4. Materials and Methods
4.1. General Information
Unless otherwise stated, all chemicals were purchased as the highest purity commercially available and were used without further purification. IR spectra were recorded on an AVATAR 370 FT-IR spectrophotometer (Thermo Nicolet, Salamanca, Spain). 1H- and 13C-NMR spectra were recorded in CDCl3 and referenced to the residual peak of CHCl3 at δ 7.26 ppm and δ 77.0 ppm, for 1H and 13C, respectively, using 200 VX (Varian, Salamanca, Spain) and DRX 400 (Bruker, Salamanca, Spain) instruments. Chemical shifts are reported in δ parts per million and coupling constants (J) are given in hertz. MS were recorded using a VG TS 250 spectrometer at 70 eV ionising voltage (Fisons, Salamanca, Spain). Data are presented as m/z (% rel. int.). HRMS were recorded on a VG Platform spectrometer using the chemical ionization (ammonia as gas) or fast atom bombardment (FAB) techniques. For some of the samples, a QSTAR XL spectrometer (Evisa, Salamanca, Spain) was employed for electrospray ionization (ESI). Optical rotations were determined on a 241 polarimeter (Perkin-Elmer, Salamanca, Spain) in 1 dm cells. Diethyl ether and THF were distilled from sodium, and dichloromethane was distilled from calcium hydride under argon atmosphere.
4.2. Preparation of 1-O-Octadecyl-2,3-isopropyliden-sn-glycerol (16)
To a solution of (R)-(−)-solketal 15 (2.6 g, 19.7 mmol) in toluene (39 mL), NaNH2 (768 mg, 19.7 mmol) was added, and the mixture was heated at 111 °C under an argon atmosphere for 1 h. Then it was cooled to rt and a solution of bromooctadecane (6.5 g, 19.7 mmol) in toluene (5 mL) was added, before heating at 111 °C for 3 h. After that time, the reaction mixture was cooled at 0 °C, crushed ice and saturated NH4Cl were added and it was extracted with Et2O. The organic layer was washed with H2O and brine. After drying over anhydrous Na2SO4, the organic layer was filtered and evaporated. The obtained residue was purified by column chromatography (Hex/EtOAc 9:1) to yield 16 (6.9 g, 92%). −8.27 (c 1.6, CHCl3); IR (film, cm−1): 2985, 2924, 2854, 1465, 1369, 1255, 1118, 1057, 849; 1H-NMR (400 MHz, CDCl3, δ ppm): 4.26 (1H, quin., J = 6.0 Hz, H-sn2), 4.06 (1H, dd, J = 8.2, 6.0 Hz, HA-sn3), 3.73 (1H, dd, J = 8.2, 6.0 Hz, HB-sn3), 3.51 (1H, dd, J = 9.9, 6.0 Hz, HA-sn1), 3.47 (2H, t, J = 6.8 Hz, H-1′), 3.41 (1H, dd, J = 9.9, 5.6 Hz, HB-sn1), 1.56 (2H, m, H-2′), 1.42, 1.36 (3H, s, each, Me2C-), 1.25 (30H, m, H-3′-17′), 0.88 (3H, t, J = 6.8 Hz, H-18′); 13C-NMR (100 MHz, CDCl3, δ ppm): 109.3 (Me2C-), 74.7 (C-sn2), 71.9 (C-1′), 71.8 (C-sn1), 66.9 (C-sn3), 31.9 (C-16′), 29.4 (C-2′), 29.4 (C-4′-15′), 26.7, 25.4 (Me2C-), 26.0 (C-3′), 22.6 (C-17′), 14.0 (C-18′); EIHRMS: calcd. for C24H48O3 [M + H]+: 385.3676, found: 385.3680.
4.3. Preparation of 1-O-Octadecyl-sn-glycerol (17)
To a solution of 16 (4.7 g, 12.24 mmol) in MeOH (36 mL), p-TsOH (2.3 g, 12.24 mmol) was added and stirred at 35 °C for 8 h. Then H2O was added, and the reaction mixture was extracted with Et2O and washed with 6% NaHCO3 and H2O. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated to give 17 (3.9 g, 11.3 mmol, 93%). +0.95 (c 0.84, CHCl3); IR (film, cm−1): 3325, 2918, 2849, 1470 1119, 1063; 1H-NMR (400 MHz, CDCl3, δ ppm): 3.85 (1H, dddd, J = 6.0, 6.0, 4.0, 4.0 Hz, H-sn2), 3.71 (1H, dd, J = 11.4, 4.0 Hz, HA-sn3), 3.63 (1H, dd, J = 11.4, 6.0 Hz, HB-sn3), 3.53 (1H, dd, J = 9.6, 4.0 Hz, HA-sn1), 3.49 (1H, dd, J = 9.6, 6.0 Hz, HB-sn1), 3.47 (1H, ddd, J = 9.3, 6.7, 6.7 Hz, HA-1′), 3.44 (1H, ddd, J = 9.3, 6.7, 6.7 Hz, HB-1′), 1.56 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.87 (3H, t, J = 6.8 Hz, H-18′); 13C-NMR (100 MHz, CDCl3, δ ppm): 72.4 (C-sn1), 71.8 (C-1′), 70.4 (C-sn2), 64.2 (C-sn3), 31.8 (C-16′), 29.5 (C-2′), 29.5 (C-4′-15′), 26.0 (C-3′), 22.6 (C-17′), 14.0 (C-18′); EIHRMS: calcd. for C21H44O3 [M + Na]+: 367.3183, found: 367.3194.
4.4. Preparation of 1-O-Octadecyl-3-O-p-methoxybenzyl-sn-glycerol (18)
To a solution of 17 (200 mg, 0.58 mmol) in toluene (3.4 mL), dibutyl tin (IV) oxide (144 mg, 0.58 mmol) was added and it was heated up to reflux for 2 h in a Dean-Stark apparatus. After this time, the solvent was evaporated to give a white solid, and CsF (167 mg, 1.1 mmol) was added to this solid. The solid mixture was dried for 1 h 30 min under high vacuum. It was then diluted in DMF (3.4 mL) and PMBCl (258 mg, 1.65 mmol) added and stirred overnight under an argon atmosphere. Then H2O (1 mL) and EtOAc (3 mL) were added, the reaction mixture was stirred vigorously for 15 min and filtered through a pad of silica gel to remove the dibutyl tin oxide. The filtrate was washed with H2O and brine. Removal of the solvents gave a residue that was purified by column chromatography (Hex/EtOAc 96:4) to obtain 18 (215 mg, 80%). +1.2 (c 0.11, CHCl3); IR (film, cm−1): 3485, 3404, 2916, 2846, 1470, 1031; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.27 (2H, d, J = 7.4 Hz, H-2′′′, H-6′′′), 6.89 (2H, d, J = 7.4 Hz, H-3′′′, H-5′′ ), 4.50 (2H, s, –OCH2Ar), 3.98 (1H, quin, J = 4.8 Hz, H-sn2), 3.82 (3H, s, –OMe), 3.65–3.55 (6H, m, H-sn1, 1′, sn3), 1.56 (2H, m, H-2′), 1.28 (30H, m, H-3′-17′), 0.89 (3H, t, J = 6.2 Hz, H-18′); 13C-NMR (100 MHz, CDCl3, δ ppm): 159.2 (C-4′′′), 130.1 (C-1′′′), 129.3 (C-2′′′), 129.3 (C-6′′′), 113.8 (C-3′′′), 113.8 (C-5′′′), 73.1 (–OCH2Ar), 71.7 (C-1′), 71.6 (C-sn1), 71.0 (C-sn3), 69.5 (C-sn2), 55.2 (C–OMe), 31.9 (C-16′), 29.5 (C-2′), 29.5 (C-4′-15′), 26.1 (C-3′), 22.7 (C-17′), 14.1 (C-18′); EIHRMS: calcd. for C29H52O4 [M + Na]+: 487.3758, found: 487.3773.
4.5. Preparation of 1-O-Octadecyl-2-chlorocarbonyl-3-O-p-methoxybenzyl-sn-glycerol (19)
To an ice cooled solution of 18 (377 mg, 0.81 mmol) in THF (1.6 mL), trichloromethyl chloroformate (diphosgene, 160 mg, 0.81 mmol) and N,N-dimethylaniline (98 mg, 0.81 mmol) were added. The mixture was stirred at 0 °C for 10 min and then at rt overnight. Then Et2O was added and the white precipitate formed was filtered. The solution washed with 0.2 M HCl, 0.2 M NaOH and H2O, then dried over anhydrous Na2SO4 and evaporated to give 19 (353 mg, 0.67 mmol, 83%). IR (film, cm−1): 2924, 2852, 1780, 1166; 1H-NMR (200 MHz, CDCl3, δ ppm): 7.24 (1H, d, J = 7.0 Hz, each, H-2′′′, H-6′′′), 6.89 (1H, d, J = 7.4 Hz, each, H-3′′′, H-5′′′), 5.16 (1H, quin, J = 4.8 Hz, H-sn2), 4.49 (2H, s, –OCH2Ar), 3.81 (3H, s, –OMe), 3.64–3.59 (4H, m, H-sn1, sn3), 3.44–3.38 (2H, m, H-1′), 1.54 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.89 (3H, t, J = 6.2 Hz, H-18′); 13C-NMR (50 MHz, CDCl3, δ ppm): 159.2 (C-4′′′), 154.9 (–O–CO–Cl), 129.8 (C-1′′′), 129.5 (C-2′′′, 6′′′), 114.1 (C-3′′′, 5′′′), 80.5 (C-sn2), 73.3 (–OCH2Ar), 72.1 (C-1′), 68.9 (C-sn1), 67.9 (C-sn3), 55.5 (C–OMe), 32.2 (C-16′), 29.8 (C-2′), 29.8 (C-4′-15′), 26.2 (C-3′), 22.9 (C-17′), 14.3 (C-18′); EIHRMS: in MeOH, calcd. for methyl ester, C31H54O6 [M + Na]+: 545.3813, found: 545.3808.
4.6. Preparation of 1-O-Octadecyl-2-O-[1,25-epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-3-p-methoxybenzyl-sn-glycerol (20)
To a solution of 1/2 (153 mg, 0.43 mmol), N,N-diisopropylethylamine (DIPEA, 71 mg, 0.55 mmol), 4-(dimethylamino) pyridine (DMAP, 26 mg, 0.21 mmol) in toluene (2.1 mL), a solution of 19 (172 mg, 0.33 mmol) in toluene (1.65 mL) was added dropwise at 0 °C . The reaction mixture was stirred at 0 °C under an argon atmosphere for 15 min and then at rt overnight. After this time the solvent was removed and the residue was purified by column chromatography (Hex/EtOAc 99:1) to obtain 20 (167 mg, 60%). IR (film, cm−1): 2924, 2853, 1744, 1514, 1464, 1258, 1115; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.42/7.41 (1H, s, H-25′′), 7.34/7.33 (1H, s, H-1′′), 7.24/7.17 (2H, d, J = 8.8 Hz, H-2′′′, H-6′′′), 6.86/6.83 (2H, d, J = 8.8 Hz, H-3′′′, H-5′′′), 6.39 (1H, s, H-2′′), 5.81/5.79 (1H, dd, J = 5.6, 3.2 Hz, H-4′′), 5.35/5.33 (1H, t, J = 3.2 Hz, H-9′′), 4.98 (1H, m, H-sn2), 4.65–4.62 (2H, m, H-20′′), 4.49/4.45 (1H, d, J = 11.6 Hz, –OCH2Ar), 4.44/4.39 (1H, d, J = 11.6 Hz, –OCH2Ar), 3.80 (3H, s, –OMe), 3.64–3.50 (2H, m, H-sn1), 3.64–3.50 (2H, m, H-sn3), 3.47–3.31 (2H, m, H-1′), 2.20–1.40 (16H, m, H-5′′, 7′′, 8′′, 11′′, 12′′, 13′′, 14′′, 16′′, 17′′), 1.70/1.67 (3H, s, Me-21′′), 1.56 (2H, m, H-2′), 1.26 (30H, m, H-3′-17′), 0.91 (3H, s, Me-22′′), 0.89 (3H, t, J = 6.2 Hz, Me-18′), 0.88 (3H, s, Me-24′′), 0.81/0.80 (3H, d, J = 7.0 Hz, Me-23′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 159.2 (C-4′′′), 154.2 (–O–CO–O–), 147.0/146.9 (C-19′′), 143.1 (C-1′′), 141.4/141.0 (C-10′′), 140.0/139.9 (C-25′′), 130.0 (C-1′′′), 129.2 (C-2′′′, 6′′′), 126.2 (C-3′′), 120.0/119.7 (C-9′′), 113.7 (C-3′′′, 5′′′), 109.1/109.0 (C-20′′), 108.8/108.7 (C-2′′), 75.5/75.4 (C-sn2), 72.9 (–OCH2Ar), 71.7/71.6 (C-1′), 70.0 (C-4′′), 69.2/69.1 (C-sn1), 68.5/68.3 (C-sn3), 55.2 (–OMe), 44.3 (C-5′′), 42.9 (C-15′′), 42.6 (C-11′′), 38.9/38.7 (C-14′′), 37.4/37.2 (C-16′′), 34.1/34.0 (C-6′′), 32.4/32.3 (C-17′′), 31.9 (C-16′), 29.6–28.9 (C-2′), 29.6–28.9 (C-4′-15′), 29.6–28.9 (C-7′′, 13′′), 26.0/25.9 (C-3′), 23.2 (C-8′′, 12′′), 22.8 (C-24′′), 22.6 (C-17′), 22.3 (C-21′′), 22.2 (C-22′′), 15.6 (C-23′′), 14.0 (C-18′); EIHRMS: calcd. for C54H86O7 [M + Na]+: 869.6266, found: 869.6233.
