
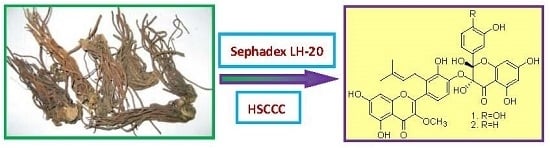
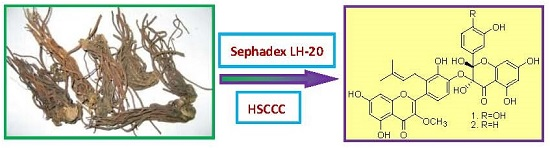
Preparative Isolation of Two Prenylated Biflavonoids from the Roots and Rhizomes of Sinopodophyllum emodi by Sephadex LH-20 Column and High-Speed Counter-Current Chromatography

Abstract
:
1. Introduction

2. Results and Discussion
2.1. Enrichment by Sephadex LH-20 Column



2.2. Selection of Two-Phase Solvent System and Other Conditions of HSCCC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent System | Ratio | K Value | α | |
|---|---|---|---|---|
| 1 | 2 | |||
| n-Hexane–ethyl acetate–methanol–water | 6:4:5:5 | 0.12 | 0.15 | 1.25 |
| n-Hexane–ethyl acetate–methanol–water | 5:5:5:5 | 0.18 | 0.23 | 1.28 |
| n-Hexane–ethyl acetate–methanol–water | 4.5:5:4.5:5 | 0.29 | 0.37 | 1.28 |
| n-Hexane–ethyl acetate–methanol–water | 4:5:4:5 | 0.60 | 0.69 | 1.15 |
| n-Hexane–ethyl acetate–methanol–water | 3.5:5:3.5:5 | 0.84 | 1.05 | 1.25 |
| n-Hexane–ethyl acetate–methanol–water | 3:5:3:5 | 1.41 | 1.64 | 1.16 |
| n-Hexane–ethyl acetate–methanol–water | 2:5:2:5 | 2.56 | 2.83 | 1.11 |
| n-Hexane–ethyl acetate–methanol–water | 1:5:1:5 | 5.93 | 6.14 | 1.04 |
2.3. Optimization of HPLC Conditions
2.4. Identification of the Separated Peaks
| Position | 1 a | 2 a | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 2 | 157.5 | 157.4 | ||
| 3 | 139.0 | 139.0 | ||
| 4 | 178.0 | 178.0 | ||
| 5 | 161.4 | 161.3 | ||
| 6 | 98.7 | 6.22 (1H, d, J = 1.6) | 98.7 | 6.22 (1H, d, J = 2.1) |
| 7 | 164.3 | 164.3 | ||
| 8 | 93.7 | 6.34 (1H, d, J = 1.6) | 93.7 | 6.34 (1H, d, J = 2.1) |
| 9 | 156.8 | 156.8 | ||
| 10 | 104.7 | 104.7 | ||
| 1′ | 124.7 | 124.3 | ||
| 2′ | 129.3 | 129.3 | ||
| 3′ | 138.3 | 138.3 | ||
| 4′ | 142.0 | 142.0 | ||
| 5′ | 115.1 | 7.01 (1H, d, J = 8.4) | 115.1 | 7.01 (1H, d, J = 8.4) |
| 6′ | 124.1 | 7.13 (1H, d, J = 8.4) | 124.1 | 7.12 (1H, d, J = 8.4) |
| 2″ | 100.1 | 100.2 | ||
| 3″ | 90.2 | 90.2 | ||
| 4″ | 187.3 | 187.3 | ||
| 5″ | 163.0 | 163.1 | ||
| 6″ | 97.1 | 5.97 (1H, s) | 97.1 | 5.98 (1H, d, J = 2.1) |
| 7″ | 167.8 | 167.8 | ||
| 8″ | 96.2 | 5.97 (1H, s) | 96.1 | 5.99 (1H, d, J = 2.1) |
| 9″ | 159.1 | 159.1 | ||
| 10″ | 99.8 | 99.7 | ||
| 1′′′ | 124.1 | 124.1 | ||
| 2′′′ | 114.7 | 7.11 (1H, d, J = 2.1) | 129.1 | 7.44 (1H, dd, J = 6.9, 2.1) |
| 3′′′ | 144.6 | 114.7 | 6.75 (1H, dd, J = 6.9, 2.1) | |
| 4′′′ | 146.8 | 158.6 | ||
| 5′′′ | 115.6 | 6.68 (1H, d, J = 8.4) | 114.7 | 6.75 (1H, dd, J = 6.9, 2.1) |
| 6′′′ | 119.1 | 6.89 (1H, dd, J = 8.4, 2.1) | 129. | 7.44 (1H, dd, J = 6.9, 2.1) |
| OCH3 | 60.1 | 3.67 (3H, s) | 60.1 | 3.66 (3H, s) |
| 1″″ | 25.5 | 3.28 (2H, d, J = 7.0) | 25.5 | 3.27 (2H, d, J = 6.8) |
| 2″″ | 121.2 | 5.01 (1H, t, J = 7.0) | 121.2 | 5.01 (1H, t, J = 6.8) |
| 3″″ | 131.7 | 131.6 | ||
| 4″″ | 17.3 | 1.27 (3H, s) | 17.3 | 1.26 (3H, s) |
| 5″″ | 25.3 | 1.48 (3H, s) | 25.2 | 1.48 (3H, s) |
| OH | 12.55 (1H, s) | 12.55 (1H, s) | ||

