2. Materials and Methods
All commercial reagents were used as received. Arylazides were prepared from the corresponding anilines following literature procedures [
23]. Anhydrous solvents were purchased from Sigma Aldrich (St. Louis, MO, USA). Flash column chromatography was performed using silica gel 200–400 Mesh. TLC analyses were performed using silica gel plates, using ultraviolet light (254 nm), phosphomolybdic acid or vanillin solution for visualization. Melting points are uncorrected and were recorded on a Buchi B-540 apparatus (Zurich, Switzerland). For NMR data, the chemical shifts are reported in δ (ppm) referenced to residual solvent protons and
13C signals in deuterated chloroform. Coupling constants (
J) are expressed in hertz (
Hz). Infrared spectra were obtained on a Thermo Scientific Nicolet 380 FT-IR apparatus (600–4000 cm
−1, Nicolet Instrument Corp., Madison, WI, USA) using attenuated total reflection (ATR). Mass spectra were obtained by GC-MS, Shimadzu QP-2010 Plus model (Shimadzu, Kyoto, Japan). SMILES notations of the xylitan derivatives were inputted into the online Molinspiration software (software version v2015.01) and only bioactive compounds were subjected to molecular properties prediction by Molinspiration software [
24].
2.1. Synthesis of 3,5-O-Isopropylidene-1,4-xylitan (2)
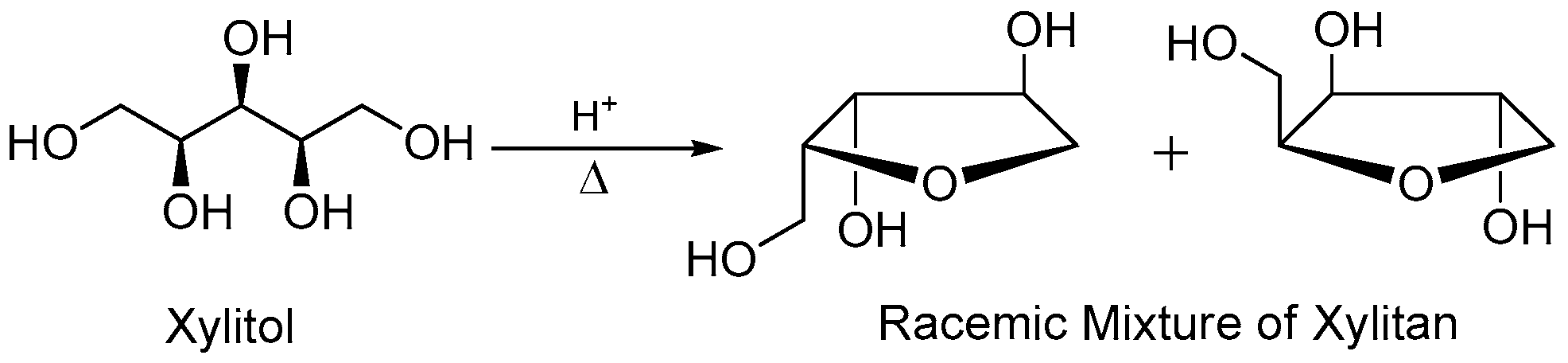
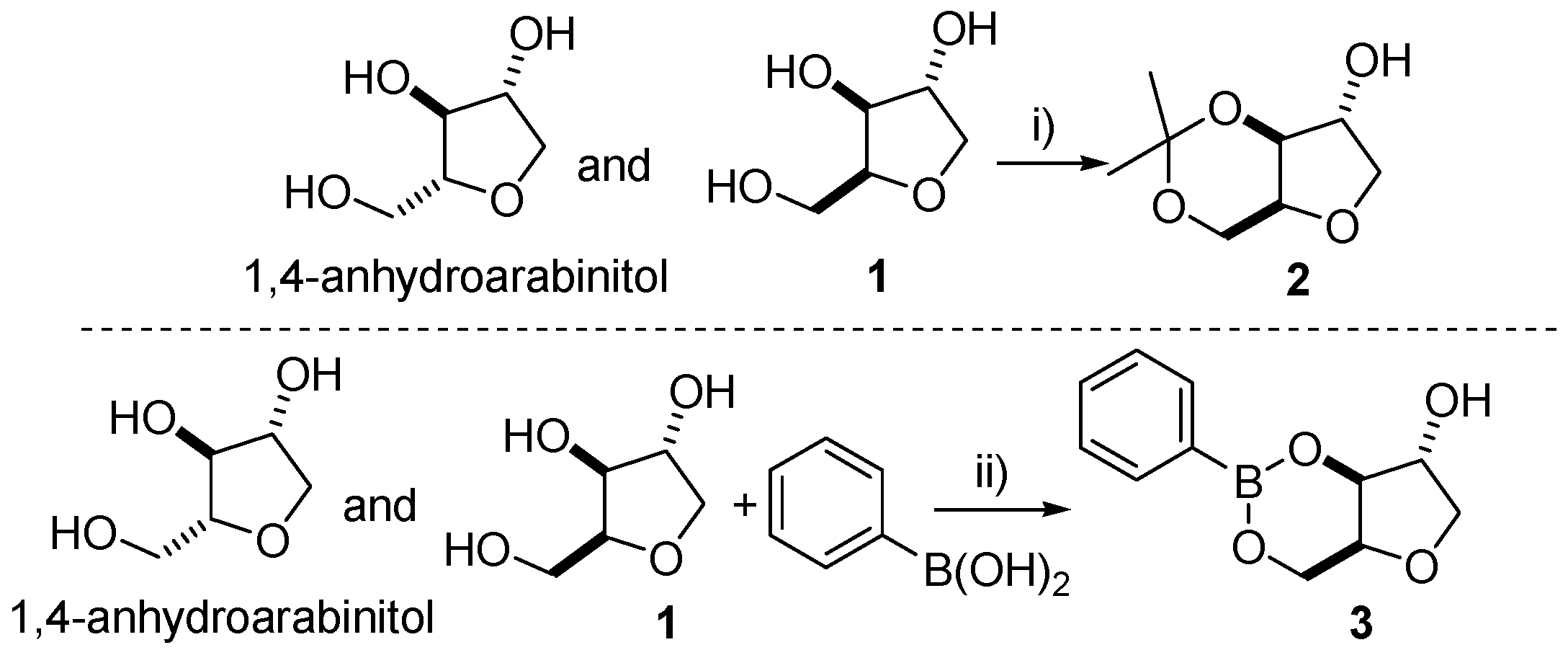
To a round bottom flask (1000 mL) equipped with stir bar was added xylitan (100 g), anhydrous copper sulfate (100 g) and dry acetone (500 mL) at room temperature under an atmosphere of nitrogen. Concentrated sulfuric acid (0.3 mL) was added to the heterogeneous mixture and the system was kept under stirring at room temperature for 12 h. After this period, the reaction mixture was filtered through a sintered glass funnel. The organic phase was neutralized with an excess of calcium hydroxide, and filtered again on a sintered glass funnel. The mixture was then concentrated in vacuo to yield a viscous liquid. Purification was performed by column chromatography on flash silica gel eluting with ethyl acetate to afford xylitan-acetonide (±)-2 (80%) as a white solid, m.p. 68–70 °C. 1H-NMR (250 MHz, CDCl3): δ 4.17–4.23 (m, 3H), 3.88–4.07 (m, 3H), 3.72 (d, J = 8.3 Hz, 1H), 2.96 (bs, OH), 1.41 (s, 3H), 1.33 (s, 3H); 13C-NMR (62.5 MHz, CDCl3): δ 97.4, 76.6, 75.4, 74.4, 72.0, 60.6, 28.5, 19.3.
