
Improved Schmidt Conversion of Aldehydes to Nitriles Using Azidotrimethylsilane in 1,1,1,3,3,3-Hexafluoro-2-propanol


Abstract
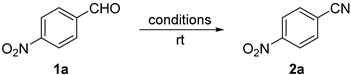
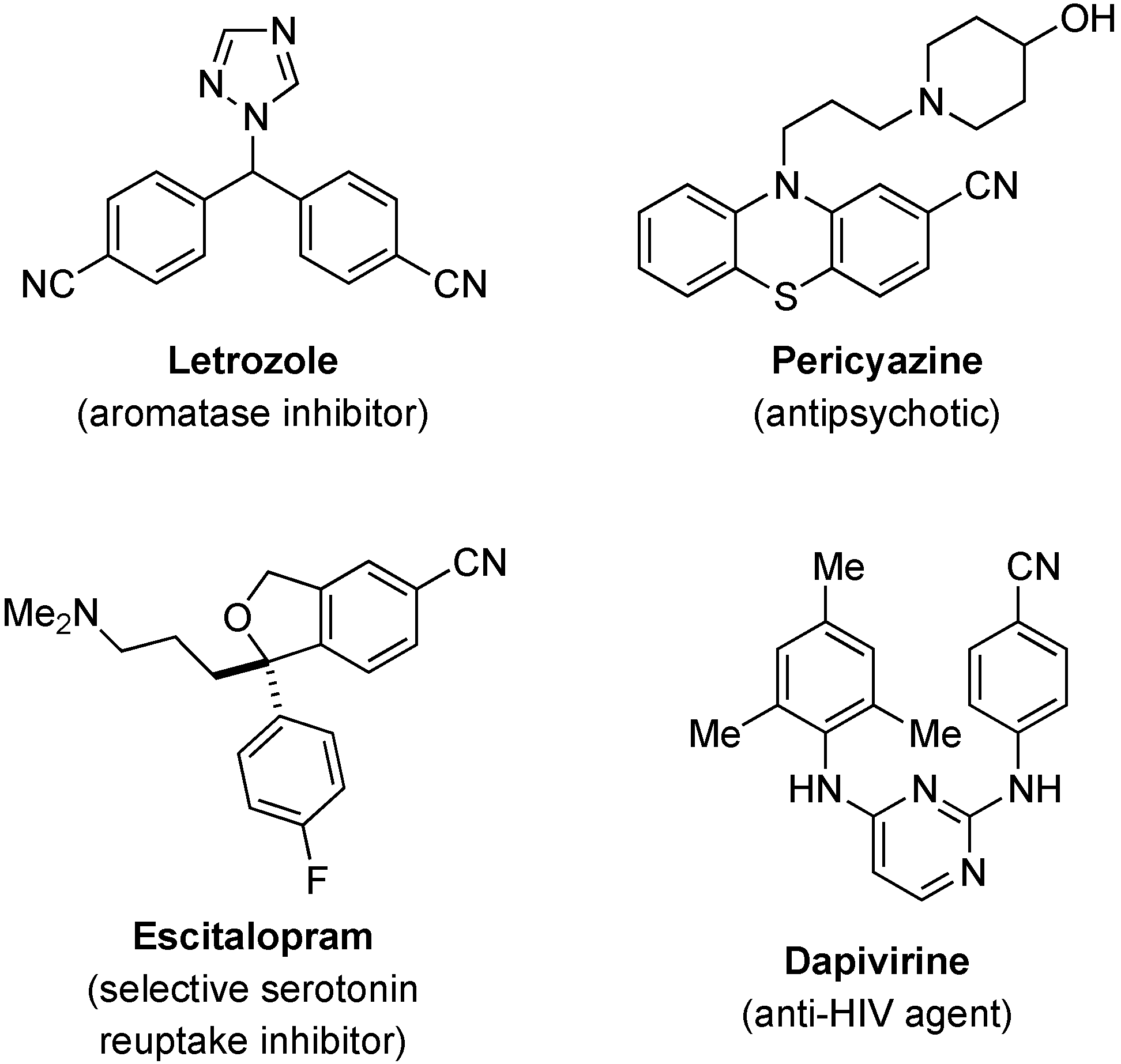
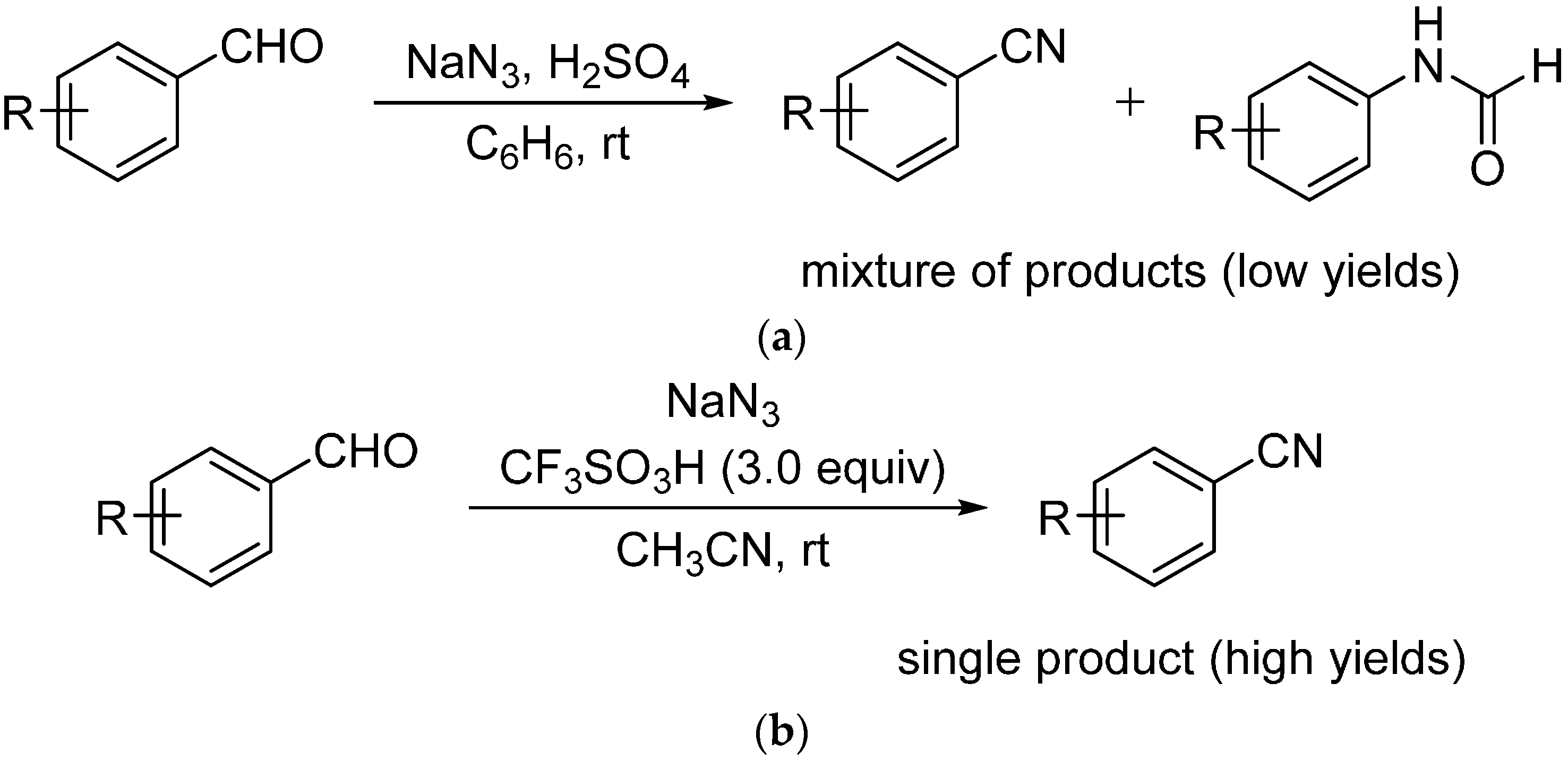
:1. Introduction


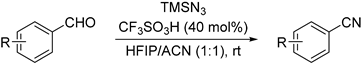
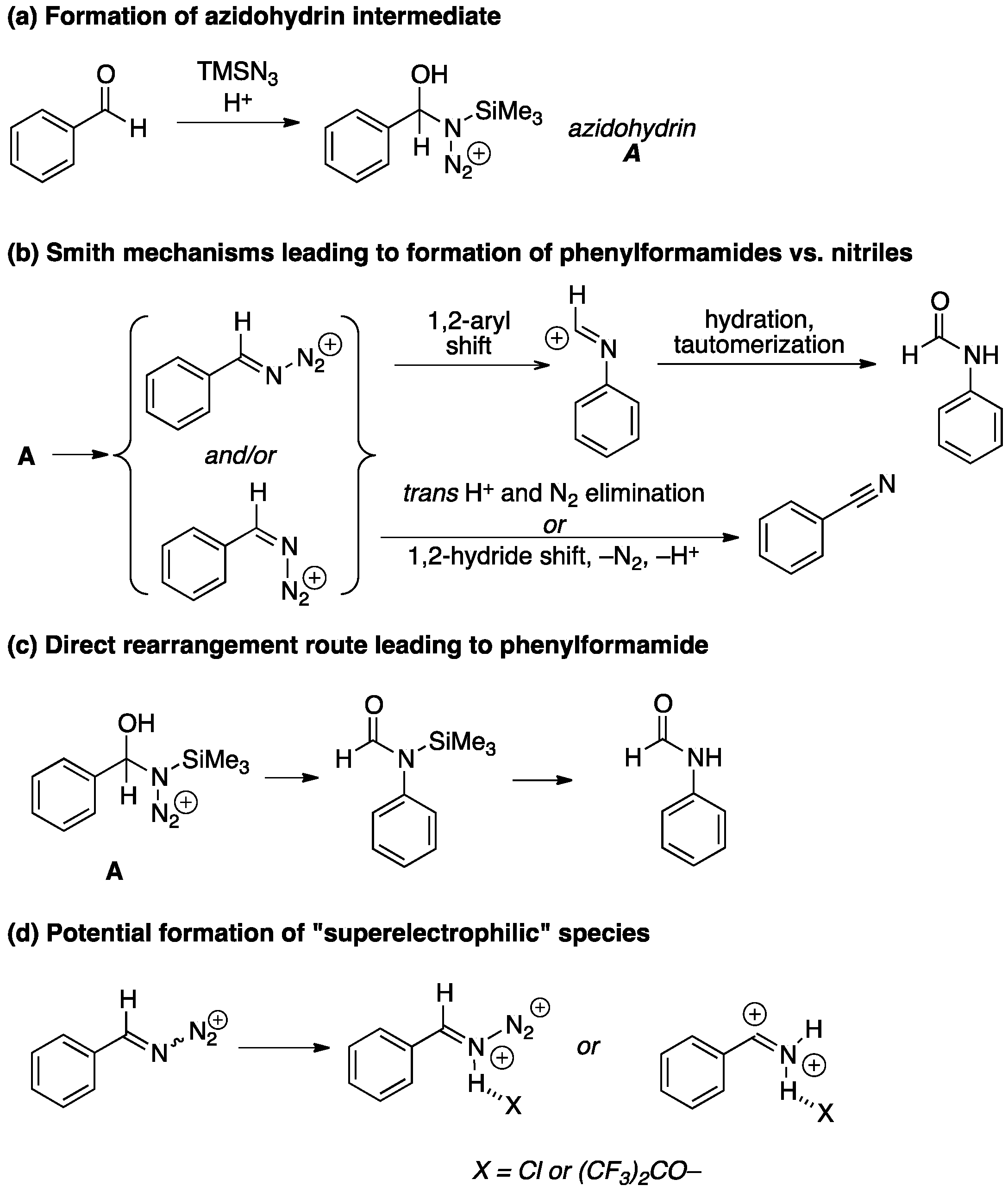
2. Results and Discussion
2.1. Optimization of Reaction Conditions

| Entry | Azide Source | Azide (equiv) | Catalyst | Catalyst (mol %) | Solvent | Time (h) | NMR Ratio (2a:1a) c | Yield (%) d 2a |
|---|---|---|---|---|---|---|---|---|
| 1 | NaN3 | 1.5 | CF3SO3H | 50 | HFIP | 16 | 30:70 e | ND |
| 2 | NaN3 | 1.5 | CH3COCl f | 80 | HFIP | 8 | 19:81 | ND |
| 3 | TMSN3 | 1.5 | TiCl4 g | 25 | HFIP | 24 | ND | 75 |
| 4 | TMSN3 | 1.5 | CF3SO3H | 25 | HFIP | 8 | ND | 68 |
| 5 | TMSN3 | 2.0 | CF3SO3H | 30 | HFIP | 2 | ND | 65 h |
| 6 | TMSN3 | 2.0 | CF3SO3H | 30 | HFIP/ACN (1:1) | 4 | ND | 81 |
| 7 | TMSN3 | 2.0 | CF3SO3H | 40 | HFIP/ACN (1:1) | 45 min | ND | 83 |
2.2. Substrate Scope
{kind=link}
{kind=link}
{kind=link}
| Entry | Aldehyde 1 | Nitrile 2 (% yield) c | Entry | Aldehyde 1 | Nitrile 2 (% yield) c |
|---|---|---|---|---|---|
| 1 | 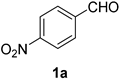 | 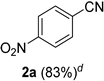 | 17 |  |  |
| 2 |  | 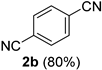 | 18 |  | 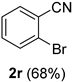 |
| 3 | 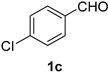 | 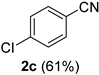 | 19 | 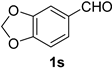 |  |
| 4 |  |  | 20 |  |  |
| 5 |  | 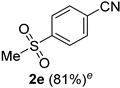 | 21 | 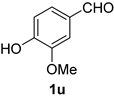 | 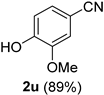 |
| 6 | 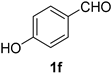 |  | 22 | 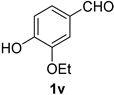 | 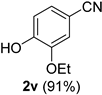 |
| 7 | 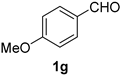 | 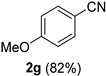 | 23 |  |  |
| 8 | 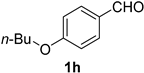 | 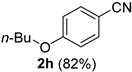 | 24 |  | 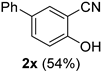 |
| 9 |  | 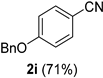 | 25 | 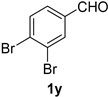 |  |