2.5. The Cytotoxic Activity of Target Compounds
| Compound | MCF-7 | HepG2 |
|---|---|---|
| 1 | 29.8 ± 2.0 | 41.6 ± 1.9 |
| 2 | 42.6 ± 3.1 | 67.5 ± 2.6 |
| etoposide | 3.17 ± 0.25 | 0.48 ± 0.03 |
3. Experimental Section
3.1. Apparatus
3.2. Materials and Reagents
3.3. Preparation of the Crude Extract
3.4. Encrichment of the Target Compounds by Sephadex LH-20 Column
3.5. Further Purification by HSCCC
3.5.1. Determination of the Partition Coefficient (K) Value
3.5.2. Preparation of Two-Phase Solvent System and Sample Solution
3.5.3. HSCCC Separation Procedure
3.5.4. HPLC Analysis and Identification of HSCCC Peaks
3.5.5. Computational Methods
3.5.6. In Vitro Cytotoxic Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, C.Q.; Cao, W.; Nagatsu, A.; Ogihara, Y. Three new glycosides from Sinopodophyllum emodi (Wall.) Ying. Chem. Pharm. Bull. 2001, 49, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Xiao, J.J.; Meng, S.C.; Dong, X.M.; Ge, Y.W.; Wang, R.F.; Shang, M.Y.; Cai, S.Q. A new cytotoxic flavonoid from the fruit of Sinopodophyllum hexandrum. Fitoterapia 2010, 81, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Q.; Zhu, Y.Y.; Chen, S.Y.; Ogihara, Y. Lignan glucoside from Sinopodophyllum emodi and its cytotoxic activity. Chin. Chem. Lett. 2011, 22, 181–184. [Google Scholar] [CrossRef]
- Yang, X.Z.; Shao, H.; Zhang, L.Q.; Zhou, C.; Xuan, Q.; Yang, C.Y. Present situation of studies on resources of podophyllotoxin. Chin. Tradit. Herb. Drugs 2001, 32, 1042–1044. [Google Scholar]
- Shi, X.L.; Li, X.W.; Liu, J.B.; Zhou, H.Y.; Zhang, H.Q.; Jin, Y.R. Lignan extraction from the roots of Sinopodophyllum emodi Wall by matrix solid-phase dispersion. Chromatographia 2010, 72, 713–717. [Google Scholar] [CrossRef]
- Zhao, C.Q.; Huang, J.; Nagatsu, A.; Ogihara, Y. Two new podophyllotoxin glycosides from Sinopodophyllum emodi (Wall.) Ying. Chem. Pharm. Bull. 2001, 49, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Q.; Nagatsu, A.; Hatano, K.; Shirai, N.; Kato, S.; Ogihara, Y. New lignan glycosides from Chinese medicinal plant, Sinopodophyllum emodi. Chem. Pharm. Bull. 2003, 51, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; Li, Z.L.; Chen, H.; Liu, X.Q.; Zhou, W.; Hua, H.M. Three new cytotoxic aryltetralin lignans from Sinopodophyllum emodi. Bioorg. Med. Chem. Lett. 2011, 21, 3794–3797. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; Li, Z.L.; Chen, H.; Liu, X.Q.; Zhou, W.; Hua, H.M. Four new cytotoxic tetrahydrofuranoid lignans from Sinopodophyllum emodi. Planta Med. 2012, 78, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; Zhou, W.; Chen, H.; Li, Z.L.; Hua, H.M. Isolation and identification of flavonoids from the roots and rhizomes of Sinopodophyllum emodi. J. Shenyang Pharm. Univ. 2012, 29, 185–189. [Google Scholar]
