
Agarose and Its Derivatives as Supports for Enzyme Immobilization




Abstract
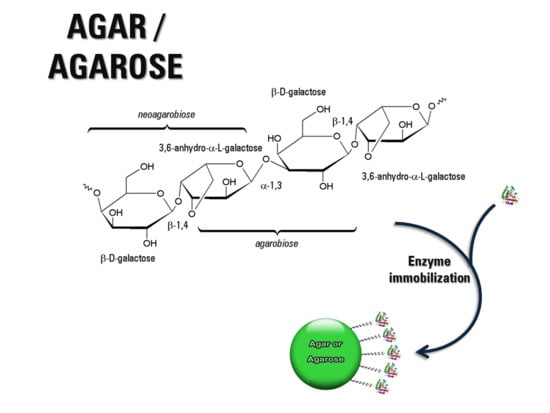
:
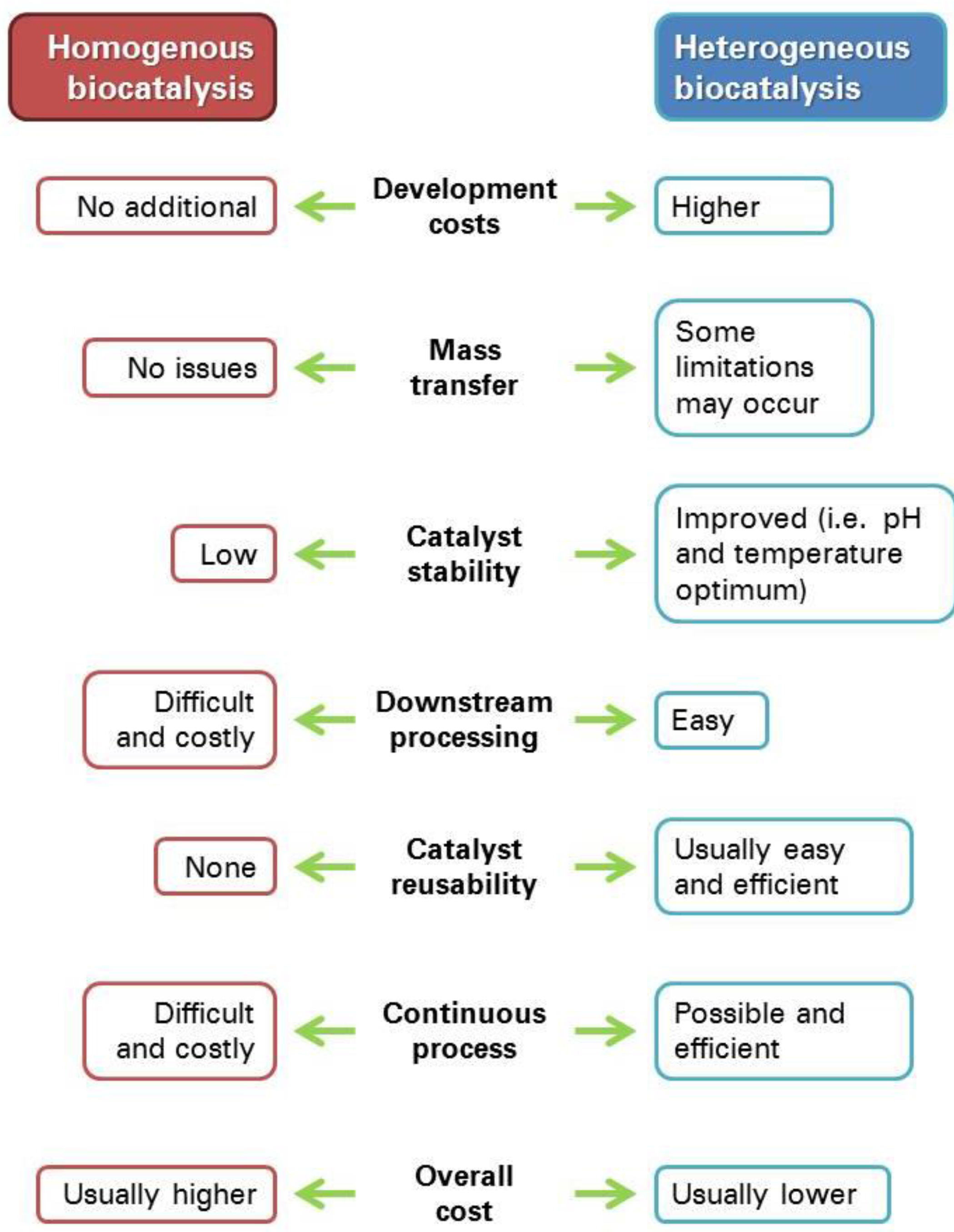
1. Enzyme Immobilization: An Overview
1.1. Choice of the Support
- Firstly, the costs of the unprocessed materials (both support and reagents needed for its possible functionalization) should be minimized.
- Supports and reagents should be harmless from both health and environmental perspective.
- Chemical and microbial inertness is usually a desirable feature.
- Mechanical properties should be compatible with practical applications.
- High surface area, large porosity and adequate particle size are basic requirements to be checked when selecting a matrix for an immobilization process.