2.2. Synthesis of (±)-3,5-O-Phenylboronate-1,4-xylitan (3)
In a round bottom flask equipped with stir bar and reflux condenser, xylitol (20 g; 131 mmol) and an aqueous solution of sulphuric acid (5.60 mL, 5% v/v) were added. The mixture was heated at 135 °C with stirring for 45 min. Upon cooling, xylitan was obtained as a viscous pale yellow liquid (16.40 g; 82%) and used in the next step without further purification. In a two necked round bottom flask equipped with stir bar, a Dean-Stark apparatus, a Liebig condenser and an addition funnel with pressure equalizing side arm, phenylboronic acid (0.874 g; 7.16 mmol) and 80 mL of benzene were added. A solution of xylitan 1 (0.800 g; 5.97 mmol) in 20 mL of methanol was added dropwise by addition funnel at 80 °C. Upon complete addition of xylitan, the temperature was raised to 100 °C and allowed to stir for 1 h. Finally, the reaction was allowed to cool to room temperature, the excess of benzene was removed under reduced pressure and the resulting liquid residue was triturated with a mixture of ethyl acetate and hexane (1:4) to afford compound (±)-3 as a white solid in 47% yield. m.p. 103–104 °C; Rf = 0.48 (ethyl acetate/hexane, 2:3); IR (cm−1): 3441, 1595, 1448, 1310; 1H-NMR (500 MHz, CDCl3): δ 1.91 (brs, 1H), 3.80 (d, 1H, J = 10 Hz), 4.22–4.51 (m, 6H), 7.34 (t, 1H, J = 7.5 Hz), 7.43 (t, 2H, J = 7.5 Hz), 7.78 (d, 2H, J = 10 Hz); 13C-NMR (125 MHz, CDCl3): δ 61.2, 74.5, 74.6, 77.0, 77.4, 127.5, 130.9, 133.8, 134.4; EI m/z (%): 57 (40%), 149 (100%).
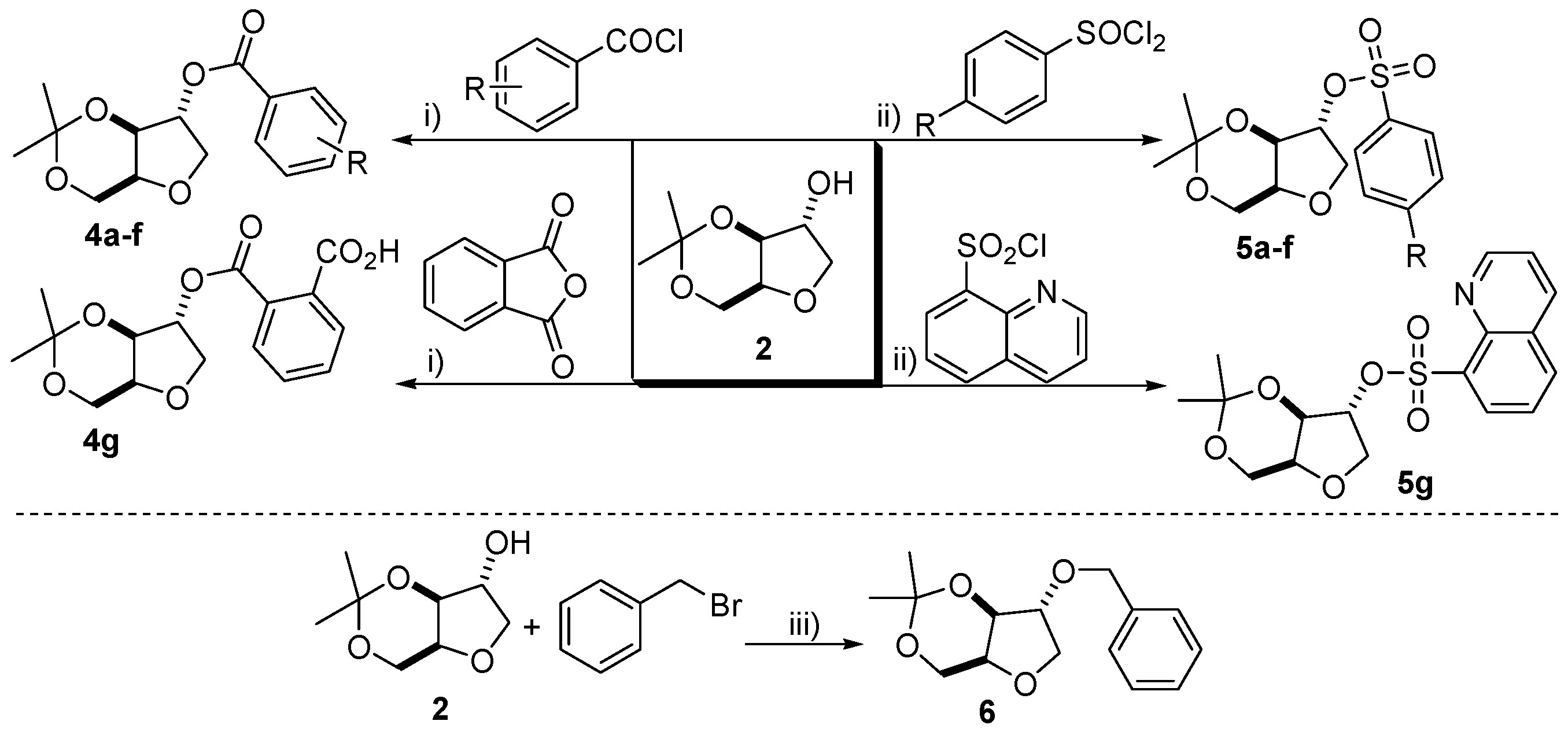
2.3. Synthesis of Benzoate Esters (±)-4a–g
A general procedure is as follows: xylitan acetonide (±)-2 (3 mmol) and a benzoyl chloride (3 mmol) or phthalic anhydride (3 mmol) were stirred in CH2Cl2 (5 mL) and pyridine (2 mL) at 0 °C for 0.5 h and then allowed to warm to room temperature. Stirring was continued for a further 16 h at room temperature. Upon completion, the reaction mixture was taken up into a separatory funnel containing a mixture of CH2Cl2 (15 mL) and a saturated aqueous solution of sodium bicarbonate (20 mL). The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo and the crude product was purified by flash chromatography.