| 10 | 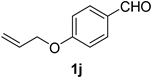 | 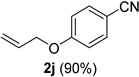 | 26 | 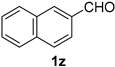 | 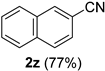 |
| 11 |  | 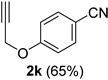 | 27 | 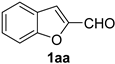 | 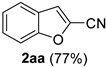 |
| 12 |  | 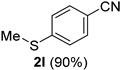 | 28 | 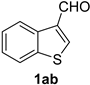 | 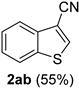 |
| 13 | 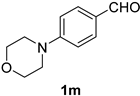 | 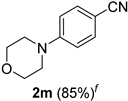 | 29 | 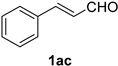 | 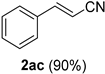 |
| 14 |  | 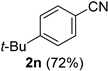 | 30 | 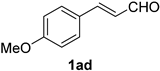 | 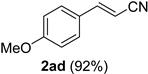 |
| 15 | 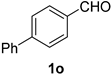 |  | 31 | 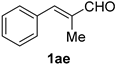 | 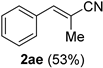 |
| 16 | 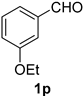 | 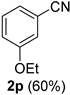 | 32 | 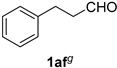 | 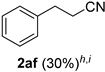 |

3. Experimental Section
3.1. General Information
3.2. General Procedure for the Optimization of Reaction Conditions for the Synthesis of 4-Nitrobenzonitrile 2a
3.3. General Procedure A for the Synthesis of Aromatic Nitriles
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fleming, F.F. Nitrile-containing natural products. Nat. Prod. Rep. 1999, 16, 597–606. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [PubMed]
- Fatiadi, A.J. Preparation and synthetic applications of cyano compounds. In Triple-Bonded Functional Groups; John Wiley & Sons, Ltd.: Chichester, UK, 1983; Volume 2, pp. 1057–1303. [Google Scholar]
- Larock, R.C. Comprehensive Organic Transformations; Wiley-VCH: New York, NY, USA, 1989. [Google Scholar]
- Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Syntheses, Patents, Applications, 4th ed.; Georg Thieme: Stuttgart, Germany, 2001. [Google Scholar]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; Wiley: Hoboken, NJ, USA, 2007; p. 2374. [Google Scholar]
- Sandmeyer, T. Substitution of the amido-group in aromatic derivatives by chlorine, bromine, and cyanogen. Chem. Ber. 1884, 17, 2650–2653. [Google Scholar] [CrossRef]
- Sandmeyer, T. Ueberführung der drei nitraniline in die nitrobenzoësäuren. Chem. Ber. 1885, 18, 1492–1496. [Google Scholar] [CrossRef]