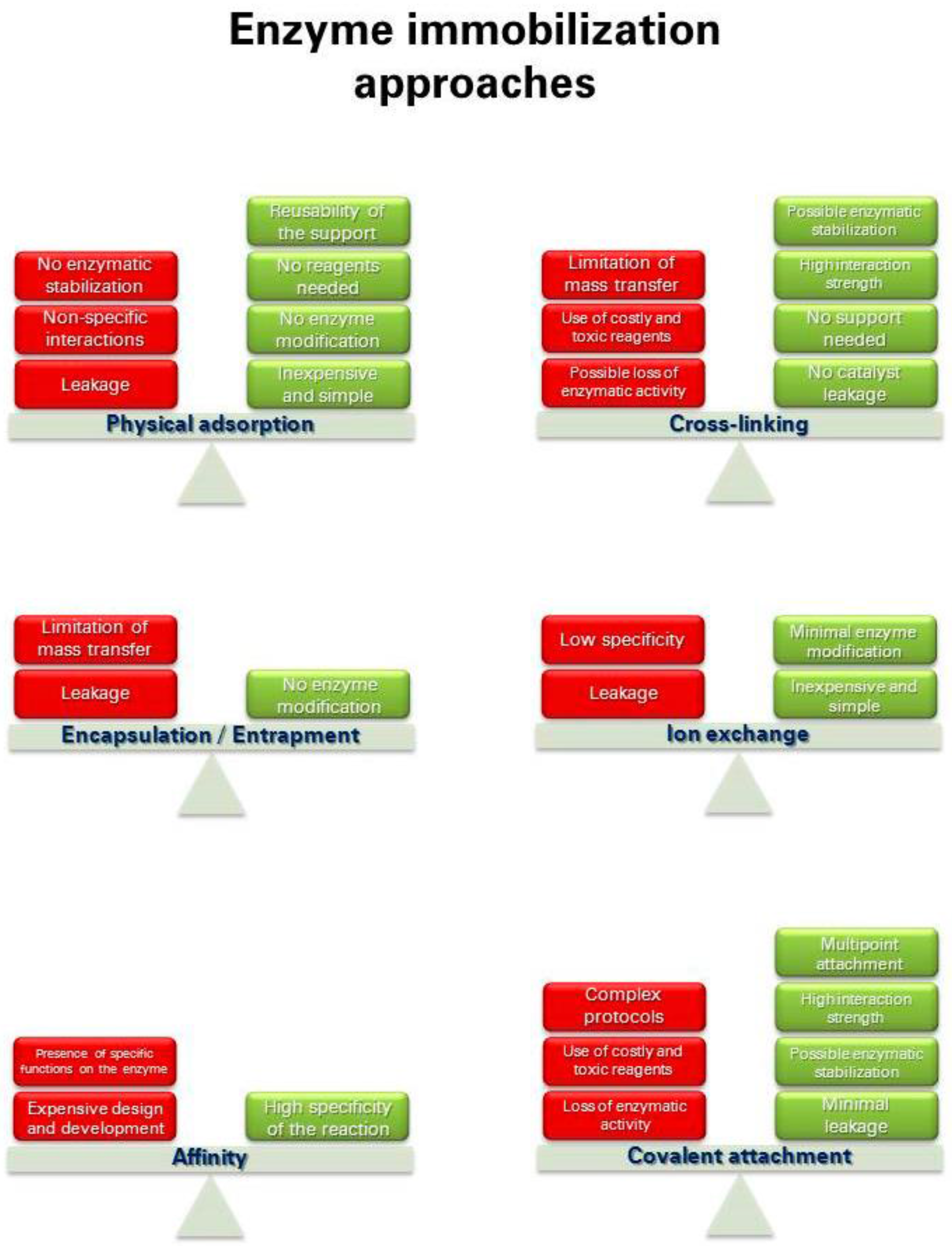
1.2. Immobilization Techniques
2. Agar-Agar and Agarose: Occurrence, Structures, Properties
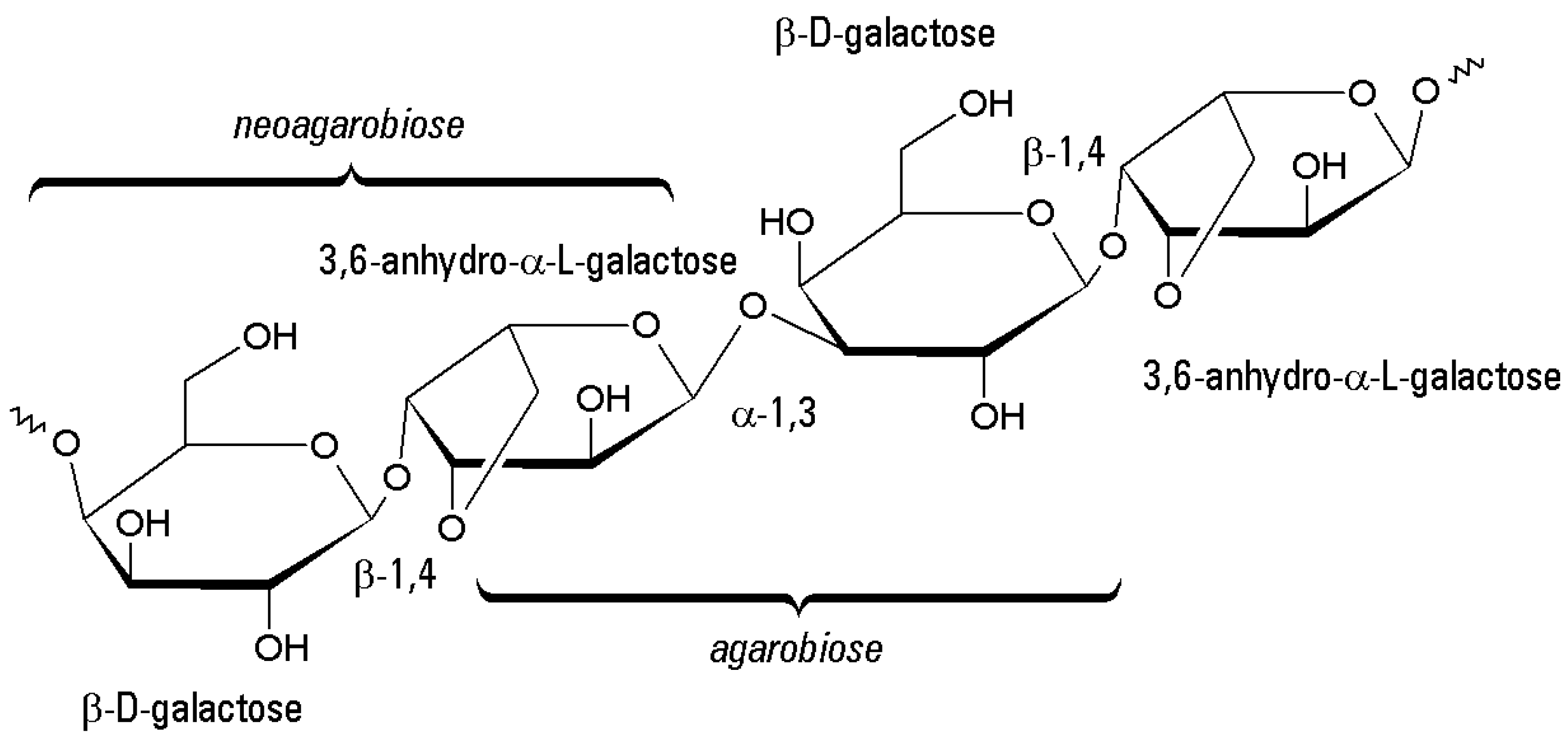
2.1. Chemical Structure
2.2. Functional Properties
3. Agarose-Based Supports: To Cross-Link or Not to Cross-Link?
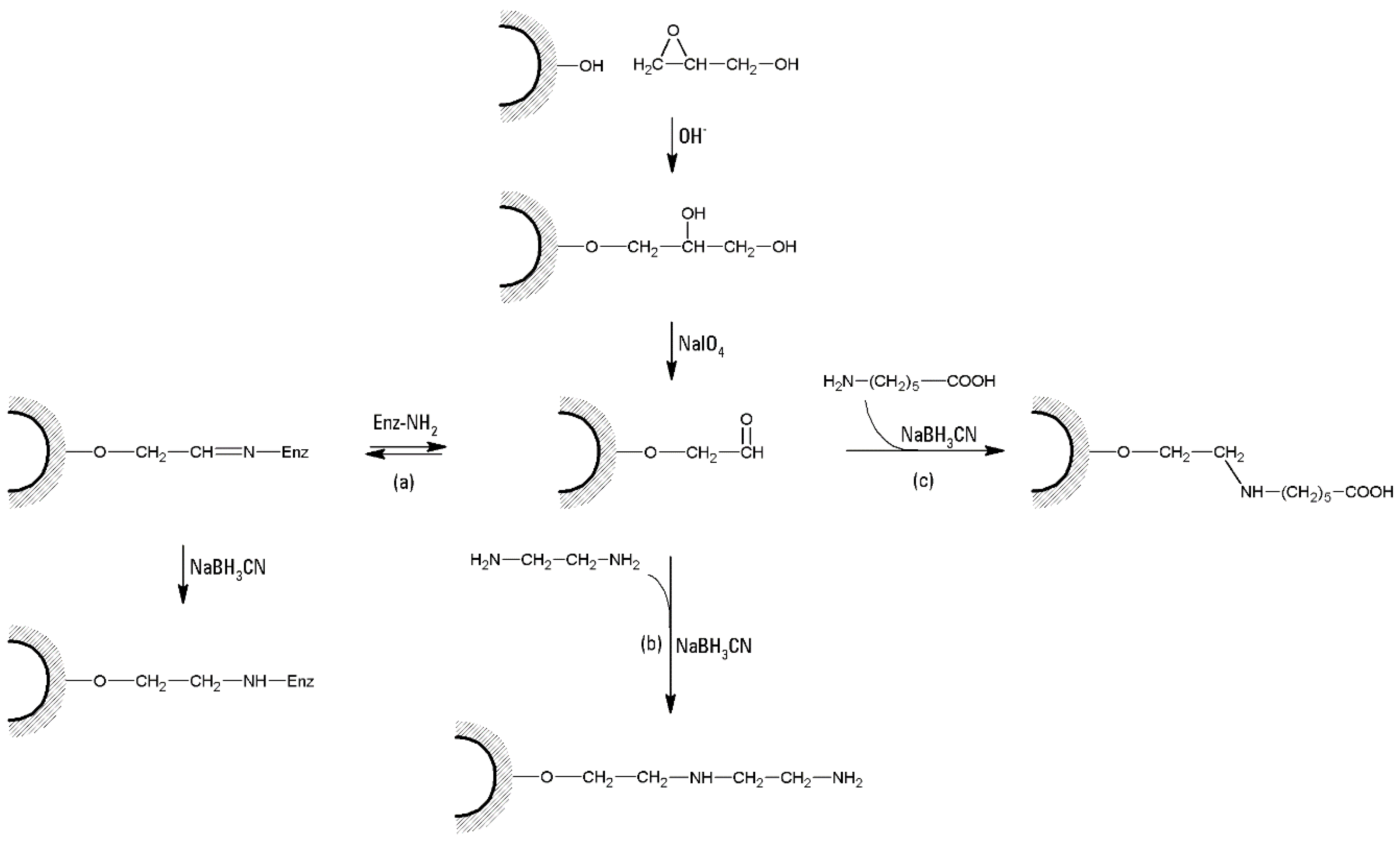
- (a)
- A typical cross-linking procedure (Figure 4) involves the suspension of 1 L of swollen agar/agarose gel with 1 L NaOH 1 M, containing 100 mL epichlorohydrin and 5 g NaBH4. After 2 h of gentle stirring at 60 °C, the suspension is washed with hot water to neutrality, and re-suspended in 500 mL NaOH 2 M, containing 2.5 g NaBH4. The suspension is then treated at 120 °C for 1 h. Several washings are then performed, including 1.5 L of hot NaOH 1 M and 0.5% NaBH4, and 1.5 L of cold NaOH 1 M and 0.5% NaBH4. Then the suspension is cooled with ice and neutralized until pH 4 with CH3COOH [39]. The suspension can be stored in 0.02% sodium azide solution.