(±)-2-Benzoyl-3,5-O-isopropylidene-1,4-xylitan (4a): Product obtained as a white solid in 58% yield. m.p. 80–82 °C; Rf = 0.15 (ethyl acetate/hexane 2:8); IR (cm−1): 1725, 1449, 1263, 1174, 1102, 924, 782, 710; 1H-NMR (300 MHz, CDCl3): δ 1.41 (s, 3H), 1.46 (s, 3H), 3.96–4.48 (m, 6H), 5.39 (d, 1H, J = 3 Hz), 7.43 (t, 2H, J = 7.5 Hz), 7.57 (t, 1H, J = 7.5 Hz), 8.01 (d, 2H, J = 6 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.4, 28.7, 60.6, 72.3, 72.6, 73.5, 79.3, 97.8, 129.6, 129.8, 133.5, 165.5; EI m/z (%): 177 (30%), 105 (100%), 77 (40%), 69 (50%).
(±)-2-(4-Methoxybenzoyl)-3,5-O-isopropylidene-1,4-xylitan (4b): Product obtained as a white solid in 25% yield. m.p. 102–104 °C; Rf = 0.48 (ethyl acetate/hexanes 2:8); IR (cm−1): 1699, 1592, 1449, 862, 756; 1H-NMR (300 MHz, CDCl3): δ 1.33 (s, 3H), 1.38 (s, 3H), 3.76–4.37 (m, 7H), 6.82 (d, 2H, J = 9 Hz), 7.87 (d, 2H, J = 9 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.1, 28.5, 55.3, 60.3, 72.0, 72.4, 73.3, 78.8, 97.5, 113.5, 121.7, 131.6, 163.6, 165.0; EI m/z (%): 207 (30%), 135 (100%), 101 (60%).
(±)-2-(3,4,5-Trimethoxybenzoyl)-3,5-O-isopropylidene-1,4-xylitan (4c): Product obtained as a white solid in 12% yield. m.p. 120–122 °C; Rf = 0.45 (ethyl acetate/hexanes 2:8); IR (cm−1): 2989, 2945, 1699, 1592, 1449, 1227, 995, 862, 756; 1H-NMR (300 MHz, CDCl3): δ 1.44 (s, 3H), 1.49 (s, 3H), 3.92–4.51 (m, 15H), 5.40 (s, 1H), 7.29 (s, 2H); 13C-NMR (75 MHz, CDCl3): δ 19.5, 28.7, 56.4, 60.6, 61.1, 72.3, 72.7, 73.5, 79.5, 97.9, 107.1, 124.5, 142.8, 153.1, 165.3; EI m/z (%): 167 (25%), 95 (100%), 68 (60%).
(±)-2-(3-Nitrobenzoyl)-3,5-O-isopropylidene-1,4-xylitan (4d): Product obtained as a white solid in 63% yield. m.p. 110–111 °C; Rf = 0.30 (ethyl acetate/hexane 2:8); IR (cm−1): 2882, 1716, 1610, 1520, 969, 827, 719; 1H-NMR (300 MHz, CDCl3): δ 1.40 (s, 3H), 1.46 (s, 3H), 3.96–4.48 (m, 6H), 5.40 (d, 1H, J = 3 Hz), 7.66 (t, 1H, J = 9 Hz), 8.33 (d, 1H, J = 9 Hz), 8.41 (d, 1H, J = 9 Hz), 8.78 (s, 1H); 13C-NMR (75 MHz, CDCl3): δ 19.4, 28.6, 60.5, 72.1, 72.7, 73.4, 80.3, 97.9, 124.6, 127.9, 129.9, 131.3, 135.4, 148.4, 163.5; EI m/z (%): 104 (30%), 81 (100%).