4.7. Reaction of Compound 20 with DDQ: Preparation of 1-O-Octadecyl-2-O-[1,25-epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4S-yloxycarbonyl]-sn-glycerol (21) and 1-O-Octadecyl-2-O-[1,25-epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R-yloxycarbonyl]-sn-glycerol (22)
To a solution of 20 (170 mg, 0.2 mmol) in CH2Cl2/H2O 18:1 (2.2 mL), DDQ (54 mg, 0.24 mmol) was added. The reaction mixture was stirred at rt under an argon atmosphere for 1 h 15 min, quenched with 6% NaHCO3 and extracted with CH2Cl2. The organic layer was washed with 6% NaHCO3 and brine and dried over anhydrous Na2SO4. Removal of the solvent gave the crude product which was purified by column chromatography on silica gel to obtain 21 (46 mg, 32%, Hex/EtOAc 97:3 as eluent) and 22 (94 mg, 65%, Hex/EtOAc 95:5 as eluent).
Compound 21: +42.2 (c 0.46, CHCl3); IR (film, cm−1): 3464, 2924, 2853, 1744, 1260; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.43 (1H, s, H-25′′), 7.35 (1H, s, H-1′′), 6.40 (1H, s, H-2′′), 5.79 (1H, dd, J = 8.5, 3.6 Hz, H-4′′), 5.33 (1H, t, J = 4.8 Hz, H-9′′), 4.81 (1H, quin, J = 5.0 Hz, H-sn2), 4.65 and 4.64 (1H, s, each, H-20′′), 3.84 (1H, dd, J = 12.0, 5.0 Hz, HA-sn3), 3.79 (1H, dd, J = 12.0, 5.0 Hz, HB-sn3), 3.60 (1H, dd, J = 10.8, 5.0 Hz, HA-sn1), 3.56 (1H, dd, J = 10.8, 5.0 Hz, HB-sn1), 3.39 (2H, m, H-1′), 2.05–1.4 (16H, m, H-5′′, 7′′, 8′′, 11′′, 12′′, 13′′ 14′′, 16′′, 17′′), 1.70 (3H, s, Me-21′′), 1.56 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.90 (3H, s, Me-22′′), 0.89 (3H, s, Me-24′′), 0.88 (3H, t, J = 7.2 Hz, Me-18′), 0.80 (3H, d, J = 7.0 Hz, Me-23′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 154.2 (–O–CO–O–), 147.1 (C-19′′), 143.2 (C-1′′), 141.4 (C-10′′), 140.0 (C-25′′), 126.0 (C-3′′), 119.7 (C-9′′), 109.1 (C-20′′), 108.6 (C-2′′), 76.6 (C-sn2), 71.9 (C-1′), 70.4 (C-4′′), 69.5 (C-sn1), 62.6 (C-sn3), 44.2 (C-5′′), 42.9 (C-15′′), 42.4 (C-11′′), 38.7 (C-14′′), 37.4 (C-16′′), 34.0 (C-6′′), 32.4 (C-17′′), 31.9 (C-16′), 31.0 (C-7′′), 29.6–29.3 (C-2′), 29.6–29.3 (C-4′-15′), 28.8 (C-13′′), 25.9 (C-3′), 22.8 (C-12′′), 22.7 (C-24′′), 22.6 (C-8′′), 22.6 (C-17′), 22.5 (C-21′′), 22.3 (C-22′′), 15.6 (C-23′′), 14.1 (C-18′); EIHRMS: calcd. for C46H78O6 [M + Na]+: 749.5691, found: 749.5706.
Compound 22: +3.5 (c 0.40, CHCl3); IR (film, cm−1): 3477, 2924, 2853, 1742, 1261; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.43 (1H, s, H-25′′), 7.35 (1H, s, H-1′′), 6.40 (1H, s, H-2′′), 5.80 (1H, dd, J = 8.4, 3.4 Hz, H-4′′), 5.36 (1H, t, J = 3.4 Hz, H-9′′), 4.83 (1H, quin, J = 5.0 Hz, H-sn2), 4.65, 4.61 (1H, s, each, H-20′′), 3.79 (2H, m, H-sn3), 3.62 (1H, dd, J = 10.6, 5.0 Hz, HA-sn1), 3.60 (1H, dd, J = 10.6, 5.0 Hz, HB-sn1), 3.44 (2H, m, H-1′), 2.15–1.4 (16H, m, H-5′′, 7′′, 8′′, 11′′, 12′′, 13′′ 14′′, 16′′, 17′′), 1.67 (3H, s, Me-21′′), 1.56 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.91 (3H, s, Me-22′′), 0.88 (3H, s, Me-24′′), 0.86 (3H, t, J = 6.2 Hz, Me-18′), 0.81 (3H, d, J = 7.0 Hz, Me-23′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 154.2 (–O–CO–O–), 147.0 (C-19′′), 143.2 (C-1′′), 141.0 (C-10′′), 140.2 (C-25′′), 125.9 (C-3′′), 120.0 (C-9′′), 109.0 (C-20′′), 108.7 (C-2′′), 76.9 (C-sn2), 71.9 (C-1′), 70.3 (C-4′′), 69.5 (C-sn1), 62.6 (C-sn3), 44.2 (C-5′′), 42.9 (C-15′′), 42.3 (C-11′′), 38.8 (C-14′′), 37.2 (C-16′′), 34.1 (C-6′′), 32.3 (C-7′′), 32.3 (C-17′′), 31.9 (C-16′), 29.6–29.3 (C-2′), 29.6–29.3 (C-4′-15′), 28.9 (C-13′′), 26.0 (C-3′), 23.2 (C-12′′), 22.8 (C-24′′), 22.6 (C-8′′), 22.6 (C-17′), 22.2 (C-21′′), 22.2 (C-22′′), 15.6 (C-23′′), 14.1 (C-18′); EIHRMS: calcd. for C46H78O6 [M + Na]+: 749.5691, found: 749.5666.
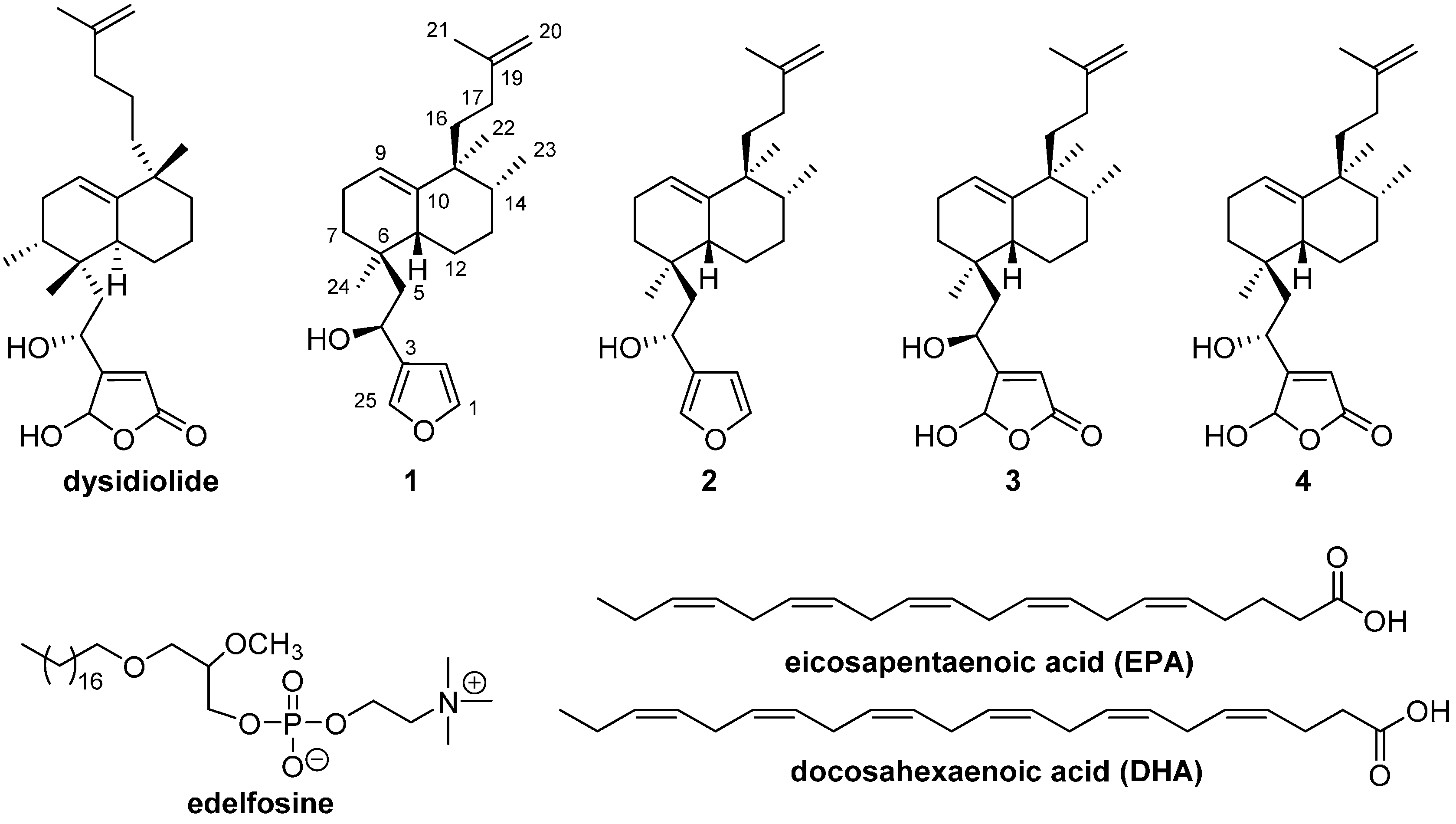
4.8. Preparaion of 1-O-Octadecyl-2-O-[1,25-epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4S-yloxycarbonyl]-3-eicosapentaenoyl-sn-glycerol (5)
To a solution of 21 (10 mg, 0.01 mmol), DMAP (3 mg, 0.02 mmol) and EDAC (3.5 mg, 0.02 mmol) in dry CH2Cl2 (0.14 mL), EPA (4.2 mg, 0.01 mmol) was added under an argon atmosphere. After stirring at rt for 12 h, the reaction mixture was passed through a short silica gel column (CH2Cl2/EtOAc 9:1 as eluent). Then the solvent was removed and the crude oil was purified by column chromatography (Hex/EtOAc 98:2) providing 5 (12 mg, 87%). +7.5 (c 0.20, CHCl3); IR (film, cm−1): 2957, 2926, 2855, 1745, 1462, 1261; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.42 (1H, s, H-25′′), 7.35 (1H, s, H-1′′), 6.39 (1H, s, H-2′′), 5.79 (1H, dd, J = 8.7, 3.1 Hz, H-4′′), 5.41–5.32 (10H, m, =CH), 5.32 (1H, m, H-9′′), 5.01 (1H, m, H-sn2), 4.65, 4.63 (1H, s, each, H-20′′), 4.36 (1H, dd, J = 12.0, 3.4 Hz, HA-sn3), 4.15 (1H, dd, J = 12.0, 6.7 Hz, HB-sn3), 3.51 (2H, d, J = 5.4 Hz, H-sn1), 3.40–3.34 (2H, m, H-1′), 2.85–2.80 (8H, m, =CCH2C=), 2.31 (2H, t, J = 7.3 Hz, H-2′′′), 2.09–2.04 (4H, m, H-4′′′, 19′′′), 2.05–1.40 (16H, m, H-5′′, 7′′, 8′′, 11′′, 12′′, 13′′, 14′′, 16′′, 17′′), 1.76–1.72 (2H, m, H-3′′′), 1.70 (3H, s, Me-21′′), 1.56 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.97 (3H, t, J = 7.5 Hz, H-20′′′), 0.90 (3H, s, Me-22′′), 0.89 (3H, t, J = 6.8 Hz, Me-18′), 0.88 (3H, s, Me-24′′), 0.80 (3H, d, J = 6.8 Hz, Me-23′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 173.4 (C-1′′′), 154.2 (–O–CO–O–), 147.3 (C-19′′), 143.5 (C-1′′), 141.7 (C-10′′), 140.0 (C-25′′), 132.3 (C-18′′′), 129.1–127.2 (=CH) × 9, 126.2 (C-3′′), 119.9 (C-9′′), 109.4 (C-20′′), 108.9 (C-2′′), 74.3 (C-sn2), 72.1 (C-1′), 70.5 (C-4′′), 68.9 (C-sn1), 63.1 (C-sn3), 44.4 (C-5′′), 43.1 (C-15′′), 42.9 (C-11′′), 39.1 (C-14′′), 37.6 (C-16′′), 34.3 (C-6′′), 33.7 (C-2′′′), 32.7 (C-17′′), 32.1 (C-16′), 31.1 (C-7′′), 29.9–29.3 (C-2′), 29.9–29.3 (C-4′-15′), 29.1 (C-13′′), 28.7–20.8 (3′′′, 4′′′, 7’’’, 10’’’, 13’’’, 16’’’, 19′′′), 26.0 (C-3′), 24.0 (C-24′′), 22.9 (C-12′′), 22.9 (C-21′′), 22.6 (C-8′′), 22.6 (C-17′), 22.5 (C-22′′), 15.9 (C-23′′), 14.5 (C-20′′′), 14.3 (C-18′); EIHRMS: calcd. for C66H106O7 [M + Na]+: 1033.7831, found: 1033.7860.