- Sun, Y.J.; Sun, Y.S.; Chen, H.; Hao, Z.Y.; Wang, J.M.; Guan, Y.B.; Zhang, Y.L.; Feng, W.S.; Zheng, X.K. Isolation of two new prenylated flavonoids from Sinopodophyllum emodi fruit by silica gel column and high-speed counter-current chromatography. J. Chromatogr. B 2014, 969, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; Hao, Z.Y.; Si, J.G.; Wang, Y.; Zhang, Y.L.; Wang, J.M.; Gao, M.L.; Chen, H. Prenylated flavonoids from the fruits of Sinopodophyllum emodi and their cytotoxic activities. RSC Adv. 2015, 5, 82736–82742. [Google Scholar] [CrossRef]
- Sun, Y.J.; Li, Z.L.; Chen, H.; Zhou, W.; Hua, H.M. Study on chemical constituents from the roots and rhizomes of Sinopodophyllum emodi. J. Chin. Med. Mat. 2012, 35, 1607–1609. [Google Scholar]
- Sun, Y.J.; Zhou, W.; Chen, H.; Li, Z.L.; Hua, H.M. Phenols from roots and rhizomes of Sinopodophyllum emodi. Chin. Tradit. Herb. Drugs 2012, 43, 226–229. [Google Scholar]
- Arens, H.; Ulbrich, B.; Fischer, H.; Parnham, M.J.; Römer, A. Novel antiinflammatory flavonoids from podophyllum versipelle cell culture. Planta Med. 1986, 52, 468–473. [Google Scholar] [CrossRef]
- Mercader, A.G.; Pomilio, A.B. Naturally-occurring dimers of flavonoids as anticarcinogens. Anticancer Agents Med. Chem. 2013, 13, 1217–1235. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.A.; Fisher, D. Role of counter-current chromatography in the modernisation of Chinese herbal medicines. J. Chromatogr. A 2009, 1216, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.; Hewitson, P.; Ignatova, S. Scale-up of counter-current chromatography: Demonstration of predictable isocratic and quasi-continuous operating modes from the test tube to pilot/process scale. J. Chromatogr. A 2009, 1216, 8787–8792. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dong, X.R.; Shuai, M.; Liang, Y.Z. Simultaneous separation and purification of calycosin and formononetin from crude extract of Astragalus membranaceus Bge. var. mongholicus (Bge.) using high speed counter current chromatography. J. Anal. Chem. 2015, 70, 92–97. [Google Scholar] [CrossRef]
- Li, S.G.; Zhao, M.F.; Li, Y.X.; Sui, Y.X.; Yao, H.; Huang, L.Y.; Lin, X.H. Preparative isolation of six anti-tumour biflavonoids from Selaginella doederleinii Hieron by high-speed counter-current chromatography. Phytochem. Anal. 2014, 25, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Shi, S.Y.; Wang, Y.X.; Huang, K.L. Target-guided isolation and purification of antioxidants from Selaginella sinensis by offline coupling of DPPH-HPLC and HSCCC experiments. J. Chromatogr. B 2011, 879, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Liu, G.X.; Xu, F.; Shang, M.Y.; Cai, S.Q. Research on chemical fingerprint chromatograms of Sinopodophyllum hexandrum. Chin. J. Chin. Mater. Med. 2013, 38, 3528–3533. [Google Scholar]