(±)-2-(4-Chlorobenzoyl)-3,5-O-isopropylidene-1,4-xylitan (4e): Product obtained as a white solid in 54% yield. m.p. 198–199 °C; Rf = 0.48 (ethyl acetate/hexane 2:8); IR (cm−1): 1681, 1583, 1414, 916, 844, 800, 765, 684; 1H-NMR (300 MHz, CDCl3): δ 1.25 (s, 3H), 1.41 (s, 3H), 3.95–5.38 (m, 7H), 7.41 (d, 2H, J = 9 Hz), 7.94 (d, 2H, J = 9 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.4, 28.7, 60.6, 72.3, 72.6, 73.5, 79.3, 97.8, 113.6, 121.7, 131.7, 163.6, 165.1; EI m/z (%): 313 (1%), 91 (100%), 68 (90%).
(±)-2-(4-Methylbenzoyl)-3,5-O-isopropylidene-1,4-xylitan (4f): Product obtained as a white solid in 55% yield. m.p. 85–87 °C; Rf = 0.25 (ethyl acetate/hexane 2:8); IR (cm−1): 2971, 1708, 1111, 969, 827, 746, 666; 1H-NMR (300 MHz, CDCl3): δ 1.41 (s, 3H), 1.46 (s, 3H), 2.40 (s, 3H), 3.95–4.48 (m, 6H), 5.37 (d, 1H, J = 6 Hz), 7.23 (d, 2H, J = 9 Hz), 7.90 (d, 2H, J = 9 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.8, 22.2, 29.2, 61.0, 72.8, 73.1, 73.1, 79.6, 98.2, 127.3, 129.7, 130.2, 144.7, 166.1; EI m/z (%): 119 (100%), 91 (30%), 81 (30%), 69 (40%).
(±)-2-(2-Carboxybenzoyl)-3,5-O-isopropylidene-1,4-xylitan (4g): Product obtained as a pale yellow liquid in 35% yield. Rf = 0.46 (ethyl acetate/hexane 2:8); 1H-NMR (300 MHz, CDCl3): δ 1.40 (s, 3H), 1.45 (s, 3H), 3.77–4.10 (m, 6H), 4.40–4.58 (m, 1H), 5.37 (d, 1H, J = 3Hz), 7.55–7.58 (m, 2H), 7.70–7.77 (m, 2H); 13C-NMR (75 MHz, CDCl3): δ 19.5, 28.7, 60.6, 71.8, 72.6, 73.2, 80.1, 97.8, 129.2, 131.6, 166.4, 167.0. EI m/z (%): 69 (80%), 81 (70%), 98 (30%), 149 (100%), 211 (40%), 282 (42%).
2.4. Synthesis of Arylsulfonate Esters (±)-5a–g
A general procedure is as follows: xylitan acetonide (±)-2 (5 mmol), the appropriate arylsulfonyl chloride (10 mmol), 4-Dimethylaminopyridine (DMAP) (5 mol %) and CH2Cl2 (10 mL) were stirred together in round bottom flask at room temperature for 12 h. Next, the reaction mixture was taken up into a separatory funnel containing a mixture of CH2Cl2 (15 mL) and a saturated aqueous solution of sodium bicarbonate (20 mL). The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo and the crude product was purified by flash chromatography.
(±)-2-Benzenesulfonyl-3,5-O-isopropylidene-1,4-xylitan (5a): Product obtained as a white solid in 74% yield. m.p. 62–64 °C; Rf = 0.19 (ethyl acetate/hexane 6:4); IR (cm−1): 1440, 1356, 910, 826, 756, 684; 1H-NMR (300 MHz, CDCl3): δ 1.31 (s, 3H), 1.35 (s, 3H), 3.76–4.04 (m, 4H), 4.20 (dd, 1H, J = 9 Hz, J = 3 Hz), 4.33 (s, 1H); 4.86 (d, 1H, J = 6 Hz), 7.54–7.70 (m, 3H), 7.90 (d, 2H, J = 9 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.3, 28.6, 60.3, 71.6, 72.1, 73.5, 84.6, 97.8, 127.9, 129.5, 134.3, 136.4; EI m/z (%): 299 (20%), 141 (30%), 81 (80%), 69 (100%), 59 (40%).
(±)-2-(4-Methylbenzenesulfonyl)-3,5-O-isopropylidene-1,4-xylitan (5b): Product obtained as a white solid in 73% yield. m.p. 82–85 °C; Rf = 0.48 (ethyl acetate/hexanes 6:4); IR (cm−1): 3004, 2912, 1592, 1355, 1188, 1099, 1050, 894, 809, 778, 667; 1H-NMR (300 MHz, CDCl3): 1.33 (s, 3H), 1.38 (s, 3H), 2.45 (s, 3H), 3.76 4.05 (m, 4H), 4.20 (dd, 1H, J = 7.5 Hz, J = 4.5 Hz), 4.35 (s, 1H), 4.84 (d, 1H, J = 3 Hz), 7.36 (d, 2H, J = 6 Hz), 7.79 (d, 2H, J = 6 Hz); 13C-NMR (75 MHz, CDCl3): 19.3, 21.8, 28.6, 60.4, 71.7, 72.2, 73.6, 84.5, 97.9, 128.0, 130.2, 133.4, 145.5; EI m/z (%): 312 (20%), 155 (60%), 91 (70%), 81 (60%), 68 (100%).
(±)-2-(4-Chlorobenzenesulfonyl)-3,5-O-isopropylidene-1,4-xylitan (5c): Product obtained as a white solid in 47% yield. m.p. 74–75 °C; Rf = 0.37 (ethyl acetate/hexane 2:8); IR (cm−1): 2909, 1699, 1574, 1485, 1360, 889, 800; 1H-NMR (300 MHz, CDCl3): δ 1.33 (s, 3H), 1.39 (s, 3H), 3.78–4.05 (m, 4H), 4.24 (dd, 1H, J = 7.5 Hz, J = 3 Hz), 4.36 (s, 1H), 4.86 (d, 1H, J = 3 Hz), 7.55 (d, 2H, J = 6 Hz), 7.85 (d, 2H, J = 6 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.3, 28.6, 60.3, 71.6, 72.2, 73.6, 84.9, 97.9, 129.4, 129.9, 134.8, 141.1; EI m/z (%): 333 (20%), 175 (30%), 111 (30%), 69 (100%), 59 (60%).
