H, s), 8.6 (4H, t, J = 7.2 Hz), 8.3 (2H, t, J = 8.0 Hz), 8.1 (2H, t, J = 7.8 Hz), 8.0 (2H, s), 7.8 (2H, d, J = 8.2 Hz), 7.62–7.7 (4H, m), 7.56 (2H, t, J = 8.0 Hz), 6.0 (2H, s), 4.8 (6H, s), 4.4 (2H, m), 4.2 (3H, s), 3.0 (2H, t, J = 7.0 Hz), 1.9 (4H, m); MALDIMS m/z 1095 ([M − OTf], 100%); HRMALDIMS calc for C50H45N8O10F6S2+ 1095.25988, found 1095.258.
3,3'-((4-((5-(1-(4-Benzoylbenzyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)pentyl)oxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) trifluoromethanesulfonate (12c). Following the general procedure above but starting with the triazole 9c afforded 10.1 mg (53% yield) of 12c as a white powder. mp 211 °C; IR (KBr) = 3122.83, 2372.10, 2344.75, 1774.14, 1735.85, 1719.20, 1701.46, 1686.39, 1655.16, 1637.81, 1560.41, 1543.41, 1475.80, 1458.82, 1399.46, 1275.61, 1156.44, 1030.19, 637.63 cm−1; 1H-NMR (400 MHz, d7-DMF) δ 11.9 (2H, s), 10.3 (2H, d, J = 2.6 Hz), 9.8 (2H, s), 9.1 (1H, s), 8.7 (2H, d, J = 9.2 Hz), 8.6 (2H, d, J = 8.4 Hz), 8.4 (2H, s), 8.3 (2H, t, J = 8.0 Hz), 8.2 (2H, t, J = 7.5 Hz), 8.02–8.05 (2H + DMF, m), 7.9 (2H, d, J = 8.1 Hz), 7.7–7.8 (5H, m), 7.6 (2H, t, J = 7.6 Hz), 6.2 (2H, s), 5.0 (6H, s), 4.5 (3H, s), 3.1 (2H, t, J = 7.6 Hz), 1.9–2.0 (4H, m), 1.7 (2H, p, J = 7.7 Hz); ESIMS m/z 270.5 (M3+, 1.5%), 398.2 ([M − Me]2+, 100%); HRESIMS calc for C49H47N8O43+ = 270.45679, found 270.45621; HRESIMS calc for C48H44N8O42+ 398.17373, found 398.17423.
3,3'-((4-((7-(1-(4-Benzoylbenzyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)heptyl)oxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) trifluoromethanesulfonate (12d). Following the general procedure above but starting with the triazole 9d afforded 2.5 mg (24% yield) of 12d as a white powder. 1H-NMR (400 MHz, d7-DMF) δ 12.0 (2H, s), 10.3 (2H, s), 9.8 (2H, s), 8.7 (2H, d, J = 8.6 Hz), 8.6 (2H, d, J = 8.6 Hz), 8.3 (2H, t, J = 8.6 Hz), 8.2 (2H, t, J = 8.1 Hz), 8.1 (2H, s), 7.9 (2H, d, J = 6.7 Hz), 7.7–7.8 (4H, m), 7.6 (2H, t, J = 7.7 Hz), 6.2 (2H, s), 5.0 (6H, s), 4.4 (3H, s), 3.1 (2H, t, J = 8.4 Hz), 1.8–1.9 (4H, m), 1.5–1.6 (6H, m). MALDIMS m/z 1137.0 ([M − OTf]+, 15%); HRMS calc for C53H51N8O10F6S2 1137.30683, found 1137.3037.