- Fang, L.; Liu, Y.Q.; Yang, B.; Wang, X.; Huang, L.Q. Separation of alkaloids from herbs using high-speed counter-current chromatography. J. Sep. Sci. 2011, 34, 2545–2558. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.J.; Liu, Q.; Xie, Y.X.; Zeng, H.L.; Zhang, L.; Jiang, X.Y.; Chen, X.Q. Separation of five flavonoids from tartary buckwheat (Fagopyrum tataricum (L.) Gaertn) grains via off-line two dimensional high-speed counter-current chromatography. Food Chem. 2015, 186, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Z.; Xiang, H.Y.; Luo, Y.Q.; Song, L.Y.; Liu, Y.; Hou, L.B.; Xie, Y. Preparative separation and purification of three flavonoids from the anti-inflammatory effective fraction of Smilax china L. by high-speed counter-current chromatography. Sep. Sci. Technol. 2014, 49, 2090–2097. [Google Scholar] [CrossRef]
- Goto, H.; Osawa, E. Corner flapping: A simple and fast algorithm for exhaustive generation of ring conformations. J. Am. Chem. Soc. 1989, 111, 8950–8951. [Google Scholar] [CrossRef]
- Goto, H.; Osawa, E. An efficient algorithm for searching low-energy conformers of cyclic and acyclic molecules. J. Chem. Soc. Perkin Trans. 1993, 2, 187–198. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Shang, M.Y.; Wang, Q.H.; Xiao, J.J.; Shang, Y.H.; Kong, Y.; Cai, S.Q. New Isopentenyl Flavone-Like Compounds and Antitumor Application Thereof. C.N. 102382092 A, 21 March 2012. [Google Scholar]
- Shang, M.Y.; Kong, Y.; Xiao, J.J.; Ma, X.J.; Ge, Y.W.; Cai, S.Q. Isopentenyl Flavone Compound Extracted from Sinopodophyllum hexandrum Fruit, Its Preparation Process and Its Application to Prepare the Medical Preparations for Treating Breast Neoplasm. C.N. 101648934 A, 24 September 2009. [Google Scholar]
- Shang, M.Y.; Wang, Q.H.; Xiao, J.J.; Shang, Y.H.; Kong, Y.; Cai, S.Q. Application of Flavonoid Compounds in Treatment of Breast Cancer. C.N. 102335165 A, 15 July 2011. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.-J.; Pei, L.-X.; Wang, K.-B.; Sun, Y.-S.; Wang, J.-M.; Zhang, Y.-L.; Gao, M.-L.; Ji, B.-Y. Preparative Isolation of Two Prenylated Biflavonoids from the Roots and Rhizomes of Sinopodophyllum emodi by Sephadex LH-20 Column and High-Speed Counter-Current Chromatography. Molecules 2016, 21, 10. https://doi.org/10.3390/molecules21010010
Sun Y-J, Pei L-X, Wang K-B, Sun Y-S, Wang J-M, Zhang Y-L, Gao M-L, Ji B-Y. Preparative Isolation of Two Prenylated Biflavonoids from the Roots and Rhizomes of Sinopodophyllum emodi by Sephadex LH-20 Column and High-Speed Counter-Current Chromatography. Molecules. 2016; 21(1):10. https://doi.org/10.3390/molecules21010010
Chicago/Turabian StyleSun, Yan-Jun, Li-Xin Pei, Kai-Bo Wang, Yin-Shi Sun, Jun-Min Wang, Yan-Li Zhang, Mei-Ling Gao, and Bao-Yu Ji. 2016. "Preparative Isolation of Two Prenylated Biflavonoids from the Roots and Rhizomes of Sinopodophyllum emodi by Sephadex LH-20 Column and High-Speed Counter-Current Chromatography" Molecules 21, no. 1: 10. https://doi.org/10.3390/molecules21010010