(±)-2-(4-Fluorobenzenesulfonyl)-3,5-O-isopropylidene-1,4-xylitan (5d): Product obtained as a pale yellow liquid in 67% yield. Rf = 0.24 (ethyl acetate/hexanes 3:7); IR (cm−1): 2918, 1699, 1592, 1502, 1369, 1114, 1102, 960, 898, 844; 1H-NMR (300 MHz, CDCl3): δ 1.27 (s, 3H), 1.33 (s, 3H), 3.73–4.00 (m, 4H), 4.18 (dd, 1H, J = 7.5 Hz, J = 4.5 Hz), 4.31 (s, 1H), 4.82 (d, 1H, J = 3 Hz), 7.21(t, 2H, J = 7.5 Hz), 7.87–7.92 (m, 2H); 13C-NMR (75 MHz, CDCl3): δ 19.2, 28.4, 60.1, 71.4, 72.0, 73.4, 84.7, 97.7, 116.8 (d, C-F, J = 23.2 Hz), 130.7 (d, C-F, J = 9.7 Hz), 132.3 (d, C-F, J = 3Hz), 165.9 (d, C-F, J = 255.7 Hz); EI m/z (%): 317 (20%), 159 (30%), 95 (40%), 69 (100%).
(±)-2-(4-Methoxybenzenesulfonyl)-3,5-O-isopropylidene-1,4-xylitan (5e): Product obtained as a pale yellow liquid in 19% yield. Rf = 0.45 (ethyl acetate/hexane 3:7); IR (cm−1): 1601, 1502, 1369, 898, 809; 1H-NMR (300 MHz, CDCl3): δ 1.31 (s, 3H), 1.36 (s, 3H), 3.75–4.33 (m, 10H), 4.80 (s, 1H), 7.00 (d, 2H, J = 6 Hz), 7.82 (d, 2H, J = 6 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.7, 29.0, 56.3, 60.8 72.1, 72.5, 74.0, 84.7, 98.2, 115.1, 128.0, 130.6, 164.6; EI m/z (%): 328 (50%), 188 (70%), 171 (100%), 107(30%), 81 (70%), 59 (60%).
(±)-2-(4-Nitrobenzenesulfonyl)-3,5-O-isopropylidene-1,4-xylitan (5f): Product obtained as a white solid in 60% yield. m.p. 116–118 °C; Rf = 0.52 (ethyl acetate/hexanes 2:8); IR (cm−1): 3117, 1602, 1529, 1356, 1314, 1193, 1099, 1039, 894, 732, 686; 1H-NMR (300 MHz, CDCl3): δ 1.34 (s, 3H), 1.40 (s, 3H), 3.82–4.07 (m, 4H), 4.27 (dd, 1H, J = 3 Hz, J = 6 Hz), 4.40 (d, 1H, J = 3 Hz), 4.95 (d, 1H, J = 3 Hz), 8.12 (d, 2H, J = 9 Hz), 8.42 (d, 2H, J = 9 Hz); 13C-NMR (75 MHz, CDCl3): δ 19.5, 28.6, 60.3, 71.6, 72.3, 73.6, 85.7, 98.1, 124.8, 129.3, 142.1, 151.1; EI m/z (%): 344 (10%), 69 (100%).
(±)-2-(8-Quinolinesulfonyl)-3,5-O-isopropylidene-1,4-xylitan (5g): Product obtained as a white solid in 32% yield. m.p. 129–131 °C; Rf = 0.24 (ethyl acetate/hexanes 4:6); IR (cm−1): 2997, 2934, 2904, 1561, 1499, 1455, 1362, 1278, 1176, 1088, 910, 840, 797, 743, 668; 1H-NMR (300 MHz, CDCl3): δ 1.34 (s, 3H), 1.36 (s, 3H), 3.84–4.04 (m, 4H), 4.28 (d, 1H, J = 3 Hz), 4.46 (s, 1H), 5.52 (s, 1H), 7.54–7.67 (m, 2H), 8.13 (d, 1H, J = 4, 5 Hz), 8.27 (d, 1H, J = 6 Hz), 8.50 (d, 1H, J = 6 Hz), 9.08 (s, 1H); 13C-NMR (75 MHz, CDCl3): δ 19.7, 29.1, 60.9, 72.4, 72.6, 74.3, 86.6, 98.2, 123.0, 125.9, 129.5, 133.7, 134.7, 135.5, 137.1, 144.2, 152.3; EI m/z (%): 210 (50%), 192 (20%), 129 (100%), 101 (20%), 81 (20%), 59 (20%).
2.5. Synthesis of (±)-2-Benzyl-3,5-O-isopropylidene-1,4-xylitan (6)
Xylitan acetonide (±)-2 (5 mmol) dissolved in anhydrous THF (15 mL) was added to sodium hydride (6 mmol) at 0 °C under an inert atmosphere of nitrogen. The mixture was stirred for 10 min and benzyl bromide (6 mmol) was added slowly to the mixture, allowed to warm to room temperature and left to stir for a further 24 h. Distilled water (15 mL) was added to quench the reaction and the organic product was extracted from the aqueous phase with ethyl acetate (2 × 20 mL) in a separatory funnel. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo and the crude product was purified by flash chromatography to afford a pale yellow liquid in 15% yield. Rf = 0.31 (ethyl acetate/hexane 2:8); 1H-NMR (300 MHz, CDCl3): δ 1.37 (s, 3H), 1.44 (s, 3H), 3.86–4.33 (m, 7H), 4.57 (s, 2H), 7.33 (s, 5H); 13C-NMR (75 MHz, CDCl3): δ 19.4, 28.8, 60.8, 71.7, 72.1, 72.4, 73.5, 84.2, 97.5, 127.7, 127.1, 128.6; EI m/z (%): 264 (10%), 249 (60%), 157 (30%), 91 (100%), 77 (10%).