- (b)
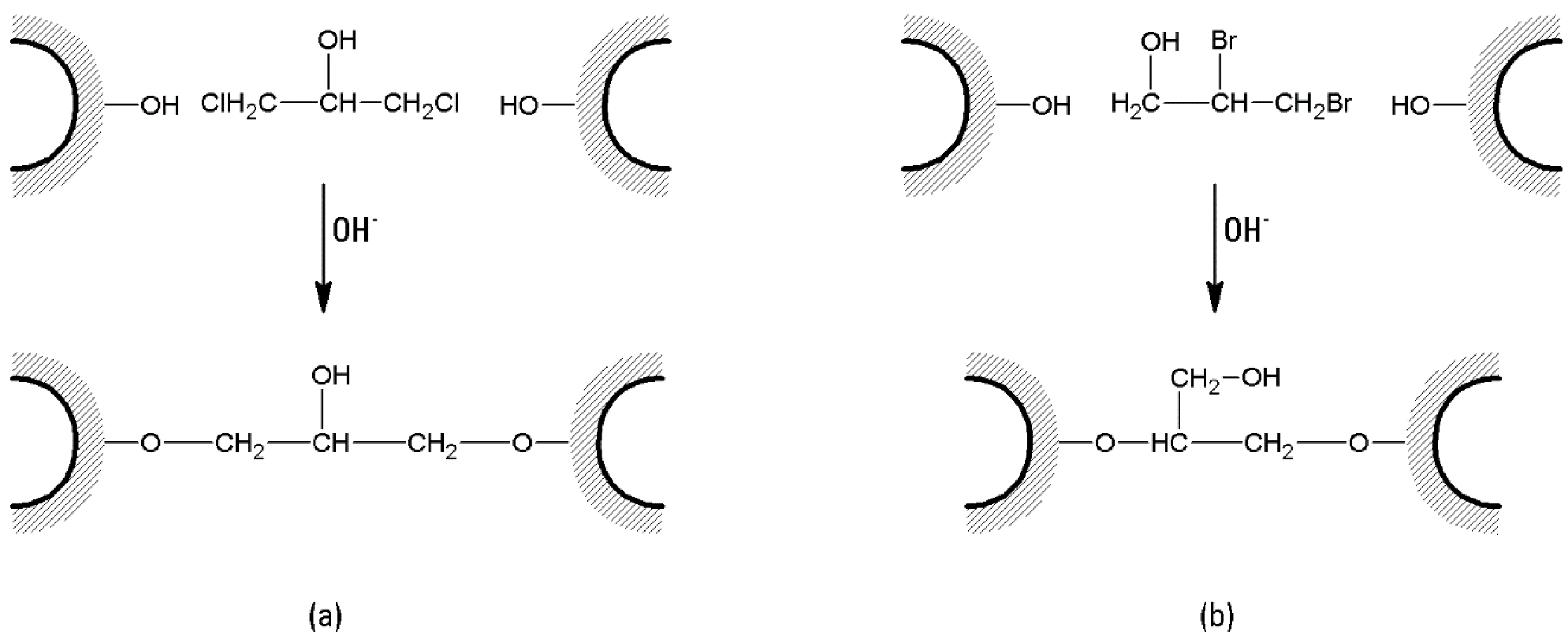
- Beads prepared from agarose powder (6% w/w) are cross-linked with 1,3-dichloro-2-propanol (DCP) under strong alkaline conditions. A net weight of 1.5 g of beads is added to 10 mL of a solution of 0.3 M NaOH in distilled water, and the mixture treated with 0.1 mL of DCP. The reaction is allowed to proceed for 1 h, with continuous agitation at ~50 °C. After that, the beads are washed with water until the effluent becomes neutral [106].
- (c)
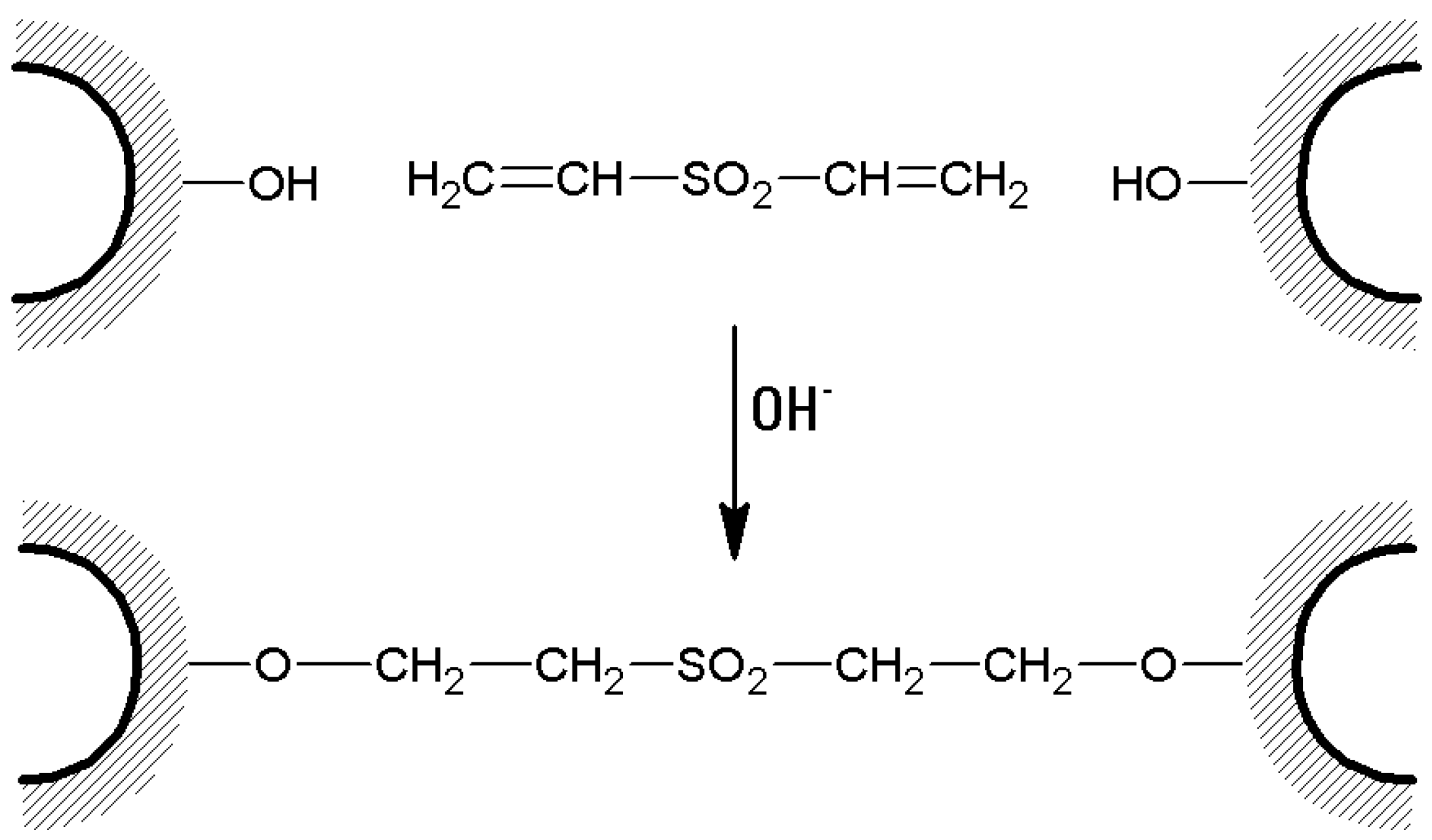