4.9. Preparation of 1-O-Octadecyl-2-O-[1,25-epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R-yloxycarbonyl]-3-eicosapentaenoyl-sn-glycerol (6)
To a solution of 22 (12.5 mg, 0.02 mmol), DMAP (3 mg, 0.02 mmol) and EDAC (4 mg, 0.02 mmol) in dry CH2Cl2 (0.2 mL), EPA (5.2 mg, 0.02 mmol) was added under an argon atmosphere. After stirring at rt for 12 h, the reaction mixture was passed through a short silica gel column (CH2Cl2/EtOAc 9:1 as eluent). Then the solvent was removed and the crude was purified by column chromatography (Hex/EtOAc 99:1) providing 6 (14 mg, 82%). +2.4 (c 0.33, CHCl3); IR (film, cm−1): 2959, 2924, 2855, 1744, 1263; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.42 (1H, s, H-25′′), 7.33 (1H, s, H-1′′), 6.39 (1H, s, H-2′′), 5.79 (1H, dd, J = 8.5, 3.0 Hz, H-4′′), 5.40–5.34 (10H, m, =CH), 5.34 (1H, m, H-9′′), 5.04–5.00 (1H, m, H-sn2), 4.65 and 4.61 (1H, s, each, H-20′′), 4.30 (1H, dd, J = 12.1, 3.6 Hz, HA-sn3), 4.15 (1H, dd, J = 12.1, 7.0 Hz, HB-sn3), 3.56 (1H, dd, J = 12.2, 5.4 Hz, HA-sn1), 3.52 (1H, dd, J = 14.2, 5.4 Hz, HB-sn1), 3.40 (2H, m, H-1′), 2.85–2.78 (8H, m, =CCH2C=), 2.21 (2H, t, J = 7.4 Hz, H-2′′′), 2.13–2.04 (4H, m, H-4′′′, 19′′′), 2.05–1.40 (16H, m, H-5′′, 7′′, 8′′, 11′′, 12′′, 13′′, 14′′, 16′′, 17′′), 1.76–1.72 (2H, m, H-3′′′), 1.68 (3H, s, Me-21′′), 1.56 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.97 (3H, t, J = 7.5 Hz, H-20′′′), 0.90 (3H, t, J = 6.8 Hz, Me-18′), 0.88 (3H, s, Me-22′′), 0.86 (3H, s, Me-24′′), 0.81 (3H, d, J = 7.0 Hz, Me-23′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 173.1 (C-1′′′), 154.2 (–O–CO–O–), 146.9 (C-19′′), 143.1 (C-1′′), 141.0 (C-10′′), 140.1 (C-25′′), 132.0 (C-18′′′), 128.9–127.0 (=CH) × 9, 126.1 (C-3′′), 120.0 (C-9′′), 109.0 (C-20′′), 108.7 (C-2′′), 74.0 (C-sn2), 71.8 (C-1′), 70.3 (C-4′′), 68.8 (C-sn1), 62.7 (C-sn3), 44.3 (C-5′′), 42.9 (C-15′′), 42.3 (C-11′′), 38.8 (C-14′′), 37.6 (C-16′′), 34.1 (C-6′′), 33.3 (C-2′′′), 32.2 (C-17′′), 31.9 (C-16′), 31.2 (C-7′′), 29.6–29.3 (C-2′), 29.6–29.3 (C-4′-15′), 28.8 (C-13′′), 28.4–22.6 (3′′′, 4′′′, 7′′′, 10′′′, 13′′′, 16′′′, 19′′′), 26.0 (C-3′), 23.7 (C-24′′), 23.2 (C-12′′), 22.8 (C-21′′), 22.6 (C-8′′), 22.6 (C-17′), 22.2 (C-22′′), 15.6 (C-23′′), 14.2 (C-20′′′), 14.0 (C-18′); EIHRMS: calcd. for C66H106O7 [M + Na]+: 1033.7831, found: 1033.7854.
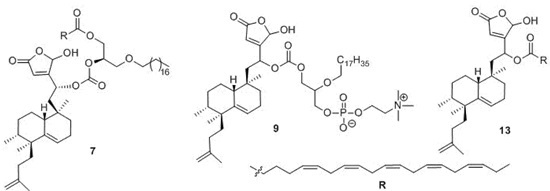
4.10. Preparation of 1-O-Octadecyl-2-O-[25-hydroxy-18-nor-ent-isodysidiola-2,9,19-trien-1,25-olide-4S-yloxycarbonyl]-3-eicosapentaenoyl-sn-glycerol (7)
Rose Bengal (1 mg) was added to a solution of 5 (6.4 mg, 6.3 × 10−3 mmol) and DIPEA (11 μL, 0.06 mmol) in dry CH2Cl2 (2 mL) at rt. Anhydrous oxygen was bubbled in for 2 min and after that, the solution was placed under an oxygen atmosphere at −78 °C and irradiated with a 200 W lamp. After 4 h irradiation was stopped, the pink solution was allowed to warm to rt, and saturated aqueous oxalic acid solution (1 mL) added. After a few minutes of vigorous stirring, the mixture was diluted with H2O and extracted with Et2O. The combined organic extracts were washed with H2O and dried over anhydrous Na2SO4. After filtration, the solvent was evaporated to give a residue that was purified by silica gel column chromatography to yield 7 (6 mg, 86%). +2.6 (c 0.2, CHCl3); IR (film, cm−1): 3427, 2924, 1747, 1259; 1H-NMR (400 MHz, CDCl3, δ ppm): 6.19/5.97 (1H, m, H-25′′), 6.01/6.00 (1H, m, H-2′′), 5.60/5.48 (1H, m, H-4′′), 5.45–5.33 (10H, m, =CH), 5.45–5.33 (1H, m, H-9′′), 5.03/4.95 (1H, m, H-sn2), 4.68–4.62 (2H, m, H-20′′), 4.35/4.18 (2H, m, H-sn3), 3.56 (2H, m, H-sn1), 3.43 (2H, m, H-1′), 2.82 (8H, m, =CCH2C=), 2.30 (2H, m, H-2′′′), 2.09 (4H, m, H-4′′′, 19′′′), 2.00–1.53 (18H, m, H-5′′, 7′′, 8′′, 11′′, 12′′, 13′′, 14′′, 16′′, 17′′, 3′′′), 1.69 (3H, s, Me-21′′), 1.54 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.97 (3H, t, J = 7.6 Hz, H-20′′′), 0.91 (3H, s, Me-22′′), 0.88 (3H, t, J = 6.8 Hz, Me-18′), 0.88 (3H, s, Me-24′′), 0.81 (3H, d, J = 6.8 Hz, Me-23′′); 13C-NMR (50 MHz, CDCl3, δ ppm): 173.7 (C-1′′′), 169.4 (C-1′′), 168.2 (C-3′′), 154.4 (–O–CO–O–), 147.5 (C-19′′), 141.8 (C-10′′), 132.3 (C-18′′′), 129.2–127.2 (=CH) × 9, 120.0 (C-9′′), 117.9 (C-2′′), 109.4 (C-20′′), 97.6 (C-25′′), 74.3 (C-sn2), 72.1 (C-1′), 71.2 (C-4′′), 68.9 (C-sn1), 63.0 (C-sn3), 43.1 (C-5′′), 42.9 (C-15′′), 42.9 (C-11′′), 38.9 (C-14′′), 37.6 (C-16′′), 34.4 (C-6′′), 33.7 (C-2′′′), 32.7 (C-17′′), 32.1 (C-16′), 31.1 (C-7′′), 29.9–29.3 (C-2′), 29.9–29.3 (C-4′-15′), 29.1 (C-13′′), 28.6–20.7 (3′′′, 4′′′, 7′′′, 10′′′, 13′′′, 16′′′, 19′′′), 25.9 (C-3′), 24.0 (C-24′′), 22.9 (C-12′′), 22.9 (C-21′′), 22.6 (C-8′′), 22.6 (C-17′), 22.5 (C-22′′), 15.9 (C-23′′), 14.5 (C-20′′′), 14.3 (C-18′); EIHRMS: calcd. for C66H106O9 [M + Na]+: 1065.7729, found: 1065.7775.