3,3'-((4-(3-(1-(3-((9,10-Dioxo-9,10-dihydroanthracene-2-carbonyl)oxy)propyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)propoxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) fluoromethanesulfonate (13a). Following the general procedure above but starting with the triazole 10a afforded 7.4 mg (66% yield) of 13a. 1H-NMR (400 MHz, d6-DMSO) δ 10.0 (2H, s), 9.6 (2H, s), 9.0 (2H, s), 8.6 (2H, s), 8.5 (2H, d, J = 8.3 Hz), 8.30–8.34 (2H, m), 8.2 (2H, t, J = 7.8 Hz), 8.18–8.23 (2H, m), 8.1 (2H, t, J = 7.4 Hz), 7.92–7.94 (2H, m), 7.90 (2H, s), 4.83 (2H, t, J = 6.5 Hz), 4.78 (6H, s), 4.5 (2H, t, J = 5.9 Hz), 4.4 (2H, t, J = 6.2 Hz), 4.2 (3H, s), 3.1 (2H, t, J = 7.4 Hz), 2.18–2.20 (2H, m). ESIMS m/z 293.8 (M3+, 60%); HRESIMS calc for C51H45N8O73+ 293.77980, found 293.78040.
3,3'-((4-(4-(1-(3-((9,10-Dioxo-9,10-dihydroanthracene-2-carbonyl)oxy)propyl)-3-methyl-1H-1,2,3-triazol-3-ium-4-yl)butoxy)pyridine-2,6-dicarbonyl)bis(azanediyl))bis(1-methylquinolin-1-ium) trifluoromethanesulfonate (13b). Following the general procedure above but starting with the triazole 10b afforded 3.5 mg (22% yield) of 13b. 1H-NMR (400 MHz, d7-DMF) δ 11.9 (2H, s), 10.2 (2H, s), 9.8 (2H, s), 9.1 (2H, s), 8.69–8.72 (3H, m), 8.6 (2H, d, J = 8.3 Hz), 8.35–8.41 (4H, m), 8.2–8.3 (4H, m), 8.1–8.2 (3H, m), 5.1 (2H, t, J = 6.7 Hz), 5.0 (6H, s), 4.6 (2H, t, J = 5.7 Hz), 4.5 (2H, t, J = 5.5 Hz), 4.4 (3H, s), 3.2 (2H, m), 2.1 (6H, m). MALDIMS m/z 1193 ([M − OTf]+, 100%). HRMALDIMS calc for C54H47N8O13F6S2+ 1193.26082, found 1193.2609.
N-(2-(Dimethylamino)ethyl)-9,10-dioxo-9,10-dihydroanthracene-2-carboxamide (14). 100 mg (1 eq, 0.37 mmol) of 9,10-dioxo-9,10-dihydroanthracene-2-carbonyl chloride was suspended in 4 mL of dichloromethane. A solution of 56 µL N,N-dimethylethylenediamine in 6 mL dichloromethane was slowly added by addition funnel over 35 min. The mixture was then stirred at 35 °C for 2 h, when an additional 50 µL of diamine were added dropwise and the mixture stirred for an additional hour, at which point the reaction appeared to be complete by TLC (75% EtOAc in Hexanes). The precipitate was isolated by filtration under reduced pressure and was washed 3X with dichloromethane, giving 46.7 mg of starting material (by TLC). The filtrate was concentrated under reduced pressure and the residue partitioned between dichloromethane and water. The aqueous layer was extracted an additional two times with dichloromethane, and the combined organic layers washed once with 2.3 M NaOH and once with brine before being dried over sodium sulfate. Removal of the solvent under reduced pressure gave 54 mg (46% yield) of product as a yellow solid. 1H-NMR (400 MHz, CDCl3) δ 8.5 (1H, s), 8.2–8.3 (4H, m), 7.6–7.7 (2H, m), 7.2 (1H, s), 3.5 (2H, q, J = 5.6 Hz), 2.5 (2H, t, J = 6.0 Hz), 2.3 (6H, s); 13C-NMR (100 MHz, CDCl3) δ 182.43, 182.36, 165.6, 139.6, 134.9, 134.31, 134.28, 133.28, 133.25, 133.0, 127.7, 127.3 125.2, 57.6, 45.1, 37.5.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