- Wet agarose gel (typically 10 or 20 g) is soaked with 10 to 20 mL of 0.5 M sodium carbonate buffer of pH 11. Then, divinylsulfone (DVS) is added in an amount expressed as % (v/w) of the wet gel, depending on the desired cross-linking degree, and the reaction is allowed to proceed for 2 h at room temperature (Figure 6). In fact, DVS influences gel stiffness and therefore water (or buffer) flow through a chromatographic column. The flow rate linearly increases as the cross-linking degree increases until about 3% cross-linking [103], which is the optimal choice to obtain rigid, stable, and fast-flow chromatographic media.
- (d)
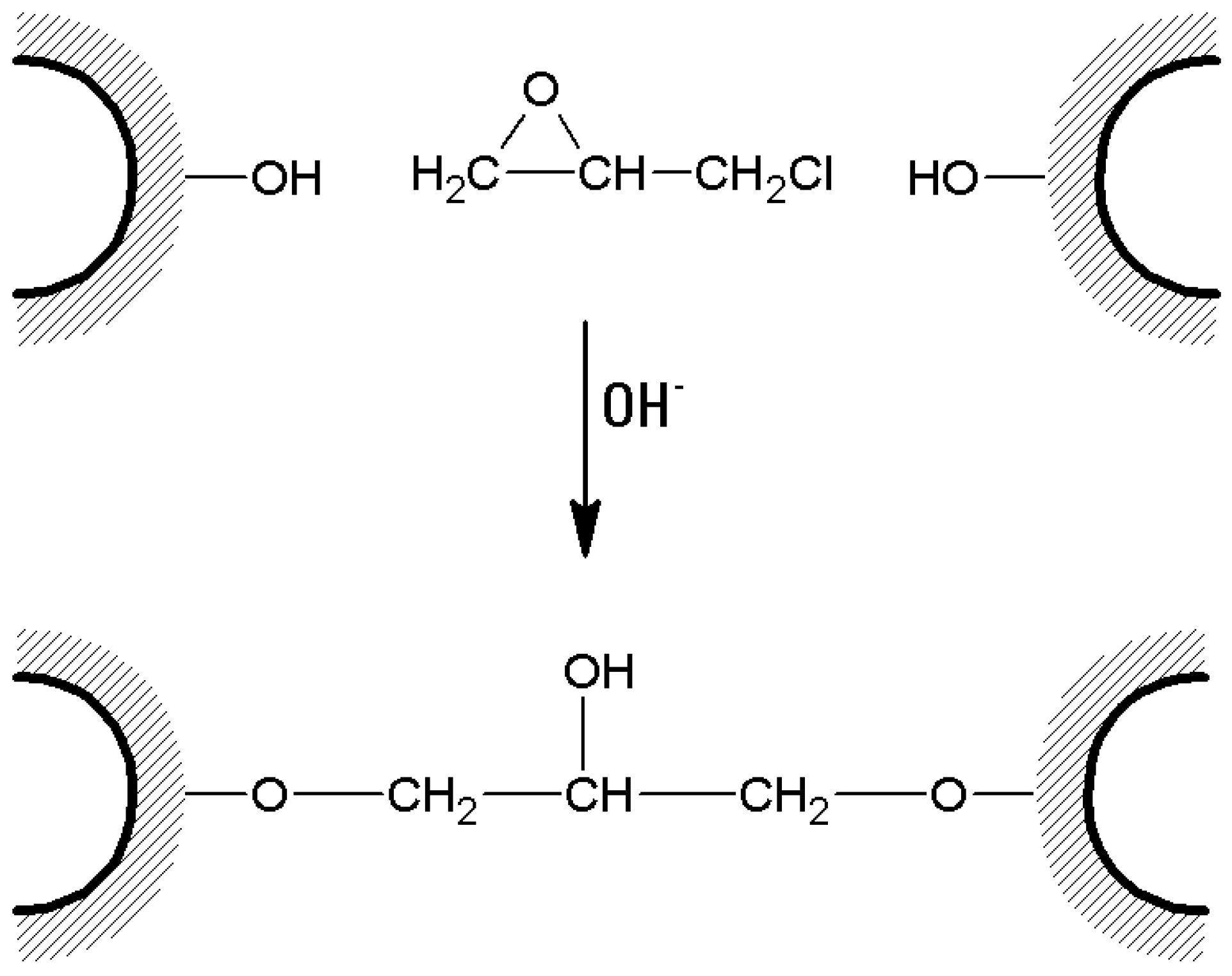
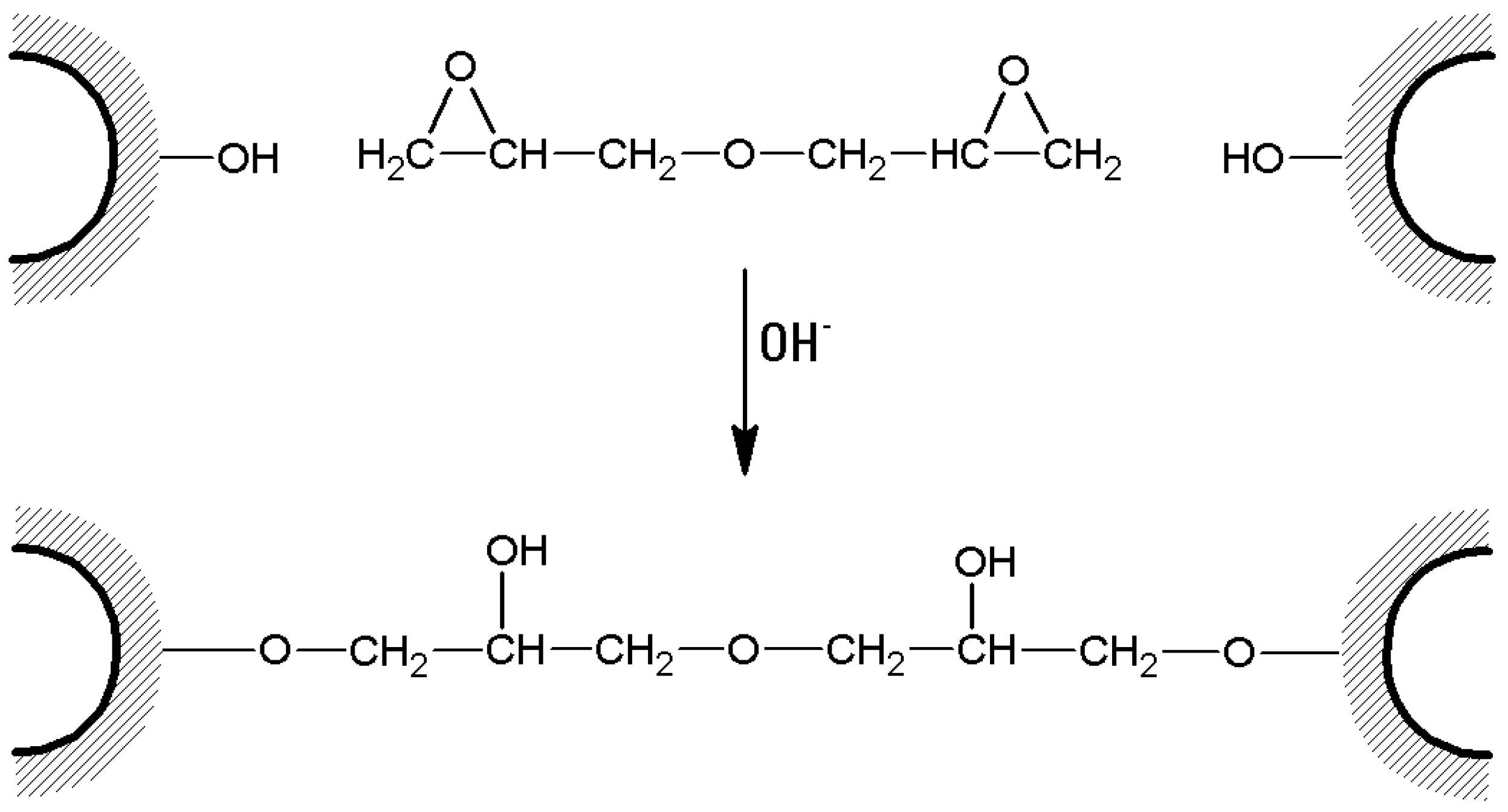
- In the case of bis-oxiranes, 1 g of dried agarose reacts for 8 h at 25 °C with 1 mL of a proper cross-linker (i.e., diglycidyl ether) and 1 mL of NaOH 0.6 M (containing 0.2% w/v NaBH4) [105], as shown in Figure 7). Deactivation of unreacted oxirane groups can be performed by treatment of the gel with 2 M glycine or ethanolamine at pH > 8.5, 23 °C for 24 h.