4.11. Preparation of 1-O-Octadecyl-2-O-[25-hydroxy-18-nor-ent-isodysidiola-2,9,19-trien-1,25-olide-4R-yloxycarbonyl]-3-eicosapentaenoyl-sn-glycerol (8)
Rose Bengal (1 mg) was added to a solution of 6 (7.6 mg, 7.5 × 10−3 mmol) and DIPEA (13 μL, 0.075 mmol) in dry CH2Cl2 (2 mL) at rt. Anhydrous oxygen was bubbled in for 2 min, then the solution was placed under an oxygen atmosphere at −78 °C and irradiated with a 200 W lamp. After 4 h irradiation was stopped, the pink solution allowed to warm to rt, and saturated aqueous oxalic acid solution (1 mL) added. After a few minutes of vigorous stirring, the mixture was diluted with H2O and extracted with Et2O. The combined organic extracts were washed with H2O and dried over anhydrous Na2SO4. After filtration, the solvent was evaporated to give a residue which was purified by silica gel column chromatography to yield 8 (7 mg, 90%). −3.0 (c 0.3, CHCl3); ); IR (film, cm−1): 3427, 2924, 1747, 1259; 1H-NMR (400 MHz, CDCl3, δ ppm): 6.13/5.96 (1H, m, H-25′′), 6.06/6.05 (1H, s, H-2′′), 5.69/5.46 (1H, m, H-4′′), 5.40–5.34 (10H, m, =CH), 5.40–5.34 (1H, m, H-9′′), 5.01/4.94 (1H, m, H-sn2), 4.67–4.63 (2H, m, H-20′′), 4.13 (1H, dd, J = 6.4, 12.4 Hz, HA-sn3), 4.01 (1H, dd, J = 4.0, 12.4 Hz, HB-sn3), 3.57 (2H, d, J = 5.2 Hz, H-sn1), 3.47–3.39 (2H, m, H-1′), 2.86–2.80 (8H, m, =CCH2C=), 2.34–2.30 (2H, m, H-2′′′), 2.13–2.04 (4H, m, H-4′′′, 19′′′), 2.00–1.52 (18H, m, H-5′′, 7′′, 8′′, 11′′, 12′′, 13′′, 14′′, 16′′, 17′′, 3′′′), 1.69 (3H, s, Me-21′′), 1.54 (2H, m, H-2′), 1.25 (30H, m, H-3′-17′), 0.97 (3H, t, J = 7.6 Hz, H-20′′′), 0.91 (3H, s, Me-22′′), 0.88 (3H, t, J = 6.8 Hz, Me-18′), 0.88 (3H, s, Me-24′′), 0.81 (3H, d, J = 6.8 Hz, Me-23′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 173.4 (C-1′′′), 169.3 (C-1′′), 168.1 (C-3′′), 154.4 (–O–CO–O–), 147.1 (C-19′′), 141.1 (C-10′′), 132.0 (C-18′′′), 128.8–127.0 (=CH) × 9, 120.0 (C-9′′), 118.0 (C-2′′), 109.0 (C-20′′), 97.5 (C-25′′), 74.0 (C-sn2), 71.8 (C-1′), 71.1 (C-4′′), 68.8 (C-sn1), 62.6 (C-sn3), 42.9 (C-5′′), 42.9 (C-15′′), 42.3 (C-11′′), 38.8 (C-14′′), 37.6 (C-16′′), 34.1 (C-6′′), 33.3 (C-2′′′), 32.2 (C-17′′), 31.9 (C-16′), 31.2 (C-7′′), 29.7–29.3 (C-2′), 29.7–29.3 (C-4′-15′), 28.8 (C-13′′), 28.3–22.6 (3′′′, 4′′′, 7′′′, 10′′′, 13′′′, 16′′′, 19′′′), 26.0 (C-3′), 23.7 (C-24′′), 23.2 (C-12′′), 22.8 (C-21′′), 22.6 (C-8′′), 22.6 (C-17′), 22.2 (C-22′′), 15.7 (C-23′′), 14.2 (C-20′′′), 14.0 (C-18′); EIHRMS: calcd. for C66H106O9 [M + Na]+: 1065.7729, found: 1065.7766.
4.12. Preparation of 1,3-Benzyliden-2-O-octadecylglycerol (24)
To a solution of 1,3-O-benzylidene glycerol 23 (3.7 g, 20.5 mmol) in toluene (21 mL), NaNH2 (800 mg, 20.5 mmol) was added and heated at 111 °C under an argon atmosphere for 1 h. The mixture was cooled to rt and a solution of bromooctadecane (6.8 g, 20.5 mmol) in toluene (20 mL) was added, then it was heated at 111 °C for 4 h. The reaction was cooled at 0 °C, crushed ice and saturated NH4Cl were added and then it was extracted with Et2O. The organic layer was washed with H2O and brine, dried over anhydrous Na2SO4, filtered and evaporated to yield 24 (8.7 g, 98%). IR (film, cm−1): 2916, 2849, 1471, 1385, 1152, 1103, 1010, 743, 695; 1H-NMR (200 MHz, CDCl3, δ ppm): 7.54–7.49 (2H, m, H-2′, 6′), 7.40–7.32 (3H, m, H-3′, 4′, 5′), 5.55 (1H, s, O–CH–O), 4.33 (2H, d, J = 12.6 Hz, HA-1, HA-3), 4.15 (1H, m, H-2), 4.06 (2H, d, J = 12.6 Hz, HB-1, HB-3), 3.55 (2H, t, J = 6.8 Hz, H-1′′), 1.65 (2H, m, H-2′′), 1.26 (30H, m, H-3′′-17′′), 0.88 (3H, t, J = 6.4 Hz, H-18′′); 13C-NMR (50 MHz, CDCl3, δ ppm): 138.5 (C-1′), 129.0 (C-4′), 128.5 (C-2′, 6′), 126.5 (C-3′, 5′), 101.5 (O–CH–O), 72.5 (C-1′′), 70.9 (C-2), 69.2 (C-1, 3), 32.2 (C-16′′), 30.1–29.2 (C-2′′, 4′′-15′′), 26.4 (C-3′′), 23.0 (C-17′′), 14.4 (C-18′′). EIHRMS: calcd. for C28H48O3 [M + Na]+: 455.3496, found: 455.3514.
4.13. Preparation of 2-O-Octadecylglycerol (25)
To a solution of 24 (8.7 g, 20 mmol) in MeOH (40 mL), p-TsOH (3.8 g, 20 mmol) was added and it was stirred at 35–40 °C for 6 h. Then H2O was added, the mixture was extracted with Et2O and washed with 6% NaHCO3 and H2O. The organic layer was dried over Na2SO4, filtered and evaporated. The residue was purified by column chromatography (EtOAc) to yield 25 (6.2 g, 90%). IR (film, cm−1): 3326, 2918, 2850, 1468, 1114, 1078, 1058, 975, 718; 1H-NMR (400 MHz, CDCl3, δ ppm): 3.78–3.66 (4H, m, H-1, H-3), 3.57 (2H, t, J = 6.7 Hz, H-1′′), 3.46 (1H, quin, J = 4.6 Hz, H-2), 1.59 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.88 (3H, t, J = 6.6 Hz, H-18′′); 13C-NMR (50 MHz, CDCl3, δ ppm): 79.9 (C-2), 70.4 (C-1′′), 62.1 (C-1), 62.1 (C-3), 32.1 (C-16′′), 30.2 (C-2′′), 29.7 (C-4′′-15′′), 26.3 (C-3′′), 22.9 (C-17′′), 14.3 (C-18′′). EIHRMS: calcd. for C21H44O3 [M + Na]+: 367.3183, found: 367.3187.
4.14. Reaction of 25 with TBDMSCl: Preparation of 1-O-tert-Butyldimethylsilyl-2-O-octadecyl-glycerol (26) and 1,3-O-di-tert-Butyldimethylsilyl-2-O-octadecylglycerol (27):
To an ice-cooled solution of 25 (3.4 g, 9.9 mmol) in DMF (99 mL), TBDMSCl (1.49 g, 9.9 mmol) and imidazole (673 mg, 9.9 mmol) were added. It was stirred overnight at rt under an argon atmosphere; the reaction mixture was cooled at 0 °C and quenched with H2O. It was extracted with Et2O and the organic layer washed with H2O. After drying over anhydrous Na2SO4 the solvent was evaporated. The crude purified by column chromatography (Hex/EtOAc 97:3) to give 26 (1.85 g, 41%); 27 (910 mg, 16%) and 25 (1.43 g, 42%).
Compound 26: IR (film, cm−1): 3450, 2925, 2854, 1475, 1100, 837; 1H-NMR (400 MHz, CDCl3, δ ppm): 3.73 (1H, dd, J = 10.2, 4.0 Hz, HA-1), 3.72 (1H, dd, J = 12.0, 5.2 Hz, HA-3), 3.62 (1H, dd, J = 12.0, 5.2 Hz, HB-3), 3.61 (1H, dd, J = 10.2, 6.8 Hz, HB-1), 3.58 (1H, ddd, J = 9.2, 6.8, 6.8 Hz, HA-1′′), 3.52 (1H, ddd, J = 9.2, 6.8, 6.8 Hz, HB-1′′), 3.42 (1H, dddd, J = 6.8, 5.2, 5.2, 4.0 Hz, H-2), 2.15 (1H, m,–OH), 1.55 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.89 (9H, s, Me3CSi-), 0.89 (3H, t, J = 6.8 Hz, H-18′′), 0.06 (6H, s, Me2Si-); 13C-NMR (100 MHz, CDCl3, δ ppm):80.1 (C-2), 70.7 (C-1′′), 63.2 (C-3), 62.9 (C-1), 32.1 (C-16′′), 30.3 (C-2′′), 29.7 (C-4′′-15′′), 26.3 (C-3′′), 26.0 (Me3CSi-), 22.9 (C-17′′), 18.4 (Me3CSi-), 14.3 (C-18′′), −5.3 (Me2Si-). EIHRMS: calcd. for C27H58O3Si [M + Na]+: 481.4047, found: 481.4025.
Compound 27: IR (film, cm−1): 2926, 2855, 1475, 1257, 1106, 836; 1H-NMR (200 MHz, CDCl3, δ ppm): 3.72–3.53 (4H, m, H-1, H-3), 3.56 (2H, t, J = 6.6 Hz, H-1′′), 3.34 (1H, quin, J = 5.4 Hz, H-2), 1.55 (2H, m, H-2′′), 1.26 (30H, m, H-3′′-17′′), 0.89 (2·9H, s, Me3CSi-), 0.89 (3H, t, J = 6.8 Hz, H-18′′), 0.05 (2·6H, s, Me2Si-); 13C-NMR (50 MHz, CDCl3, δ ppm): 81.4 (C-2), 70.9 (C-1′′), 63.1 (C-3), 63.1 (C-1), 32.2 (C-16′′), 30.4 (C-2′′), 29.9–29.6 (C-4′′-15′′), 26.4 (C-3′′), 26.1 (2·Me3CSi-), 22.9 (C-17′′), 18.5 (2·Me3CSi-), 14.3 (C-18′′), −5.2 (2·Me2Si-). EIHRMS: calcd. for C33H72O3Si2 [M + Na]+: 595.4912, found: 595.4927.
4.15. Preparation of 1-Chlorocarbonyl-2-O-octadecyl-3-O-tert-butyldimethylsilylglycerol (28)
To an ice cooled solution of 26 (500 mg, 1 mmol) in THF (2 mL), trichloromethyl chloroformate (diphosgene, 0.12 mL, 1 mmol) and N,N-dimethylaniline (0.13 mL, 1 mmol) were slowly added. The mixture was stirred at 0 °C for 10 min and then at rt for 4 h. Then Et2O was added and the solution washed with 0.2 M HCl, 0.2 M NaOH and H2O, dried over anhydrous Na2SO4 and evaporated. The reaction bulk was purified by column chromatography to separate 28 (370 mg, 71%). IR (film, cm−1): 2925, 2854, 1781, 1462, 1254, 1164, 838, 780; 1H-NMR (200 MHz, CDCl3, δ ppm): 4.49 (1H, dd, J = 11.4, 3.4 Hz, HA-1), 4.33 (1H, dd, J = 11.4, 5.0 Hz, HB-1), 3.83–3.60 (2H, m, H-3), 3.60–3.50 (1H, m, H-2), 3.54 (2H, t, J = 6.6 Hz, H-1′′), 1.55 (2H, m, H-2′′), 1.26 (30H, m, H-3′′-17′′), 0.89 (9H, s, Me3CSi-), 0.88 (3H, t, J = 6.8 Hz, H-18′′), 0.06 (6H, s, Me2Si-); 13C-NMR (50 MHz, CDCl3, δ ppm): 150.9 (O–CO–Cl), 77.6 (C-2), 71.2 (C-1′′), 71.0 (C-1), 61.9 (C-3), 32.2 (C-16′′), 30.3 (C-2′′), 29.9–29.6 (C-4′′-15′′), 26.2 (C-3′′), 26.0 (Me3CSi-), 22.9 (C-17′′), 18.4 (Me3CSi-), 14.3 (C-18′′), −5.3 (Me2Si-). EIHRMS: in MeOH, calcd. for the methyl ester, C29H60O5Si [M + Na]+: 539.4102, found: 539.4122.