- Mowry, D.T. The preparation of nitriles. Chem. Rev. 1948, 42, 189–283. [Google Scholar] [CrossRef] [PubMed]
- Rosenmund, K.W.; Struck, E. Halogen attached to a ring carbon atom and its replacement by other substituents. I. Replacement of the halogen by the carboxyl group. Chem. Ber. 1919, 52, 1749–1756. [Google Scholar] [CrossRef]
- V. Braun, J.; Manz, G. Fluoranthen und seine derivate. III. Mitteilung. Justus Liebigs Ann. Chem. 1931, 488, 111–126. [Google Scholar] [CrossRef]
- Takagi, K.; Okamoto, T.; Sakakibara, Y.; Oka, S. Palladium(II) catalyzed synthesis of aryl cyanides from aryl halides. Chem. Lett. 1973, 2, 471–474. [Google Scholar] [CrossRef]
- Ellis, G.P.; Romney-Alexander, T.M. Cyanation of aromatic halides. Chem. Rev. 1987, 87, 779–794. [Google Scholar] [CrossRef]
- Anbarasan, P.; Schareina, T.; Beller, M. Recent developments and perspectives in palladium-catalyzed cyanation of aryl halides: Synthesis of benzonitriles. Chem. Soc. Rev. 2011, 40, 5049–5067. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Jin, J.; Mei, Y.; Lu, P.; Wang, Y. Copper-mediated cyanation of aryl halides by activation of benzyl Cyanide as the cyanide source. Eur. J. Org. Chem. 2013, 2013, 4032–4036. [Google Scholar] [CrossRef]
- Kim, J.; Chang, S. A new combined source of “CN” from N,N-dimethylformamide and ammonia in the palladium-catalyzed cyanation of aryl C–H bonds. J. Am. Chem. Soc. 2010, 132, 10272–10274. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Kuang, C.; Zhang, Y.; Wang, J. Palladium-catalyzed direct cyanation of indoles with K4[Fe(CN)6]. Org. Lett. 2010, 12, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Do, H.-Q.; Daugulis, O. Copper-catalyzed cyanation of heterocycle carbon-hydrogen bonds. Org. Lett. 2010, 12, 2517–2519. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Wang, J. Lewis acid catalyzed direct cyanation of indoles and pyrroles with N-cyano-N-phenyl-p-toluenesulfonamide (NCTS). Org. Lett. 2011, 13, 5608–5611. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Jiao, N. Direct transformation of N,N-dimethylformamide to –CN: Pd-catalyzed cyanation of heteroarenes via C–H functionalization. J. Am. Chem. Soc. 2011, 133, 12374–12377. [Google Scholar] [CrossRef] [PubMed]