4. Chemical Functionalization of Agarose-Based Supports: Chemistries and Protocols
5. Outstanding Examples of Enzymes Immobilized on to Agarose-Based Supports
5.1. Systems for Enzyme Co-Immobilization
5.2. Studying the Immobilized Enzyme
6. Problems and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Eş, I.; Vieira, J.D.G.; Amaral, A.C. Principles, techniques, and applications of biocatalyst immobilization for industrial application. Appl. Microbiol. Biotechnol. 2015, 99, 2065–2082. [Google Scholar] [CrossRef] [PubMed]
- Zucca, P.; Littarru, M.; Rescigno, A.; Sanjust, E. Cofactor Recycling for Selective Enzymatic Biotransformation of Cinnamaldehyde to Cinnamyl Alcohol. Biosci. Biotechnol. Biochem. 2009, 73, 1224–1226. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Kostrov, X. Immobilization of enzymes on porous silicas-benefits and challenges. Chem. Soc. Rev. 2013, 42, 6277–6289. [Google Scholar] [CrossRef] [PubMed]
- Faber, K. Biotransformations in Organic Chemistry: A Textbook; Springer: Berlin, Germany, 1995. [Google Scholar]
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszyńska, D. Immobilization as a Strategy for Improving Enzyme Properties-Application to Oxidoreductases. Molecules 2014, 19, 8995–9018. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.; Pedro, A.; Santos, F.; Martins, L.; Maia, C.; Queiroz, J.; Passarinha, L. Trends in Protein-Based Biosensor Assemblies for Drug Screening and Pharmaceutical Kinetic Studies. Molecules 2014, 19, 12461–12485. [Google Scholar] [CrossRef] [PubMed]
- Zucca, P.; Rescigno, A.; Pintus, M.; Rinaldi, A.C.; Sanjust, E. Degradation of textile dyes using immobilized lignin peroxidase-like metalloporphines under mild experimental conditions. Chem. Cent. J. 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Zucca, P.; Rescigno, A.; Rinaldi, A.C.; Sanjust, E. Biomimetic metalloporphines and metalloporphyrins as potential tools for delignification: Molecular mechanisms and application perspectives. J. Mol. Catal. A Chem. 2014, 388, 2–34. [Google Scholar] [CrossRef]
- Zucca, P.; Rescigno, A.; Olianas, A.; MacCioni, S.; Sollai, F.A.; Sanjust, E. Induction, purification, and characterization of a laccase isozyme from Pleurotus sajor-caju and the potential in decolorization of textile dyes. J. Mol. Catal. B Enzym. 2011, 68, 216–222. [Google Scholar] [CrossRef]
- Laveille, P.; Falcimaigne, A.; Chamouleau, F.; Renard, G.; Drone, J.; Fajula, F.; Pulvin, S.; Thomas, D.; Bailly, C.; Galarneau, A. Hemoglobin immobilized on mesoporous silica as effective material for the removal of polycyclic aromatic hydrocarbons pollutants from water. New J. Chem. 2010, 34, 2153–2165. [Google Scholar] [CrossRef]
- Riva, S. Laccases: Blue enzymes for green chemistry. Trends Biotechnol. 2006, 24, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Jung, D. Biocatalysis with enzymes immobilized on mesoporous hosts: The status quo and future trends. J. Mater. Chem. 2010, 20, 844–857. [Google Scholar] [CrossRef]
- Magner, E. Immobilisation of enzymes on mesoporous silicate materials. Chem. Soc. Rev. 2013, 42, 6213–6222. [Google Scholar] [CrossRef] [PubMed]
- Salis, A.; Casula, M.F.; Bhattacharyya, M.S.; Pinna, M.; Solinas, V.; Monduzzi, M. Physical and chemical lipase adsorption on SBA-15: Effect of different interactions on enzyme loading and catalytic performance. ChemCatChem 2010, 2, 322–329. [Google Scholar] [CrossRef]
- Salis, A.; Pinna, M.; Monduzzi, M.; Solinas, V. Comparison among immobilised lipases on macroporous polypropylene toward biodiesel synthesis. J. Mol. Catal. B Enzym. 2008, 54, 19–26. [Google Scholar] [CrossRef]
- Cesarini, S.; Pastor, F.I.J.; Diaz, P. Improvement of P. aeruginosa 42A2 lipase preparations for FAMEs production, both in immobilized and soluble form. J. Mol. Catal. B Enzym. 2014, 99, 1–7. [Google Scholar] [CrossRef]
- Liu, Y.; Hua, X. Production of biodiesel using a nanoscaled immobilized lipase as the catalyst. Catal. Lett. 2014, 144, 248–251. [Google Scholar] [CrossRef]
- Vasic-Racki, D. History of Industrial Biotransformations—Dreams and Realities. In Industrial Biotransformations; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 1–36. [Google Scholar]
- Zhou, Z.; Hartmann, M. Progress in enzyme immobilization in ordered mesoporous materials and related applications. Chem. Soc. Rev. 2013, 42, 3894–3912. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lafuente, R. Editorial: Special Issue—Enzyme Immobilization. Molecules 2014, 19, 20671–20674. [Google Scholar] [CrossRef] [PubMed]
- Zucca, P.; Sanjust, E. Inorganic materials as supports for covalent enzyme immobilization: Methods and mechanisms. Molecules 2014, 19, 14139–14194. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M. Ordered mesoporous materials for bioadsorption and biocatalysis. Chem. Mater. 2005, 17, 4577–4593. [Google Scholar] [CrossRef]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Immobilization strategies to develop enzymatic biosensors. Biotechnol. Adv. 2012, 30, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Dicosimo, R.; McAuliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–6474. [Google Scholar] [CrossRef] [PubMed]
- Liese, A.; Hilterhaus, L. Evaluation of immobilized enzymes for industrial applications. Chem. Soc. Rev. 2013, 42, 6236–6249. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Asgher, M.; Parra-Saldivar, R.; Hu, H.; Wang, W.; Zhang, X.; Iqbal, H.M.N. Immobilized ligninolytic enzymes: An innovative and environmental responsive technology to tackle dye-based industrial pollutants—A review. Sci. Total Environ. 2017, 576, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lorente, G.; Godoy, C.A.; Mendes, A.A.; Lopez-Gallego, F.; Grazu, V.; de las Rivas, B.; Palomo, J.M.; Hermoso, J.; Fernandez-Lafuente, R.; Guisan, J.M. Solid-phase chemical amination of a lipase from Bacillus thermocatenulatus to improve its stabilization via covalent immobilization on highly activated glyoxyl-agarose. Biomacromolecules 2008, 9, 2553–2561. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.N.; Balkus, K.J. Perspective of recent progress in immobilization of enzymes. ACS Catal. 2011, 1, 956–968. [Google Scholar] [CrossRef]