4.16. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecyl-3-O-tert-butyldimethylsilylglycerol (29)
To a solution of 1/2 (324 mg, 0.91 mmol), N,N-diisopropylethylamine (DIPEA, 0.22 mL, 1.27 mmol), 4-(dimethylamino) pyridine (DMAP, 56 mg, 0.46 mmol) in toluene (4.6 mL), a solution of 28 (364 mg, 0.7 mmol) in toluene (3.5 mL) was added dropwise at 0 °C . The reaction mixture was stirred at 0 °C under an argon atmosphere for 15 min and then at rt overnight. After this time the solvent was removed and the residue was purified by column chromatography (Hex/Et2O 99.9:0.1) to obtain 29 (330 mg, 56%). IR (film, cm−1): 2926, 2855, 1745, 1464, 1256, 1107, 837, 777; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.43/7.42 (1H, s, H-25′), 7.33 (1H, s, H-1′), 6.39 (1H, s, H-2′), 5.79–5.76 (1H, m, H-4′), 5.35–5.32 (1H, m, H-9′), 4.66–4.60 (2H, m, H-20′), 4.32–4.06 (2H, m, H-1), 3.64–3.57 (2H, m, H-3), 3.54–3.49 (1H, m, H-2), 3.54–3.49 (2H, m, H-1′′), 2.20–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.70/1.66 (3H, s, H-21′), 1.54 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.89 (3H, s, H-22′), 0.89 (3H, s, H-24′), 0.88 (3H, t, J = 6.8 Hz, H-18′′), 0.87 (9H, s, Me3CSi-), 0.81 (3H, d, J = 6.9 Hz, H-23′), 0.04 (6H, s, Me2Si-); 13C-NMR (50 MHz, CDCl3, δ ppm): 154.6/154.5 (O–CO–O), 147.0 (C-19′), 143.2/143.1 (C-1′), 142.6 (C-10′), 140.1 (C-25′), 126.1 (C-3′), 120.0/119.7 (C-9′), 109.1/109.0 (C-20′), 108.7/108.6 (C-2′), 78.0 (C-2), 70.7 (C-1′′), 70.0/69.9 (C-4′), 67.1/66.8 (C-1), 62.2 (C-3), 44.2 (C-5′), 42.9 (C-15′), 42.4 (C-11′), 38.8 (C-14′), 37.4 (C-16′), 34.1/34.0 (C-6′), 32.4/32.3 (C-17′), 31.9 (C-16′′), 30.0–29.3 (C-2′′, 4′′-15′′, 7′, 13′), 26.0 (C-3′′), 25.8 (Me3CSi-), 22.8 (C-12′), 22.6 (C-8′), 22.6 (C-17′′), 22.4 (C-24′), 22.2 (C-21′), 22.2 (C-22′), 18.2 (Me3CSi-), 15.6 (C-23′), 14.1 (C-18′′), −5.2 (Me2Si-). EIHRMS: calcd. for C52H92O6Si [M + Na]+: 863.6555, found: 863.6542.
4.17. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecylglycerol (30)
To a solution of 29 (126 mg, 0.15 mmol) in THF (1.7 mL), 1 M TBAF in THF (0.22 mL, 0.22 mmol) was added under an argon atmosphere. The mixture was stirred for 2 h at rt, then the reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with H2O and brine, dried over anhydrous Na2SO4, filtered and removed the solvent. The residue was purified by column chromatography (EtOAc) providing 30 (97 mg, 89%). IR (film, cm−1): 3335, 2922, 2853, 1744, 1466, 1258, 1078, 1059, 1024; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.42 (1H, s, H-25′), 7.34 (1H, s, H-1′), 6.39 (1H, s, H-2′), 5.78 (1H, m, H-4′), 5.35–5.32 (1H, m, H-9′), 4.66–4.61 (2H, m, H-20′), 4.22–4.14 (2H, m, H-1), 3.75–3.67 (2H, m, H-3), 3.56 (2H, t, J = 6.4 Hz, H-1′′), 3.49–3.43 (1H, m, H-2), 2.20–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.69/1.66 (3H, s, H-21′), 1.55 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.89 (3H, s, H-22′), 0.88 (3H, t, J = 6.8 Hz, H-18′′), 0.87 (3H, s, H-24′), 0.80/0.79 (3H, d, J = 6.8 Hz, H-23′); 13C-NMR (50 MHz, CDCl3, δ ppm): 154.8 (O–CO–O), 147.3 (C-19′), 143.5 (C-1′), 141.6/141.3 (C-10′), 140.4 (C-25′), 126.2 (C-3′), 120.2/120.0 (C-9′), 109.4/109.3 (C-20′), 108.9/108.8 (C-2′), 79.8 (C-2), 70.9/70.4 (C-1′′), 70.5 (C-4′), 66.5/66.4 (C-1), 62.4/62.1 (C-3), 44.4 (C-5′), 43.1 (C-15′), 42.7/42.6 (C-11′), 39.1 (C-14′), 37.6/37.4 (C-16′), 34.4/34.3 (C-6′), 32.7/32.6 (C-17′), 32.1 (C-16′′), 31.5 (C-7′), 30.3–29.6 (C-2′′, 4′′-15′′), 29.1 (C-13′), 26.3/26.2 (C-3′′), 23.4 (C-24′), 23.1 (C-21′), 22.9 (C-8′), 22.9 (C-12′), 22.9 (C-17′′), 22.5 (C-22′), 15.9 (C-23′), 14.3 (C-18′′). EIHRMS: calcd. for C46H78O6 [M + Na]+: 749.5691, found: 749.5665.
4.18. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecyl-glycero-3-phosphate (31)
To a solution of 30 (37 mg, 0.05 mmol) and anhydrous pyridine (8 μL) in THF (0.3 mL), POCl3 (5 μL, 0.06 mmol) was added dropwise under an argon atmosphere with stirring at 0 °C for 5 h. Then 6% NaHCO3 was added and the mixture stirred for an additional 15 min. at 0 °C. After that time, crushed ice was added, the mixture was acidified with 2 M HCl to pH = 2 and extracted with EtOAc. The organic layer was washed with H2O and the solvent removed to give 31 (39 mg, 97%). IR (film, cm−1): 2924, 2853, 1744, 1466, 1258 1H-NMR (200 MHz, CDCl3, δ ppm): 7.39 (1H, broad s, H-25′), 7.3 (1H, broad s, H-1′), 6.37 (1H, broad s, H-2′), 5.76 (1H, m, H-4′), 5.33 (1H, m, H-9′), 4.64–4.61 (2H, m, H-20′), 4.34–3.83 (4H, m, H-1, H-3), 3.64–3.31 (3H, m, , H-1′′, H-2), 2.10–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.69/1.65 (3H, s, H-21′), 1.55 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.89 (3H, s, H-22′), 0.88 (3H, t, J = 6.8 Hz, H-18′′), 0.87 (3H, s, H-24′), 0.80/0.79 (3H, d, J = 6.8 Hz, H-23′).
4.19. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecyl-glycero-3-phosphocoline (9)
Compound 31 (39 mg, 0.05 mmol), choline tetraphenyl borate (21 mg, 0.05 mmol) and TPS (18 mg, 0.06 mmol) were dissolved in anhydrous pyridine (0.4 mL). The mixture was heated at 70 °C for 1 h and then it was stirred at rt overnight. After addition of H2O (0.1 mL), the solvents were removed by rotary evaporation. The crude mixture was dissolved in Et2O and stirred for a few minutes. The solid formed was eliminated by filtration. The organic solution was evaporated and the reaction bulk purified by column chromatography (CHCl3/MeOH/NH4OH 65:30:5) to yield 9 (16 mg, 35%). IR (film, cm−1): 2920, 2851, 1738, 1467; 1H-NMR (400 MHz, CDCl3, δ ppm):7.42 (1H, s, H-25′), 7.37 (1H, s, H-1′), 6.39 (1H, s, H-2′), 5.75 (1H, m, H-4′), 5.36–5.34 (1H, m, H-9′), 4.66–4.61 (2H, m, H-20′), 4.37 (2H, m, H-1′′′), 4.33–4.10 (2H, m, H-1), 4.00–3.80 (2H, m, H-3), 3.98–3.75 (2H, m, H-2′′′), 3.65 (1H, m, H-2), 3.51 (2H, m, H-1′′), 3.35 (9H, s, Me3N–), 2.05–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.70/1.67 (3H, s, H-21′), 1.54 (2H, m, H-2′′), 1.26 (30H, m, H-3′′-17′′), 0.90 (3H, s, H-22′), 0.88 (3H, t, J = 6.8 Hz, H-18′′), 0.87 (3H, s, H-24′), 0.82/0.81 (3H, d, J = 6.9 Hz, H-23′); 13C-NMR (100 MHz, CDCl3, δ ppm): 154.5 (O–CO–O), 146.9 (C-19′), 143.3 (C-1′), 141.4/141.0 (C-10′), 140.0 (C-25′), 126.2 (C-3′), 120.0/119.7 (C-9′), 109.1/109.0 (C-20′), 108.7 (C-2′), 76.2 (C-2), 70.8/70.6 (C-1′′), 70.2 (C-4′), 66.9 (C-1), 66.2 (C-2′′′), 64.4 (C-3), 59.7 (C-1′′′), 54.4 (Me3N-), 44.3 (C-5′), 42.9 (C-15′), 42.6/42.5 (C-11′), 38.8/38.7 (C-14′), 37.3/37.2 (C-16′), 34.1 (C-6′), 32.4/32.3 (C-17′), 31.9 (C-16′′), 31.2 (C-7′), 29.9–29.3 (C-2′′, 4′′-15′′), 28.9/28.8 (C-13′), 25.9 (C-3′′), 23.2 (C-12′), 22.8 (C-24′), 22.7 (C-21′), 22.6 (C-8′), 22.6 (C-17′′), 22.2 (C-22′), 15.6 (C-23′), 14.0 (C-18′′). EIHRMS: calcd. for C51H90NO9P [M + Na]+: 914.6245, found: 914.6229.
4.20. Preparation of 1-O-[25-Hydroxy-18-nor-ent-isodysidiola-2,9,19-trien-1,25-olide-4R/S-yloxycarbonyl]-2-O-octadecyl-glycero-3-phosphocoline (10)
Rose Bengal (4 mg) was added to a solution of 9 (20 mg, 0.02 mmol) and DIPEA (38 μL, 0.22 mmol) in dry CH2Cl2 (10 mL) at rt. Anhydrous oxygen was bubbled in for 10 min and the solution was placed under an oxygen atmosphere at −78 °C and irradiated with a 200 W lamp. After 4 h irradiation was stopped, the pink solution allowed to warm to rt, and saturated aqueous oxalic acid solution (1.7 mL) added. After 30 min of vigorous stirring, the mixture was diluted with H2O and extracted with Et2O. The combined organic extracts were washed with H2O and brine. The solvent was evaporated to give a residue which was purified by silica gel column chromatography (CHCl3/MeOH/H2O 65:35:1) to yield 10 (6 mg, 30%). 1H-NMR (200 MHz, CDCl3, δ ppm): 6.28/6.10 (1H, s, H-25′), 5.91/5.83 (1H, m, H-2′), 5.65 (1H, m, H-4′), 5.36 (1H, m, H-9′), 4.63 (2H, m, H-20′), 4.33 (2H, m, H-1′′′), 4.32–4.10 (2H, m, H-1), 4.05–3.95 (2H, m, H-3), 4.00–3.80 (2H, m, H-2′′′), 3.65–3.55 (1H, m, H-2), 3.51 (2H, m, H-1′′), 3.34 (9H, s, Me3N–), 2.05–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.69 (3H, s, H-21′), 1.54 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.87 (6H, s, H-22′, 24′), 0.88 (3H, t, J = 7.0 Hz, H-18′′), 0.81 (3H, d, J = 6.9 Hz, H-23′); EIMS found for C51H90 NO11P [M + Na]+: 946.7.