- Pollak, P.; Romeder, G.; Hagedorn, F.; Gelbke, H.-P. Nitriles. In Ullmann's Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Lücke, B.; Martin, A. Fine Chemicals through Heterogeneous Catalysis; VCH-Wiley: Weinheim, Germany, 2001. [Google Scholar]
- Lücke, B.; Narayana, K.V.; Martin, A.; Jähnisch, K. Oxidation and ammoxidation of aromatics. Adv. Synth. Catal. 2004, 346, 1407–1424. [Google Scholar] [CrossRef]
- Hatsuda, M.; Seki, M. A practical synthesis of highly functionalized aryl nitriles through cyanation of aryl bromides employing heterogeneous Pd/C: In quest of an industrially viable process. Tetrahedron 2005, 61, 9908–9917. [Google Scholar] [CrossRef]
- Nakajima, N.; Saito, M.; Ubukata, M. Activated dimethyl sulfoxide dehydration of amide and its application to one-pot preparation of benzyl-type perfluoroimidates. Tetrahedron 2002, 58, 3561–3577. [Google Scholar] [CrossRef]
- Kuo, C.-W.; Zhu, J.-L.; Wu, J.-D.; Chu, C.-M.; Yao, C.-F.; Shia, K.-S. A convenient new procedure for converting primary amides into nitriles. Chem. Commun. 2007, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Narang, S.C.; Fung, A.P.; Balaram Gupta, B.G. Synthetic Methods and Reactions; 82. Cyanuric Chloride, a Mild Dehydrating Agent in the Preparation of Nitriles from Amides. Synthesis 1980, 1980, 657–658. [Google Scholar]
- Zhou, S.; Junge, K.; Addis, D.; Das, S.; Beller, M. A general and convenient catalytic synthesis of nitriles from amides and silanes. Org. Lett. 2009, 11, 2461–2464. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Chang, S. Highly efficient and catalytic conversion of aldoximes to nitriles. Org. Lett. 2001, 3, 4209–4211. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M.; An, J.; Lindovska, P.; Lewis, W. Phosphonium salt-catalysed synthesis of nitriles from in situ activated oximes. Tetrahedron 2012, 68, 2899–2905. [Google Scholar] [CrossRef]
- Choi, E.; Lee, C.; Na, Y.; Chang, S. [RuCl2(p-cymene)]2 on carbon: An efficient, selective, reusable, and environmentally versatile heterogeneous catalyst. Org. Lett. 2002, 4, 2369–2371. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Giacomelli, G.; Porcheddu, A. Beckmann rearrangement of oximes under very mild conditions. J. Org. Chem. 2002, 67, 6272–6274. [Google Scholar] [CrossRef] [PubMed]