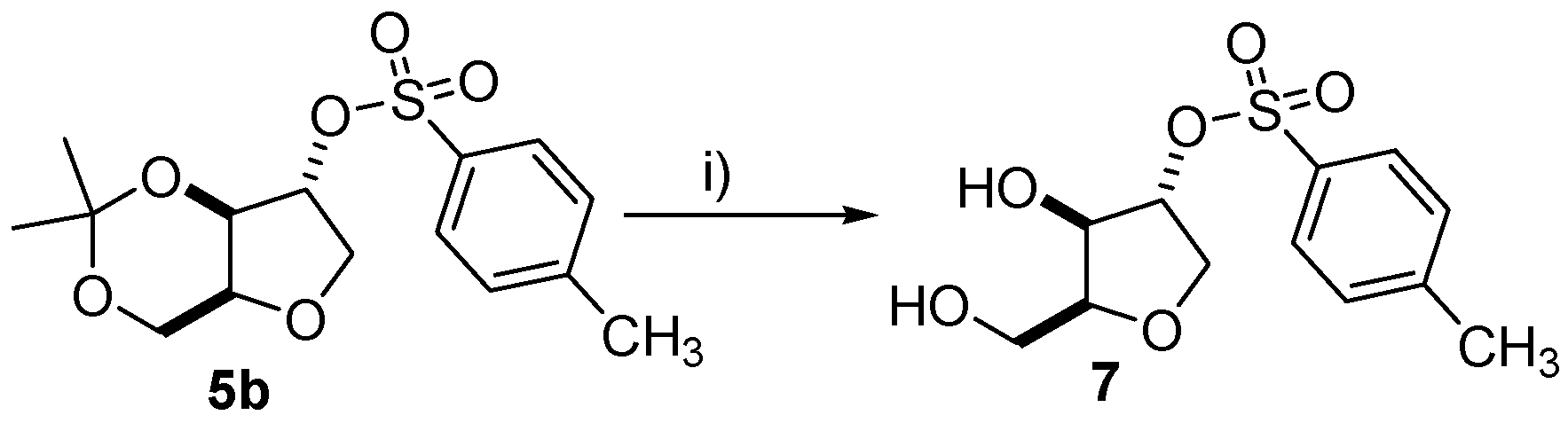
2.6. Synthesis of (±)-2-(4-Methylbenzenosulfonyl)-1,4-xylitan (7)
To a round bottom flask equipped with stir bar was added (±)-5b (0.3 mmol), CH2Cl2 (5 mL) and TFA (0.9 mL). The reaction was stirred for 1 h at room temperature and after this time, the solvent was removed under reduced pressure and the resultant liquid residue was transferred directly to a column and purified by flash chromatography. Upon purification, compound (±)-7 was obtained as a transparent liquid in 15% yield. Rf = 0.55 (ethyl acetate/hexane 3:7); 1H-NMR (300 MHz, CDCl3): δ 2.46 (s, 3H), 3.83–4.23 (m, 5H), 4.41 (s, 1H), 4.81 (s, 1H), 7.36 (d, 2H, J = 9 Hz), 7.79 (d, 2H, J = 9 Hz); 13C-NMR (75 MHz, CDCl3): δ 21.4, 61.2, 70.6, 78.6, 85.1, 109.7, 127.6, 129.8, 132.8, 145.1; EI m/z (%): 91 (100%), 81 (80%), 68 (80%).
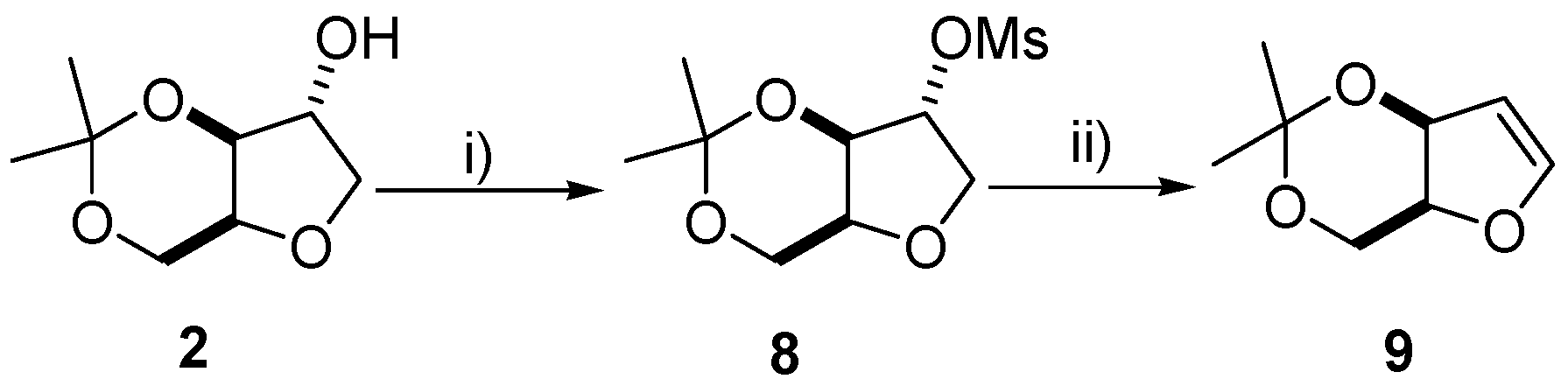
2.7. Synthesis of (±)-3,5-Isopropylidene-1,4-anidro-2-deoxy-pent-1-enitol (9)
Xylitan-acetonide (±)-2 (2.4 g, 13.9 mmol), anhydrous CH2Cl2 (20 mL) and triethylamine (5.8 mL, 41.7 mmol), were stirred in a round bottom flask under an atmosphere of nitrogen at −10 °C. Next, methanesulfonyl chloride (2.2 mL, 27.7 mmol), was added dropwise by addition funnel and the mixture was allowed to stir at room temperature for 2 h. The reaction was quenched by the addition of distilled water (20 mL) and then taken up into a separatory funnel. The organic layer was washed with HCl (1 M, 10 mL), saturated solution of Na2CO3 (20 mL) and finally the organic layer was evaporated under reduced pressure to afford (±)-8 as a light brown solid in 92% yield. An analytical sample of (±)-8 was obtained by recrystallization with EtOAc/Hex. White solid, m.p. 93–94 °C. Rf = 0.75 (ethyl acetate/hexane 3:7); IR (cm−1): 2999, 2934, 1349, 1179, 1095, 1042; 1H-NMR (400 MHz, CDCl3): δ 1.35 (s, 3H), 1.43 (s, 3H), 3.05 (s, 3H), 3.88–4.09 (m, 4H), 4.35 (dd, 1H, J = 12 Hz, J = 8 Hz), 4.48 (s, 1H), 5.03 (d, 1H, J = 4 Hz); 13C-NMR (100 MHz, CDCl3): δ 19.3, 28.5, 38.5, 60.3, 71.8, 72.1, 73.5, 83.7, 97.8; EI m/z (%): 81 (90%), 69 (100%), 59 (60%), 57 (40%). Compound (±)-8 (4 mmol) was stirred in 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (10 mL) at 150 °C for 45 min then cooled to room temperature and extracted with water (30 mL) and ethyl acetate (40 mL) in a seperatory funnel. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo and the crude product was purified by flash chromatography to provide a viscous pale yellow liquid in 56% yield Rf = 0.36 (Hexane/EtOAc, 5:1) after visualization by vanillin; 1H-NMR (250 MHz, CDCl3): δ 6.67 (d, J = 2.8 Hz, 1H), 5.17 (t, J = 2.5 Hz, 1H), 4.93 (dd, J = 2.8, 6.5 Hz, 1H), 4.37 (dd, J = 6.0, 12.5 Hz, 1H), 4.06 (dd, J = 6.0, 12.0 Hz, 1H), 3.87 (dd, J = 6.5, 12.0 Hz, 1H), 1.41 (s, 3H), 1.38 (s, 3H)); EI m/z (%) = 81 (100%), 68 (35%).
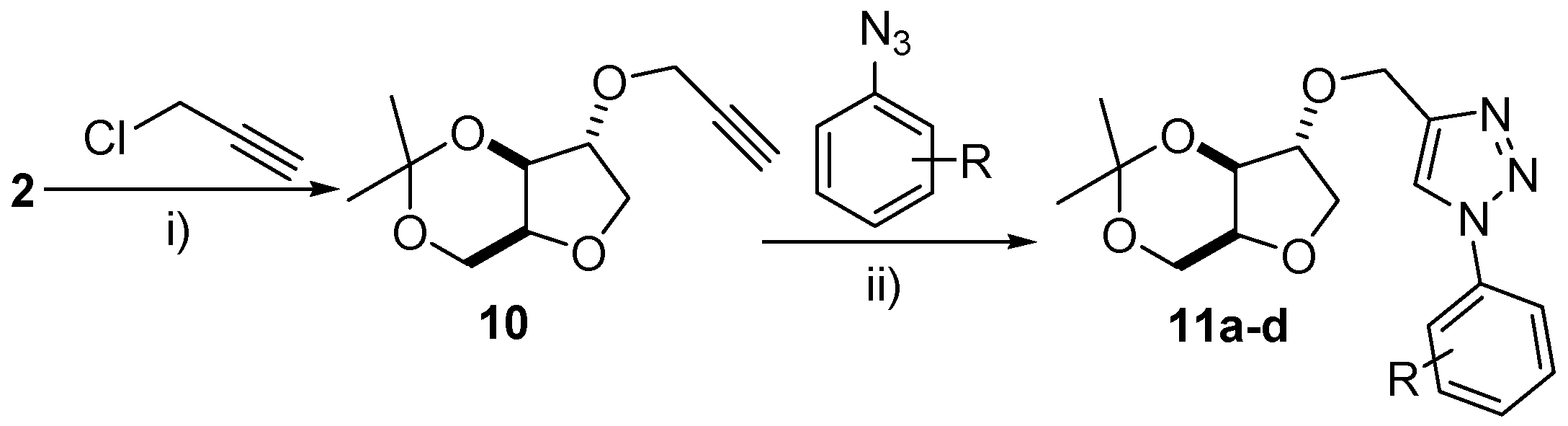
2.8. Synthesis of (±)-2-Propargyl-3,5-O-isopropylidene-1,4-xylitan (10)
To a round bottom flask equipped with stir bar and 4 Å molecular sieves (1 g), was added xylitan acetonide (±)-2 (3.2 mmol), sodium tert-butoxide (6.3 mmol) and acetonitrile (35 mL). The solution was cooled to 0 °C and a solution of propargyl chloride (3.2 mmol in 7 mL of CH3CN) was added dropwise by addition funnel. Upon addition of propargyl chloride, the solution was allowed to warm to room temperature and stirred for a further 12 h. Next, the solution was filtered to remove the molecular sieves and the filtrate was taken up into a separatory funnel containing distilled water (20 mL). The product was extracted from the aqueous phase with ethyl acetate (3× 20 mL) and the organic phase extractions were combined, washed with brine, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography to furnish a (±)-10 as a transparent liquid in 86% yield. Rf = 0.36 (ethyl acetate/hexane 1:9); 1H-NMR (300 MHz, CDCl3): δ 1.34 (s, 3H), 1.42 (s, 3H), 2.45 (s, 1H), 3.80–4.31 (m, 8H); 13C-NMR (75 MHz, CDCl3): δ 19.3, 28.7, 56.9, 60.6, 71.8, 72.3, 73.2, 75.1, 79.3, 83.8, 97.6; EI m/z (%): 197 (30%), 101 (65%), 82 (100%).
2.9. Synthesis of Triazoles 11a–d
To a one necked round bottom flask equipped with stir bar was added freshly prepared arylazide (2.7 mmol), a solution of CuSO4·5H2O (0.44 mmol in 3 mL of H2O) and a solution of sodium ascorbate (0.56 mmol in 3 mL of H2O). The mixture was stirred vigorously and a solution of (±)-10 (1.4 mmol in 6 mL of CH2Cl2) was added dropwise and allowed to stir at room temperature for 24 h. The reaction mixture was transferred to a separatory funnel and extracted with CH2Cl2 (2 × 25 mL). The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated in vacuo and the crude product was purified by flash chromatography.