4.21. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecyl-3-eicosa-pentaenoylglycerol (32)
To a solution of 30 (23 mg, 0.03 mmol), DMAP (5 mg, 0.04 mmol) and EDAC (8 mg, 0.04 mmol) in dry CH2Cl2 (0.3 mL), EPA (9.6 μL, 0.03 mmol) was added under an argon atmosphere. After stirring at rt for 13 h, the reaction mixture was passed through a short silica gel column (CH2Cl2/EtOAc 9:1 as eluent). Then the solvent was removed and the crude was purified by column chromatography (Hex/EtOAc 98:2) providing 32 (13 mg, 64%). IR (film, cm−1): 2959, 2926, 1740, 1560, 1383, 1261; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.43/7.42 (1H, s, H-25′), 7.35/7.34 (1H, s, H-1′), 6.39 (1H, s, H-2′), 5.77 (1H, m, H-4′), 5.42–5.30 (10H, m, =CH), 5.42–5.30 (1H, m, H-9′), 4.64–4.61 (2H, m, H-20′), 4.21–4.07 (2H, m, H-1), 4.21–4.07 (2H, m, H-3), 3.67 (1H, m, H-2), 3.57–3.49 (2H, m, H-1′′), 2.84–2.80 (8H, m, =CCH2C=), 2.35–2.30 (2H, m, H-2′′′), 2.14–2.04 (4H, m, H-4′′′, H-19′′′), 2.00–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.74 (2H, m, H-3′′′), 1.70/1.67 (3H, s, H-21′), 1.54 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.97 (3H, t, J = 7.5 Hz, H-20′′′), 0.90 (3H, s, H-22′), 0.88 (3H, t, J = 7.0 Hz, H-18′′), 0.88 (3H, s, H-24′), 0.81/0.80 (3H, d, J = 6.9 Hz, H-23′); 13C-NMR (100 MHz, CDCl3, δ ppm): 173.1 (C-1′′′), 154.4 (O–CO–O), 147.0 (C-19′), 143.2 (C-1′), 141.0 (C-10′), 140.1 (C-25′), 132.0 (C-18′′′), 128.8–127.0 (=CH) × 9, 126.0 (C-3′), 120.0 (C-9′), 109.1/109.0 (C-20′), 108.7/108.6 (C-2′), 75.1/75.0 (C-2), 70.8/70.7 (C-1′′), 70.2 (C-4′), 66.7/66.4 (C-1), 63.0/62.8 (C-3), 44.2 (C-5′), 42.9 (C-15′), 42.5/42.3 (C-11′), 38.8 (C-14′), 37.3/37.2 (C-16′), 34.1/34.0 (C-6′), 33.5 (C-2′′′), 32.4/32.3 (C-17′), 31.9 (C-16′′), 31.2 (C-7′), 29.8–28.8 (C-2′′, 4′′-15′′), 29.9–29.8 (C-13′), 26.5–20.5 (3′′′, 4′′′, 7′′′, 10′′′, 13′′′, 16′′′, 19′′′), 25.9 (C-3′′), 22.8 (C-12′), 22.6 (C-8′), 22.6 (C-17′′), 22.3 (C-24′), 22.2 (C-21′), 22.2 (C-22′), 15.6 (C-23′), 14.2 (C-20′′′), 14.0 (C-18′′); EIHRMS: calcd. for C66H106O7 [M + Na]+: 1033.7831, found: 1033.7865.
4.22. Preparation of 1-O-[25-Hydroxy-18-nor-ent-isodysidiola-2,9,19-trien-1,25-olide-4R/S-yloxycarbonyl]-2-O-octadecyl-3-eicosapentaenoylglycerol (11)
Rose Bengal (1 mg) was added to a solution of 32 (9 mg, 0.009 mmol) and DIPEA (16 μL, 0.09 mmol) in dry CH2Cl2 (0.7 mL) at rt. Anhydrous oxygen was bubbled in for 10 min, the solution placed under oxygen atmosphere at −78 °C and irradiated with a 200 W lamp. After 4 h irradiation was stopped, the pink solution allowed to warm to rt, and saturated aqueous oxalic acid solution (0.7 mL) added. After 30 min of vigorous stirring, the mixture was diluted with H2O and extracted with CH2Cl2. The combined organic extracts were washed with H2O and brine, and dried over anhydrous Na2SO4. The solvent was evaporated to give a residue that was purified by silica gel column chromatography (Hex/EtOAc 9:1) to yield 11 (5 mg, 53%). IR (film, cm−1): 3387, 2924, 2855, 1747, 1454, 1258, 1134; 1H-NMR (400 MHz, CDCl3, δ ppm): 6.21/5.96 (1H, m, H-25′), 6.03/6.02 (1H, s, H-2′), 5.63 (1H, m, H-4′), 5.38–5.36 (10H, m, =CH), 5.38–5.36 (1H, m, H-9′), 4.67–4.61 (2H, m, H-20′), 4.33–4.06 (2H, m, H-1), 4.33–4.06 (2H, m, H-3), 3.70–3.67 (1H, m, H-2), 3.52 (2H, t, J = 6.7 Hz, H-1′′), 2.84–2.78 (8H, m, =CCH2C=), 2.34 (2H, t, J = 7.5 Hz, H-2′′′), 2.14–2.04 (4H, m, H-4′′′, H-19′′′), 2.00–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.74 (2H, m, H-3′′′), 1.70/1.69 (3H, s, H-21′), 1.54 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.97 (3H, t, J = 7.5 Hz, H-20′′′), 0.91 (3H, s, H-22′), 0.87 (3H, t, J = 6.8 Hz, H-18′′), 0.88 (3H, s, H-24′), 0.81 (3H, d, J = 6.9 Hz, H-23′); 13C-NMR (100 MHz, CDCl3, δ ppm): 173.4 (C-1′′′), 169.3 (C-1′), 168.2 (C-3′), 154.2 (O–CO–O), 147.2 (C-19′), 141.0 (C-10′), 132.0 (C-18′′′), 129.0–127.0 (=CH) × 9, 119.4 (C-9′), 118.4 (C-2′), 109.3/109.2 (C-20′), 97.4 (C-25′), 75.0 (C-2), 70.9 (C-1′′), 70.8 (C-4′), 66.9 (C-1), 62.3 (C-3), 43.1 (C-5′), 42.8 (C-15′), 42.6 (C-11′), 38.7 (C-14′), 37.4 (C-16′), 34.6 (C-6′), 33.5 (C-2′′′), 32.4 (C-17′), 31.9 (C-16′′), 29.6–29.4 (C-2′′, 4′′-15′′), 28.9 (C-13′), 29.3 (C-7′), 26.5–20.5 (3′′′, 4′′′, 7′′′, 10′′′, 13′′′, 16′′′, 19′′′), 25.9 (C-3′′), 22.9 (C-12′), 22.9 (C-8′), 22.8 (C-24′), 22.7 (C-21′),22.6 (C-17′′), 22.5 (C-22′), 15.6 (C-23′), 14.2 (C-20′′′), 14.1 (C-18′′). EIMS found for C66H106O9 [M + Na]+: 1065.7.
4.23. Preparation of 3-O-p-Methoxybenzyl-sn-glycerol (35)
To an ice cooled solution of (S)-(+)-solketal 33 (2.5 g, 18.9 mmol) in THF (94 mL), 60% NaH (756 mg, 31.5 mmol) and PMBCl (2.56 mL, 18.9 mmol) were added. The mixture was stirred at 0 °C for 10 min and at rt for 1 h. Then it was refluxed overnight, cooled to rt, and crushed ice and saturated NH4Cl added. The aqueous layer was extracted with EtOAc and the organic layer was washed with H2O and brine, dried over anhydrous Na2SO4, filtered and evaporated. The bulk reaction mixture was purified by column chromatography on silica gel (EtOAc) to obtain 35 (3.6 g, 90%). −2.43 (c 0.7, CHCl3); IR (film, cm−1): 3395, 2934, 2866, 1612, 1514, 1248, 1082, 1034; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.25 (2H, d, J = 7.2 Hz, H-2′, H-6′), 6.89 (2H, d, J = 8.6 Hz, H-3′, H-5′), 4.48 (2H, s, –OCH2Ar), 3.88 (1H, m, H-sn2), 3.81 (3H, s, –OCH3), 3.70 (1H, dd, J = 11.4, 3.9 Hz, HA-sn1), 3.63 (1H, dd, J = 11.4, 5.3 Hz, HB-sn1), 3.56 (1H, dd, J = 9.6, 3.9 Hz, HA-sn3), 3.52 (1H, dd, J = 9.6, 6.2 Hz, HB-sn3); 13C-NMR (50 MHz, CDCl3, δ ppm): 159.6 (C-4′′′), 130.0 (C-1′′′), 129.7 (C-2′′′, C-6′′′), 114.1 (C-3′′′, C-5′′′), 73.4 (–OCH2Ar), 71.6 (C-sn3), 71.0 (C-sn2), 64.2 (C-sn1), 55.5 (–OCH3). EIHRMS: calcd. for C11H16O4 [M + Na]+: 235.0941, found: 235.0946.
4.24. Preparation of 3-O-p-Methoxybenzyl-1-O-trityl-sn-glycerol (36)
To a solution of 35 (2.4 g, 11 mmol) in pyridine (23 mL), TrCl (3.1 g, 11 mmol) was added and the mixture was heated to boiling for 15 h. The reaction mixture was allowed to cool to rt and H2O was added, then it was extracted with EtOAc and washed with 2 M HCl, 6% NaHCO3 and brine, dried over anhydrous Na2SO4 and filtered. The removal of the solvent led to a crude which was purified by column chromatography (Hex/EtOAc 9:1) to obtain 36 (4.6 g, 92%). −0.7 (c 0.8, CHCl3); IR (film, cm−1) 3449, 2932, 2870, 1512, 1491, 1448, 1248, 1076, 1034, 765, 706, 633; 1H-NMR (400 MHz, CDCl3) δ 7.46–7.42 (6H, m, H-2′, 6′), 7.32–7.24 (9H, m, H-3′-5′), 7.22 (2H, d, J = 8.6 Hz, H-2′′′, H-6′′′), 6.87 (2H, d, J = 8.6 Hz, H-3′′′, H-5′′′), 4.48 (2H, s, –OCH2Ar), 3.99 (1H, m, H-sn2), 3.81 (3H, s, –OCH3), 3.59 (1H, dd, J = 9.7, 4.3 Hz, HA-sn3), 3.54 (1H, dd, J = 9.7, 6.2 Hz, HB-sn3) 3.25 (1H, dd, J = 9.4, 5.7 Hz, HA-sn1), 3.21 (1H, dd, J = 9.4, 5.7 Hz, HB-sn1); 13C-NMR (100 MHz, CDCl3) δ 159.2 (C-4′′′), 143.8 (C-1′), 130.1 (C-1′′′), 129.3 (C-2′′′, C-6′′′), 128.6 (C-3′, C-5′), 127.8 (C-2′, C-6′), 127.0 (C-4′), 113.8 (C-3′′′, C-5′′′), 86.6 (–CPh3), 73.0 (–OCH2Ar), 71.2 (C-sn3), 69.9 (C-sn2), 64.4 (C-sn1), 55.2 (–OCH3). EIHRMS: calcd. for C30H30O4 [M + Na]+ 477.2036, found [M + Na]+ 477.2022.
4.25. Preparation of 3-O-p-Methoxybenzyl-2-O-octadecyl-1-O-trityl-sn-glycerol (37)
To a solution of 36 (570 mg, 1.26 mmol) in toluene (2.5 mL), NaNH2 (491 mg, 12.6 mmol) was added, it was heated at 111 °C under an argon atmosphere for 1 h. The mixture was cooled to rt and a solution of bromooctadecane (1.7 g, 5 mmol) in toluene (2 mL) added, then heated at 111 °C overnight. The reaction was allowed to cool to 0 °C, crushed ice and saturated NH4Cl added and extracted with EtOAc. The organic layer was washed with H2O and brine. After drying over anhydrous Na2SO4, filtering and evaporating, the reaction mixture was purified by column chromatography (Hex/EtOAc 98:2) to yield 37 (865 mg, 97%). −3.6 (c 1.2, CHCl3); 1H-NMR (400 MHz, CDCl3, δ ppm): 7.47–7.44 (6H, m, H-2′, 6′), 7.31–7.24 (9H, m, H-3′-5′), 7.19 (1H, d, each, J = 8.6 Hz, H-2′′′, H-6′′′), 6.84 (1H, d, each, J = 8.6 Hz, H-3′′′, H-5′′′), 4.48, 4.43 (1H, d, each, J = 11.7 Hz, –OCH2Ar), 3.80 (3H, s, –OCH3), 3.61–3.54 (1H, m, H-sn2), 3.61–3.54 (2H, m, H-sn3), 3.53 (2H, t, J = 6.6 Hz, H-1′′), 3.21 (2H, d, J = 4.6 Hz, H-sn1), 1.56 (2H, m, H-2′′), 1.27 (30H, m, H-3′′-17′′), 0.89 (3H, t, J = 6.8 Hz, H-18′′); 13C-NMR (50 MHz, CDCl3, δ ppm): 159.3 (C-4′′′), 144.4 (3·C-1′), 130.8 (C-1′′′), 129.4 (C-2′′′, C-6′′′), 129.0/128.2 (3·C-3′, 3·C-5′), 128.0/127.5 (3·C-2′, 3·C-6′), 127.1 (3·C-4′), 113.9 (C-3′′′, C-5′′′), 86.8 (–CPh3), 78.6 (C-sn2), 73.1 (–OCH2Ar), 70.9 (C-1′′), 70.4 (C-sn3), 63.7 (C-sn1), 55.5 (–OCH3), 32.2 (C-16′′), 30.4–29.6 (C-2′′, 4′′-15′′), 26.4 (C-3′′), 22.9 (C-17′′), 14.4 (C-18′′); EIHRMS: calcd. for C48H66O4 [M + Na]+: 729.4853, found: 729.4854.