- Baxendale, I.R.; Ley, S.V.; Sneddon, H.F. A clean conversion of aldehydes to nitriles using a solid-supported hydrazine. Synlett 2002, 2002, 775–777. [Google Scholar] [CrossRef]
- Chill, S.T.; Mebane, R.C. A facile one-pot conversion of aldehydes into nitriles. Synth. Commun. 2009, 39, 3601–3606. [Google Scholar] [CrossRef]
- Srinivas, K.V.N.S.; Reddy, E.B.; Das, B. Highly convenient and efficient one-pot conversions of aldehydes into nitriles and ketones into amides using HY-zeolite. Synlett. 2002, 2002, 0625–0627. [Google Scholar] [CrossRef]
- Laulhé, S.; Gori, S.S.; Nantz, M.H. A chemoselective, one-pot transformation of aldehydes to nitriles. J. Org. Chem. 2012, 77, 9334–9337. [Google Scholar] [CrossRef] [PubMed]
- Augustine, J.K.; Bombrun, A.; Atta, R.N. A practical and cost-efficient, one-pot conversion of aldehydes into nitriles mediated by “activated DMSO”. Synlett 2011, 2011, 2223–2227. [Google Scholar] [CrossRef]
- Telvekar, V.N.; Patel, K.N.; Kundaikar, H.S.; Chaudhari, H.K. A novel system for the synthesis of nitriles from aldehydes using aqueous ammonia and sodium dichloroiodate. Tetrahedron Lett. 2008, 49, 2213–2215. [Google Scholar] [CrossRef]
- Kelly, C.B.; Lambert, K.M.; Mercadante, M.A.; Ovian, J.M.; Bailey, W.F.; Leadbeater, N.E. Access to nitriles from aldehydes mediated by an oxoammonium salt. Angew. Chem. Int. Ed. 2015, 54, 4241–4245. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani-Choghamarani, A.; Zolfigol, M.A.; Hajjami, M.; Sardari, S. Direct synthesis of nitriles from alcohols or aldehydes using H5IO6/KI in aqueous ammonia. Synth. Commun. 2012, 43, 52–58. [Google Scholar] [CrossRef]
- Patil, U.B.; Shendage, S.S.; Nagarkar, J.M. One-pot synthesis of nitriles from aldehydes catalyzed by deep eutectic solvent. Synthesis 2013, 45, 3295–3299. [Google Scholar]
- Augustine, J.K.; Atta, R.N.; Ramappa, B.K.; Boodappa, C. Propylphosphonic anhydride (T3P®): A remarkably efficient reagent for the one-pot transformation of aromatic, heteroaromatic, and aliphatic aldehydes to nitriles. Synlett 2009, 2009, 3378–3382. [Google Scholar] [CrossRef]
- Zhu, J.-L.; Lee, F.-Y.; Wu, J.-D.; Kuo, C.-W.; Shia, K.-S. An efficient new procedure for the one-pot conversion of aldehydes into the corresponding nitriles. Synlett. 2007, 2007, 1317–1319. [Google Scholar] [CrossRef]