(±)-2-[(1’-(4-Methoxyphenyl)-2-O-metil-1H-1,2,3-triazol]-3,5-O-isopropylidene-1,4-xylitan (11a): Product obtained as a light brown solid in 85% yield. m.p. 106–108 °C; Rf = 0.29 (ethyl acetate/hexane 2:8); IR (cm−1): 2936, 1494, 1201, 1102, 1031, 969, 836, 746; 1H-NMR (300 MHz, CDCl3): δ 1.35 (s, 3H), 1.43 (s, 3H), 3.84–4.37 (m, 7H), 4.74 (s, 2H), 6.99 (d, 2H, J = 9 Hz), 7.58 (d, 2H, J = 9 Hz), 7.88 (s, 1H); 13C-NMR (75 MHz, CDCl3): δ 19.4, 28.8, 55.7, 60.7, 63.1, 72.0, 72.3, 73.2, 84.4, 97.6, 114.8, 121.1, 122.3, 130.4, 144.1 159.9; EI m/z (%): 361 (1%), 167 (25%), 149 (100%), 57 (60%).
(±)-2-[(1’-(4-Chlorophenyl)-2-O-metil-1H-1,2,3-triazol]-3,5-O-isopropylidene-1,4-xylitan (11b): Product obtained as a white solid in 41% yield. m.p. 134–136 °C; Rf = 0.52 (ethyl acetate/hexane 1:9); IR (cm−1): 3007, 2909, 1520, 1449, 1272, 1209, 1111, 1040, 978, 827; 1H-NMR (300 MHz, CDCl3): δ 1.37 (s, 3H), 1.45 (s, 3H), 3.86–4.38 (m, 7H), 4.77 (s, 2H), 7.48–7.52 (m, 2H), 7.66–7.70 (m, 2H), 7.95 (s, 1H); 13C-NMR (75 MHz, CDCl3): δ 19.5, 28.7, 60.7, 63.2; 72.0, 72.5, 73.3, 83.9, 84.7, 97.5, 120.7, 121.7, 129.1, 134.7, 135.4; EI m/z (%): 167 (30%), 149 (80%), 71 (60%), 57 (100%).
(±)-2-[(1’-(2-Acylphenyl)-2-O-metil-1H-1,2,3-triazol]-3,5-O-isopropylidene-1,4-xylitan (11c): Product obtained as a white solid in 29% yield. m.p. 135–136 °C; Rf = 0.29 (ethyl acetate/hexane 2:8); IR (cm−1): 2900, 1502, 1440, 960, 809, 756; 1H-NMR (400 MHz, CDCl3): δ 1.37 (s, 3H), 1.45 (s, 3H), 2.65 (s, 3H), 3.75–4.39 (m, 7H), 4.78 (s, 2H), 7.86 (d, 2H, J = 8 Hz), 8.05 (s, 1H), 8.12 (d, 2H, J = 8 Hz); 13C-NMR (100 MHz, CDCl3): δ 19.9, 27.1, 29.1, 61.0, 63.5, 72.3, 72.8, 73.7, 85.0, 98.0, 120.5, 121.0, 130.6, 137.4, 140.4, 146.3, 196.1; EI m/z (%): 207 (50%), 73 (100%).
(±)-2-[(1’-(3,4-Dimethylphenyl)-2-O-metil-1H-1,2,3-triazol]-3,5-O-isopropylidene-1,4-xylitan (11d): Product obtained as a white solid in 34% yield. m.p. 110–111 °C; Rf = 0.13 (ethyl acetate/hexane 2:8); IR (cm−1): 2936, 1601, 1440, 1378, 1201, 1066, 960, 827, 756; 1H-NMR (400 MHz, DMSO): δ 1.23 (s, 3H), 1.39 (s, 3H), 2.28 (s, 3H), 2.31 (s, 3H), 3.68–4.07 (m, 6H), 4.39 (m, 1H), 4.68 (s, 2H); 7.33–7.41 (m, 2H), 7.59 (d, 1H, J = 4 Hz), 8.74 (s, 1H); 13C-NMR (100 MHz, DMSO): δ 19.1, 19.6, 28.8, 60.1, 62.1, 71.4, 71.9, 72.7, 83.7, 96.9, 117.5, 121.1, 122.3, 130.7, 134.7, 137.2, 138.3, 144.8; EI m/z (%): 202 (40%), 158 (100%), 145 (51%), 105 (70%).
2.10. Anti-Trypanosoma cruzi Activity Assay
The in vitro anti-
T. cruzi activity was evaluated on L929 cells (mouse fibroblasts) infected with Tulahuen strain of the parasite expressing the
Escherichia coli β-galactosidase as reporter gene according to the method described previously [
16]. Briefly, for the bioassay, 4000 L929 cells were added to each well of a 96-well microtiter plate. After an overnight incubation, 40,000 trypomastigotes were added to the cells and incubated for 2 h. Then the medium containing extracellular parasites was replaced with 200 μL of fresh medium and the plate was incubated for an additional 48 h to establish the infection. For IC
50 determination, the cells were exposed to each synthesized compound at serial decreasing dilutions and the plate was incubated for 96 h. After this period, 50 μL of 500 μM chlorophenol red beta-
d-galactopyranoside (CPRG) in 0.5% Nonidet P40 was added to each well, and the plate was incubated for 16–20 h, after which the absorbance at 570 nm was measured. Controls with uninfected cells, untreated infected cells, infected cells treated with benznidazole at 3.8 μM (positive control) or DMSO 1% were used. The results were expressed as the percentage of
T. cruzi growth inhibition in compound-tested cells as compared to the infected cells and untreated cells. The IC
50 values were calculated by linear interpolation. Quadruplicates were run in the same plate, and the experiments were repeated at least once.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}