4.26. Preparation of 3-O-p-Methoxybenzyl-2-O-octadecyl-sn-glycerol (38)
To a mixture of 37 (865 mg, 1.22 mmol) in MeOH (12 mL) and CHCl3 (1 mL), p-TsOH (232 mg, 1.22 mmol) was added, and the mixture was stirred under an argon atmosphere at rt for 2 h 30 min., H2O added and the mixture extracted with EtOAc. The organic layer washed with 6% NaHCO3 and H2O, dried over anhydrous Na2SO4, filtered and evaporated. The residue was purified by column chromatography (Hex/EtOAc 95:5) to yield 38 (464 mg, 82%). +8.0 (c 0.9, CHCl3); IR (film, cm−1): 3451, 2922, 2853, 1514, 1248, 1094, 1040; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.25 (2H, d, J = 8.6 Hz, H-2′′′, H-6′′′), 6.88 (2H, d, J = 8.6 Hz, H-3′′′, H-5′′′), 4.49, 4.45 (1H, d, each, J = 11.9 Hz, –OCH2Ar), 3.80 (3H, s, –OCH3), 3.74–3.55 (2H, m, H-sn1), 3.55–3.47 (1H, m, H-sn2), 3.55–3.47 (2H, m, H-sn3), 3.53 (2H, t, J = 4.4 Hz, H-1′′), 1.56 (2H, m, H-2′′), 1.26 (30H, m, H-3′′-17′′), 0.88 (3H, t, J = 6.8 Hz, H-18′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 159.5 (C-4′′′), 130.3 (C-1′′′), 129.5 (C-2′′′, C-6′′′), 114.0 (C-3′′′, C-5′′′), 78.6 (C-sn2), 73.4 (–OCH2Ar), 70.6 (C-1′′), 69.9 (C-sn3), 63.2 (C-sn1), 55.5 (–OCH3), 32.2 (C-16′′), 30.3–29.6 (C-2′′, 4′′-15′′), 26.3 (C-3′′), 22.9 (C-17′′), 14.4 (C-18′′); EIHRMS: calcd. for C29H52O4 [M + Na]+: 487.3758, found: 487.3755.
4.27. Preparation of 1-Chlorocarbonyl-3-O-p-methoxybenzyl-2-O-octadecyl-sn-glycerol (39)
To an ice cooled solution of 38 (261 mg, 0.56 mmol) in THF (1.1 mL), trichloromethyl chloroformate (diphosgene, 67 μL, 0.56 mmol) and N,N-dimethylaniline (71 μL, 0.56 mmol) were slowly added. The mixture was stirred at 0 °C for 15 min and then at rt for 2 h. Then Et2O was added and the white precipitate filtered. The solution washed with 0.2 M HCl, 0.2 M NaOH and H2O, dried over anhydrous Na2SO4 and evaporated to give 39 (244 mg, 82%). IR (film, cm−1): 2924, 2853, 1774, 1248, 1167, 1101; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.24 (2H, d, J = 8.4 Hz, H-2′′′, H-6′′′), 6.88 (2H, d, J = 8.4 Hz, H-3′′′, H-5′′′), 4.47 (2H, s, –OCH2Ar), 4.45 (1H, dd, J = 11.2, 3.7 Hz, HA-sn1), 4.36 (1H, dd, J = 11.2, 6.2 Hz, HB-sn1), 3.80 (3H, s, –OCH3), 3.68 (1H, m, H-sn2), 3.56–3.52 (2H, m, H-1′′), 3.55–3.51 (1H, dd, J = 10.0, 4.7 Hz, HA-sn3), 3.49–3.45 (1H, dd, J = 10.0, 6.2 Hz, HB-sn3) 1.54 (2H, m, H-2′′), 1.26 (30H, m, H-3′′-17′′), 0.88 (3H, t, J = 6.8 Hz, H-18′′); 13C-NMR (100 MHz, CDCl3, δ ppm): 159.3 (C-4′′′), 150.7 (–O–CO–Cl), 129.8 (C-1′′′), 129.3 (C-2′′′, C-6′′′), 113.8 (C-3′′′, C-5′′′), 75.8 (C-sn2), 73.1 (–OCH2Ar), 71.1 (C-sn1), 70.8 (C-1′′), 68.2 (C-sn3), 55.2 (–OCH3), 31.9 (C-16′′), 29.8–29.3 (C-2′′, 4′′-15′′), 25.9 (C-3′′), 22.6 (C-17′′), 14.1 (C-18′′); EIHRMS: in MeOH, calcd. for methyl ester, C31H54O6 [M + Na]+: 545.3813, found: 545.3794.
4.28. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecyl-3-O-p-methoxybenzyl-sn-glycerol (40)
To a solution of 1/2 (176 mg, 0.49 mmol), N,N-diisopropylethylamine (DIPEA, 0.14 mL, 0.79 mmol), 4-(dimethylamino) pyridine (DMAP) (30 mg, 0.25 mmol) in toluene (2.5 mL), a solution of 39 (244 mg, 0.46 mmol) in toluene (2.3 mL) was added dropwise at 0 °C . The reaction mixture was stirred at 0 °C under an argon atmosphere for 15 min and then at rt for 20 h. Then the solvent was removed and the residue was purified by column chromatography (Hex/EtOAc 98:2) to obtain 40 (226 mg, 58%). −7.1 (c 0.8, CHCl3); IR (film, cm−1): 2924, 2853, 1745, 1514, 1250; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.42/7.41 (1H, broad s, H-25′), 7.34/7.33 (1H, broad s, H-1′), 7.24/7.22 (2H, d, J = 8.6 Hz, H-2′′′, H-6′′′), 6.87/6.86 (2H, d, J = 8.6 Hz, H-3′′′, H-5′′′), 6.39 (1H, s, H-2′), 5.79/5.77 (1H, dd, J = 5.4, 3.3 Hz, H-4′), 5.35/5.33 (1H, t, J = 4.0 Hz, H-9′), 4.65–4.60 (2H, m, H-20′), 4.47/4.44 (2H, s, –OCH2Ar), 4.38/4.37 (1H, dd, each, J = 11.2, 4.0 Hz, HA-sn1), 4.30–4.25/4.20–4.10 (1H, m, each, HB-sn1), 3.80 (3H, s, –OMe), 3.70–3.60 (1H, m, H-sn2), 3.56–3.41 (2H, m, H-sn3), 3.48 (2H, t, J = 5.0 Hz, H-1′′), 2.00–1.49 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.70/1.66 (3H, s, Me-21′), 1.56 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.91 (3H, s, Me-22′), 0.89 (3H, t, J = 7.0 Hz, Me-18′′), 0.88 (3H, s, Me-24′), 0.81/0.80 (3H, d, J = 7.0 Hz, Me-23′); 13C-NMR (50 MHz, CDCl3, δ ppm): 159.5/159.4 (C-4′′′), 154.8 (–O–CO–O–), 147.3 (C-19′), 143.5 (C-1′), 141.6/141.3 (C-10′), 140.3 (C-25′), 130.4/130.2 (C-1′′′), 129.5 (C-2′′′, 6′′′), 126.3 (C-3′), 120.3/120.0 (C-9′), 114.0 (C-3′′′, 5′′′), 109.4/109.3 (C-20′), 109.0/108.9 (C-2′), 76.6 (C-sn2), 73.3 (–OCH2Ar), 71.0 (C-sn1), 70.3 (C-4′), 69.3/69.0 (C-1′′), 68.5 (C-sn3), 55.5 (–OMe), 44.4 (C-5′), 43.1 (C-15′), 42.8/42.6 (C-11′), 39.1 (C-14′), 37.6/37.4 (C-16′), 34.4/34.3 (C-6′), 32.6 (C-17′), 32.2 (C-16′′), 30.2–29.0 (C-2′′), 29.6–28.9 (C-4′′-15′′), 29.6–28.9 (C-7′, 13′), 26.2/26.1 (C-3′′), 23.1 (C-8′, 12′), 24.2 (C-24′), 23.0 (C-21′), 22.9 (C-17′′), 22.5 (C-22′), 15.9 (C-23′), 14.4 (C-18′′); EIHRMS: calcd. for C54H86O7 [M + Na]+: 869.6266, found: 869.6260.
4.29. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecyl-sn-glycerol (41)
To a solution of 40 (167 mg, 0.20 mmol) in CH2Cl2/H2O 18:1 (2.2 mL), DDQ (10 mg, 0.045 mmol) was added. It was stirred at rt under an argon atmosphere for 7 h, then quenched with 6% NaHCO3 and extracted with CH2Cl2. The organic layer was washed with 6% NaHCO3 and brine and dried over anhydrous Na2SO4. Removal of the solvent gave a crude product which was purified by column chromatography (Hex/EtOAc 97:3) on silica gel to obtain 41 (103 mg, 71%). +15.2 (c 1, CHCl3); IR (film, cm−1): 3506, 2924, 2853, 1746, 1258; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.42 (1H, s, H-25′), 7.34 (1H, s, H-1′), 6.39 (1H, s, H-2′), 5.80/5.75 (1H, m, H-4′), 5.36/5.33 (1H, t, J = 3.4 Hz, H-9′), 4.65–4.61 (2H, m, H-20′), 4.21/4.20 (1H, dd, each, J = 11.4, 4.8 Hz, HA-sn3), 4.16/4.13 (1H, dd, each, J = 11.4, 4.8 Hz, HB-sn3) 3.68–3.46 (3H, m, H-sn1, H-sn2), 3.56 (2H, t, J = 6.6 Hz, H-1′′), 2.20–1.32 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.70/1.67 (3H, s, Me-21′), 1.54 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.90 (3H, s, Me-22′), 0.88 (3H, t, J = 7.0 Hz, Me-18′′), 0.88 (3H, s, Me-24′), 0.81/0.80 (3H, d, J = 7.0 Hz, Me-23′); 13C-NMR (100 MHz, CDCl3, δ ppm): 154.6 (–O–CO–O–), 147.0 (C-19′), 143.2 (C-1′), 141.4/141.1 (C-10′), 140.2/140.1 (C-25′), 125.9 (C-3′), 120.0/119.7 (C-9′), 109.1/109.0 (C-20′), 108.6 (C-2′), 77.5/77.4 (C-sn2), 70.6 (C-1′′), 70.3 (C-4′), 66.2/66.1 (C-sn1), 61.8 (C-sn3), 44.2 (C-5′), 42.9 (C-15′), 42.4/42.3 (C-11′), 38.8 (C-14′), 37.3/37.2 (C-16′), 34.1/34.0 (C-6′), 32.4/32.3 (C-17′), 31.9 (C-16′′), 29.9–28.8 (C-2′′), 29.9–28.8 (C-4′′-15′′), 29.9–28.8 (C-7′, 13′), 26.0 (C-3′′), 22.8 (C-24′), 22.7 (C-8′, 12′), 22.6 (C-17′′), 22.2 (C-21′), 22.2 (C-22′), 15.6 (C-23′), 14.0 (C-18′′); EIHRMS: calcd. for C46H78O6 [M + Na]+: 749.5691, found: 749.5718.