- Wrobleski, A.; Coombs, T.C.; Huh, C.W.; Li, S.-W.; Aubé, J. The Schmidt reaction. Org. React. 2012, 78, 1–320. [Google Scholar]
- McEwen, W.E.; Conrad, W.E.; VanderWerf, C.A. The Schmidt reaction applied to aldehydes and epoxides. J. Am. Chem. Soc. 1952, 74, 1168–1171. [Google Scholar] [CrossRef]
- Rokade, B.V.; Prabhu, K.R. Chemoselective Schmidt reaction mediated by triflic acid: Selective synthesis of nitriles from aldehydes. J. Org. Chem. 2012, 77, 5364–5370. [Google Scholar] [CrossRef] [PubMed]
- Nandi, G.C.; Laali, K.K. Schmidt reaction in ionic liquids: Highly efficient and selective conversion of aromatic and heteroaromatic aldehydes to nitriles with [BMIM(SO3H)][OTf] as catalyst and [BMIM][PF6] as solvent. Tetrahedron Lett. 2013, 54, 2177–2179. [Google Scholar] [CrossRef]
- Hazarika, N.; Baishya, G. One-pot sequential Schmidt and Ritter reactions for the synthesis of N-tert-butyl amides. Eur. J. Org. Chem. 2014, 2014, 5686–5690. [Google Scholar] [CrossRef]
- Motiwala, H.F.; Fehl, C.; Li, S.-W.; Hirt, E.; Porubsky, P.; Aubé, J. Overcoming Product Inhibition in Catalysis of the Intramolecular Schmidt Reaction. J. Am. Chem. Soc. 2013, 135, 9000–9009. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A.S. The Schmidt reaction: Experimental conditions and mechanism. J. Am. Chem. Soc. 1948, 70, 320–323. [Google Scholar] [CrossRef]
- Bach, R.D.; Wolber, G.J. Theoretical study of the barrier to nitrogen inversion in N-cyano- and N-diazoformimine. Mechanism of the Schmidt reaction. J. Org. Chem. 1982, 47, 239–245. [Google Scholar] [CrossRef]
- Ostrovskii, V.A.; Koldobskii, G.I.; Shirokova, N.P.; Gidaspov, B.V. Mechanism of the Schmidt reaction. XVIII. Kinetics of reaction of substituted benzaldehydes with hydrazoic acid in aqueous solutions of sulfuric acid. Zh. Org. Khim. 1977, 13, 339–343. [Google Scholar]
- Ostrovskii, V.A.; Kuznetsov, A.V.; Koldobskii, G.I.; Shirokova, N.P.; Gidaspov, B.V. Mechanism of the Schmidt reaction. XIV. Determination of the ratio of reaction products of substituted benzaldehydes with hydrazoic acid. Zh. Org. Khim. 1975, 11, 1027–1030. [Google Scholar]
- Raja, E.K.; Klumpp, D.A. Superelectrophilic chemistry of amino-nitriles and related substrates. Tetrahedron 2011, 67, 4494–4497. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, D.; Kawagoe, Y.; Moriyama, K.; Togo, H. Direct oxidative conversion of methylarenes into aromatic nitriles. Org. Lett. 2013, 15, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, H.; Zhang, X.; Ye, J.; Liu, J.; Xu, Q.; Lautens, M. Organoselenium-catalyzed mild dehydration of aldoximes: An unexpected practical method for organonitrile synthesis. Org. Lett. 2014, 16, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Guo, C.-X.; Zhong, C.-L.; Diao, Z.-F.; He, L.-N. Metal-free chemoselective oxidation of sulfides by in situ generated Koser’s reagent in aqueous media. Tetrahedron Lett. 2014, 55, 1818–1821. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, K.T.; Jeon, H.B. Deoxygenation of sulfoxides to sulfides with thionyl chloride and triphenylphosphine: Competition with the Pummerer reaction. J. Org. Chem. 2013, 78, 6328–6331. [Google Scholar] [CrossRef] [PubMed]
- Al-Amin, M.; Arai, S.; Hoshiya, N.; Honma, T.; Tamenori, Y.; Sato, T.; Yokoyama, M.; Ishii, A.; Takeuchi, M.; Maruko, T.; et al. Development of second generation gold-supported palladium material with low-leaching and recyclable characteristics in aromatic amination. J. Org. Chem. 2013, 78, 7575–7581. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Srivastava, V.P.; Yadav, L.D.S. Visible-light-mediated efficient conversion of aldoximes and primary amides into nitriles. RSC Adv. 2014, 4, 4181–4186. [Google Scholar] [CrossRef]
- Saha, D.; Saha, A.; Ranu, B.C. Ionic liquid-promoted dehydration of aldoximes: A convenient access to aromatic, heteroaromatic and aliphatic nitriles. Tetrahedron Lett. 2009, 50, 6088–6091. [Google Scholar] [CrossRef]
- Dev, D.; Palakurthy, N.B.; Kumar, N.; Mandal, B. An unexpected involvement of ethyl-2-cyano-2-(hydroxyimino) acetate cleaved product in the promotion of the synthesis of nitriles from aldoximes: A mechanistic perception. Tetrahedron Lett. 2013, 54, 4397–4400. [Google Scholar] [CrossRef]
- Rudenko, A.P.; Salfetnikova, Y.N.; Vasil’ev, A.V. Oxidation of aromatic compounds. V. Oxidation of substituted benzonitriles and 2,4,6-triaryl-1,3,5-triazines in the HSO3F-PbO2 system. Zh. Org. Khim. 1996, 32, 1499. [Google Scholar]