4.30. Preparation of 1-O-[1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yloxycarbonyl]-2-O-octadecyl-3-eicosapentaenoyl-sn-glycerol (42)
To a solution of 41 (18 mg, 0.025 mmol), DMAP (4 mg, 0.032 mmol) and EDAC (6 mg, 0.032 mmol) in dry CH2Cl2 (0.24 mL), EPA (8 mg, 0.026 mmol) was added under an argon atmosphere. After stirring at rt for 14 h, the reaction mixture was passed through a short silica gel column (CH2Cl2/EtOAc 9:1 as eluent), the solvent removed and the crude purified by column chromatography (Hex/EtOAc 99:1) providing 42 (16 mg, 63%). IR (film, cm−1): 2959, 2924, 2853, 1744, 1258; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.42 (1H, broad s, H-25′), 7.34 (1H, broad s, H-1′), 6.39 (1H, s, H-2′), 5.79–5.76 (1H, m, H-4′), 5.40–5.30 (10H, m, =CH), 5.40–5.30 (1H, m, H-9′), 4.65/4.61 (2H, m, H-20′), 4.23–4.06 (2H, m, H-sn3), 4.23–4.06 (2H, m, H-sn1), 3.66 (1H, m, H-sn2), 3.50 (2H, t, J = 6.6 Hz, H-1′′), 2.84–2.80 (8H, m, =CCH2C=), 2.32 (2H, t, J = 7.5 Hz, H-2′′′), 2.13–2.06 (4H, m, H-4′′′, 19′′′), 2.04–1.40 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.82–1.79 (2H, m, H-3′′′), 1.69/1.66 (3H, s, Me-21′), 1.56 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.97 (3H, t, J = 7.6 Hz, H-20′′′), 0.89 (3H, s, Me-22′), 0.88 (3H, t, J = 7.0 Hz, Me-18′′), 0.88 (3H, s, Me-24′), 0.80 (3H, d, J = 6.8 Hz, Me-23′); 13C-NMR (50 MHz, CDCl3, δ ppm): 173.4 (C-1′′′), 154.7 (–O–CO–O–), 147.3 (C-19′), 143.5 (C-1′), 141.6/141.3 (C-10′), 140.4 (C-25′), 132.3 (C-18′′′), 129.1–127.2 (=CH) × 9, 126.2 (C-3′), 120.3/120.0 (C-9′), 109.4/109.3 (C-20′), 108.9 (C-2′), 75.2 (C-sn2), 71.0 (C-1′′), 70.5 (C-4′), 66.9/66.7 (C-sn3), 63.1 (C-sn1), 44.4 (C-5′), 43.1 (C-15′), 42.8/42.6 (C-11′), 39.1/38.9 (C-14′), 37.6/37.4 (C-16′), 34.4/34.3 (C-6′), 33.7 (C-2′′′), 32.7/32.6 (C-17′), 32.2 (C-16′′), 30.6 (C-7′), 30.1–29.2 (C-2′′), 30.1–29.2 (C-4′′-15′′), 30.1–29.2 (C-13′), 26.8–20.8 (3′′′, 4′′′, 7′′′, 10′′′, 13′′′, 16′′′, 19′′′, 17′′, 8′, 12′), 25.8 (C-3′′), 23.9 (C-24′), 22.9 (C-21′), 22.5 (C-22′), 15.9 (C-23′), 14.5 (C-20′′′), 14.3 (C-18′′); EIHRMS: calcd. for C66H106O7 [M + Na]+: 1033.7831, found: 1033.7866.
4.31. Preparation of 1-O-[25-Hydroxy-18-nor-ent-isodysidiola-2,9,19-trien-1,25-olide-4R/S-yloxycarbonyl]--2-O-octadecyl-3-eicosapentaenoyl-sn-glycerol (12)
Rose Bengal (2 mg) was added to a solution of 42 (16 mg, 0.016 mmol) and DIPEA (28 μL, 0.16 mmol) in dry CH2Cl2 (2 mL) at rt. Anhydrous oxygen was bubbled in for 5 min and, the solution placed under an oxygen atmosphere at −78 °C and irradiated with a 200 W lamp. After 4 h irradiation was stopped, the pink solution allowed to warm to rt, and saturated aqueous oxalic acid solution (2.5 mL) added. After a few minutes of vigorous stirring, the mixture was diluted with H2O (2 mL) and extracted with Et2O. The combined organic extracts were washed with H2O and dried over anhydrous Na2SO4. After filtration, the solvent was evaporated to give a residue which was purified by silica gel column chromatography to yield 12 (9 mg, 54%). IR (film, cm−1): 3402, 2959, 2924, 2853, 1751, 1256, 1136, 1126; 1H-NMR (400 MHz, CDCl3, δ ppm): 6.19/5.95 (1H, m, H-25′), 6.02/6.01 (1H, s, H-2′), 5.64–5.58 (1H, m, H-4′), 5.38–5.34 (10H, m, =CH), 5.38–5.34 (1H, m, H-9′), 4.67–4.63 (2H, m, H-20′), 4.34–3.98 (4H, m, H-sn1, H-sn3), 3.70–3.66 (1H, m, H-sn2), 3.53 (2H, t, J = 6.4 Hz, H-1′′), 2.85–2.78 (8H, m, =CCH2C=), 2.34 (2H, t, J = 7.2 Hz, H-2′′′), 2.13–2.04 (4H, m, H-4′′′, 19′′′), 2.00–1.49 (16H, m, H-5′, 7′, 8′, 11′, 12′, 13′, 14′, 16′, 17′), 1.84 (2H, m, H-3′′′), 1.70/1.69 (3H, s, Me-21′), 1.54 (2H, m, H-2′′), 1.25 (30H, m, H-3′′-17′′), 0.97 (3H, t, J = 7.6 Hz, H-20′′′), 0.91 (3H, s, Me-22′), 0.87 (3H, t, J = 6.8 Hz, Me-18′′), 0.87 (3H, s, Me-24′), 0.80 (3H, d, J = 6.9 Hz, Me-23′); 13C-NMR (100 MHz, CDCl3, δ ppm): 173.3 (C-1′′′), 169.0 (C-1′), 167.5 (C-3′), 154.2 (O–CO–O), 147.2 (C-19′), 141.4 (C-10′), 132.0 (C-18′′′), 128.9–127.0 (=CH) × 9, 120.0 (C-9′), 118.4 (C-2′), 109.2 (C-20′), 97.4 (C-25′), 76.5 (C-sn2), 70.8 (C-1′′), 70.8 (C-4′), 66.5 (C-sn1), 62.3 (C-sn3), 43.1 (C-5′), 42.8 (C-15′), 42.3 (C-11′), 38.4 (C-14′), 37.2 (C-16′), 34.6 (C-6′), 33.5 (C-2′′′), 32.4 (C-17′), 31.9 (C-16′′), 29.8–29.3 (C-2′′, 4′′-15′′), 28.9 (C-13′), 29.3 (C-7′), 26.5–20.5 (3′′′, 4′′′, 7′′′, 10′′′, 13′′′, 16′′′, 19′′′, 3′′), 22.8 (C-12′), 22.8 (C-8′), 22.7 (C-24′), 22.7 (C-21′), 22.6 (C-17′′), 22.5 (C-22′), 15.6 (C-23′), 14.2 (C-20′′′), 14.0 (C-18′′); EIHRMS: calcd. for C66H106O9 [M + Na]+: 1065.7729, found: 1065.7725.
4.32. Preparation of 1,25-Epoxy-18-nor-ent-isodysidiola-1,3(25),9,19-tetraen-4R/S-yl eicosapentaenoate (13)
To a solution of 1/2 (36 mg, 0.1 mmol), DMAP (16 mg, 0.13 mmol) and EDAC (25 mg, 0.13 mmol) in dry CH2Cl2 (1 mL), EPA (30 mg, 0.1 mmol) was added under an argon atmosphere. After stirring at rt for 20 h, the reaction mixture was passed through a short silica gel column ( CH2Cl2/EtOAc/ 9:1 as eluent), the solvent removed and the crude product purified by column chromatography to give 13 (40 mg, 63%). 1H-NMR (400 MHz, CDCl3, δ ppm): 7.38 (1H, s, H-25), 7.32 (1H, s, H-1), 6.35 (1H, s, H-2), 5.98/5.96 (1H, dd, J = 5.1, 3.4 Hz, H-4), 5.39–5.32 (10H, m, =CH), 5.39–5.32 (1H, m, H-9), 4.64 (1H, broad s, HA-20), 4.61 (1H, broad s, HB-20), 2.85–2.75 (8H, m, =CCH2C=), 2.25 (2H, t, J = 7.6 Hz, H-2′), 2.12–2.03 (4H, m, H-4′, 19′), 2.03–1.30 (16H, m, H-5, 7, 8, 11, 12, 13, 14, 16, 17), 1.87–1.78 (2H, m, H-3′), 1.70/1.67 (3H, s, Me-21), 0.97 (3H, t, J = 7.5 Hz, H-20′), 0.90 (3H, s, Me-22), 0.88 (3H, s, Me-24), 0.81/0.80 (3H, d, J = 6.9 Hz, Me-23); 13C-NMR (100 MHz, CDCl3, δ ppm): 172.8 (C-1′), 147.0 (C-19), 143.0 (C-1), 141.3 (C-10), 140.0 (C-25), 132.0 (C-18′), 129.0–127.0 (=CH) × 9, 126.7 (C-3), 119.9/119.8 (C-9), 109.0 (C-20), 108.8 (C-2), 65.4 (C-4), 44.0 (C-5), 42.9 (C-15), 42.4 (C-11), 38.9/38.8 (C-14), 37.2 (C-16), 34.2/34.1 (C-6), 34.0 (C-2′), 32.3 (C-17), 29.7 (C-7), 29.6 (C-13), 28.9–20.5 (3′, 4′, 7′, 10′, 13′, 16′, 19′), 22.8 (C-24), 22.7 (C-8, 12), 22.3 (C-21), 22.3 (C-22), 15.6 (C-23), 14.2 (C-20′); EIHRMS: calcd. for C44H64O3 [M + Na]+: 663.4748, found: 663.4747.
4.33. Preparation of 4-Eicosapentaenoyl-25-hydroxy-18-nor-ent-isodysidiola-2,9,19-trien-1,25-olide (14)
Rose Bengal (2 mg) was added to a solution of 13 (18 mg, 0.028 mmol) and DIPEA (36 mg, 0.28 mmol) in dry CH2Cl2 (4 mL) at rt. Anhydrous oxygen was bubbled in for 5 min., the solution placed under an oxygen atmosphere at −78 °C and irradiated with a 200 W lamp. After 4 h irradiation was stopped, the pink solution allowed to warm to rt, and saturated aqueous oxalic acid solution (3 mL) added. After a few minutes of vigorous stirring, the mixture was diluted with H2O (3 mL) and extracted with Et2O. The combined organic extracts were washed with H2O and dried over anhydrous Na2SO4. After filtration, the solvent was evaporated to give a residue which was purified by silica gel column chromatography (Hex/EtOAc 95:5) to yield 14 (10 mg, 54%). IR (film, cm−1): 3389, 2961, 2926, 2872, 1744, 1142; 1H-NMR (400 MHz, CDCl3, δ ppm): 6.19 (1H, m, H-25)/5.96 (1H, s, H-25), 5.99/5.93 (1H, s, H-2), 5.58 (1H, d, J = 9.0 Hz, H-4), 5.44–5.32 (10H, m, =CH), 5.44–5.32 (1H, m, H-9), 4.66 (1H, broad s, HA-20), 4.61 (1H, broad s, HB-20), 2.86–2.78 (8H, m, =CCH2C=), 2.37–2.30 (2H, m, H-2′), 2.12–2.06 (4H, m, H-4′, 19′), 2.06–1.30 (16H, m, H-5, 7, 8, 11, 12, 13, 14, 16, 17), 1.89–1.83 (2H, m, H-3′), 1.70 (3H, s, Me-21), 0.97 (3H, t, J = 7.5 Hz, H-20′), 0.92 (3H, s, Me-22), 0.89 (3H, s, Me-24), 0.81 (3H, d, J = 6.9 Hz, Me-23); 13C-NMR (50 MHz, CDCl3, δ ppm): 173.3 (C-1′), 169.5 (C-1), 168.7 (C-3), 147.3 (C-19), 141.4 (C-10), 132.3 (C-18′), 129.5–127.2 (=CH) × 9, 120.1 (C-9), 118.3 (C-2), 109.5 (C-20), 97.9 (C-25), 67.2 (C-4), 43.1 (C-15), 42.9 (C-11), 42.8 (C-5), 39.0/38.6 (C-14), 37.6 (C-16), 34.9/34.6 (C-6), 33.9 (C-2′), 32.7 (C-17), 31.6 (C-7), 28.9 (C-13), 26.7–20.8 (3′, 4′, 7′, 10′, 13′, 16′, 19′, 8, 12), 23.0 (C-24), 22.4 (C-21), 22.4 (C-22), 15.8 (C-23), 14.5 (C-20′); EIHRMS: calcd. for C44H64O5 [M + Na]+: 695.4646, found: 695.4669.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}