- Luo, Y.; Wen, Q.; Wu, Z.; Jin, J.; Lu, P.; Wang, Y. Copper-mediated cyanation of aryl boronic acids using benzyl cyanide. Tetrahedron 2013, 69, 8400–8404. [Google Scholar] [CrossRef]
- Cohen, D.T.; Buchwald, S.L. Mild palladium-catalyzed cyanation of (hetero)aryl halides and triflates in aqueous media. Org. Lett. 2015, 17, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Powell, K.J.; Han, L.-C.; Sharma, P.; Moses, J.E. Chemoselective palladium-catalyzed cyanation of alkenyl halides. Org. Lett. 2014, 16, 2158–2161. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, T.; Sankranti, R.; Vaughan, T.G.; Maejima, T.; Yanase, T. An α-diaminoboryl carbanion assisted stereoselective single-pot preparation of α,β-disubstituted acrylonitriles. J. Org. Chem. 2011, 76, 8053–8058. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xu, J.; Zhang, L.; Jiao, N. An efficient transformation from benzyl or allyl halides to aryl and alkenyl nitriles. Org. Lett. 2010, 12, 2888–2891. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, K.; Kelleher, S.; Evans, P. An investigation into the one-pot Heck olefination-hydrogenation reaction. J. Org. Chem. 2011, 76, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Mirjafari, A.; Mohammadpoor-Baltork, I.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Khosropour, A.R. Microwave-promoted, one-pot conversion of alkoxymethylated protected alcohols into their corresponding nitriles, bromides, and iodides using [bmim][InCl4] as a green catalyst. Tetrahedron Lett. 2010, 51, 3274–3276. [Google Scholar] [CrossRef]
- Sample Availability: Not avaiable.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motiwala, H.F.; Yin, Q.; Aubé, J. Improved Schmidt Conversion of Aldehydes to Nitriles Using Azidotrimethylsilane in 1,1,1,3,3,3-Hexafluoro-2-propanol. Molecules 2016, 21, 45. https://doi.org/10.3390/molecules21010045
Motiwala HF, Yin Q, Aubé J. Improved Schmidt Conversion of Aldehydes to Nitriles Using Azidotrimethylsilane in 1,1,1,3,3,3-Hexafluoro-2-propanol. Molecules. 2016; 21(1):45. https://doi.org/10.3390/molecules21010045
Chicago/Turabian StyleMotiwala, Hashim F., Qin Yin, and Jeffrey Aubé. 2016. "Improved Schmidt Conversion of Aldehydes to Nitriles Using Azidotrimethylsilane in 1,1,1,3,3,3-Hexafluoro-2-propanol" Molecules 21, no. 1: 45. https://doi.org/10.3390/molecules21010045