- Minteer, S.D. Enzyme Stabilization and Immobilization: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2011; p. 679. [Google Scholar]
- Garcia-Galan, C.; Berenguer-Murcia, A.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of different enzyme immobilization strategies to improve enzyme performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R. Stabilization of multimeric enzymes: Strategies to prevent subunit dissociation. Enzyme Microb. Technol. 2009, 45, 405–418. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Strategies for the one-step immobilization-purification of enzymes as industrial biocatalysts. Biotechnol. Adv. 2015, 33, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Grazú, V.; Pessela, B.C.C.; Montes, T.; Palomo, J.M.; Torres, R.; López-Gallego, F.; Fernández-Lafuente, R.; Guisán, J.M. Advances in the design of new epoxy supports for enzyme immobilization-stabilization. Biochem. Soc. Trans. 2007, 35, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Porath, J.; Axén, R. Immobilization of enzymes to agar, agarose, and sephadex support. Methods Enzymol. 1976, 44, 19–45. [Google Scholar] [PubMed]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the Support Properties for Immobilization or Purification of Enzymes. ChemCatChem 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Epton, R.; Hibbert, B.L.; Thomas, T.H. Enzymes covalently bound to polyacrylic and polymethacrylic copolymers. Methods Enzymol. 1976, 44, 84–107. [Google Scholar] [PubMed]
- Mosbach, R.; Koch-Schmidt, A.C.; Mosbach, K. [5] Immobilization of enzymes to various acrylic copolymer. Methods Enzymol. 1976, 44, 53–65. [Google Scholar] [PubMed]
- Nan, C.; Zhang, Y.; Zhang, G.; Dong, C.; Shuang, S.; Choi, M.M.F. Activation of nylon net and its application to a biosensor for determination of glucose in human serum. Enzyme Microb. Technol. 2009, 44, 249–253. [Google Scholar] [CrossRef]
- Aranaz, I.; Mengíbar, M.; Harris, R.; Paños, I.; Miralles, B.; Acosta, N.; Galed, G.; Heras, Á. Functional characterization of chitin and chitosan. Curr. Chem. Biol. 2009, 3, 203–230. [Google Scholar] [CrossRef]
- Krajewska, B. Application of chitin- and chitosan-based materials for enzyme immobilizations: A review. Enzyme Microb. Technol. 2004, 35, 126–139. [Google Scholar] [CrossRef]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Liebert, T. Cellulose Solvents-Remarkable History, Bright Future; Oxford University Press: Oxford, UK, 2010; pp. 3–54. [Google Scholar]
- Aschenbrenner, E.; Bley, K.; Koynov, K.; Makowski, M.; Kappl, M.; Landfester, K.; Weiss, C.K. Using the polymeric ouzo effect for the preparation of polysaccharide-based nanoparticles. Langmuir 2013, 29, 8845–8855. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.M.; Griffin, E.G. Adsorption of invertase. J. Am. Chem. Soc. 1916, 38, 1109–1115. [Google Scholar] [CrossRef]
- Sheldon, R.A. Cross-linked enzyme aggregates (CLEA®s): Stable and recyclable biocatalysts. Biochem. Soc. Trans. 2007, 35, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Górecka, E.; Jastrzębska, M. Immobilization techniques and biopolymer carriers. Biotechnol. Food Sci. 2011, 75, 65–86. [Google Scholar]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Fernandez-Lafuente, R.; Guisan, J.M. Reversible enzyme immobilization via a very strong and nondistorting ionic adsorption on support-polyethylenimine composites. Biotechnol. Bioeng. 2000, 68, 98–105. [Google Scholar] [CrossRef]
- Rimola, A.; Costa, D.; Sodupe, M.; Lambert, J.-F.; Ugliengo, P. Silica surface features and their role in the adsorption of biomolecules: Computational modeling and experiments. Chem. Rev. 2013, 113, 4216–4313. [Google Scholar] [CrossRef] [PubMed]
- Bucur, B.; Danet, A.F.; Marty, J.L. Cholinesterase immobilisation on the surface of screen-printed electrodes based on concanavalin a affinity. Anal. Chim. Acta 2005, 530, 1–6. [Google Scholar] [CrossRef]
- Mattiasson, B. Affinity immobilization. Methods Enzymol. 1988, 137, 647–656. [Google Scholar] [PubMed]
- Sollai, F.; Noli, B.; Floris, G.; Sanjust, E. Irreversible affinity immobilization of lentil seedling amine oxidase with activity retention. Environ. Eng. Manag. J. 2007, 6, 31–35. [Google Scholar]
- Secundo, F. Conformational changes of enzymes upon immobilisation. Chem. Soc. Rev. 2013, 42, 6250–6261. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Cruz, J.; Rueda, N.; Dos Santos, J.C.S.; Torres, R.; Ortiz, C.; Villalonga, R.; Fernandez-Lafuente, R. Inactivation of immobilized trypsin under dissimilar conditions produces trypsin molecules with different structures. RSC Adv. 2016, 6, 27329–27334. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R.; Resell, C.M.; Rodriguez, V.; Guisan, J.M. Strategies for enzyme stabilization by intramolecular crosslinking with bifunctional reagents. Enzyme Microb. Technol. 1995, 17, 517–523. [Google Scholar] [CrossRef]
- Grazú, V.; Abian, O.; Mateo, C.; Batista-Viera, F.; Fernández-Lafuente, R.; Guisán, J.M. Stabilization of enzymes by multipoint immobilization of thiolated proteins on new epoxy-thiol supports. Biotechnol. Bioeng. 2005, 90, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Godoy, C.A.; Volpato, G.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Guisan, J.M. Immobilization-stabilization of the lipase from Thermomyces lanuginosus: Critical role of chemical amination. Process Biochem. 2009, 44, 963–968. [Google Scholar] [CrossRef]
- Fu, X.T.; Kim, S.M. Agarase: Review of major sources, categories, purification method, enzyme characteristics and applications. Mar. Drugs 2010, 8, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Araki, C. Acetylation of agar like substance of Gelidium amansii. J. Chem. Soc. Jpn. 1937, 58, 1338–1350. [Google Scholar]
- Armisén, R.; Galatas, F. Agar. In Handbook of Hydrocolloids, 2nd ed.; Woodhead Publishing Limited: Cambridge, UK; pp. 82–107.
- Armisen, R.; Galatas, F. Production, properties and uses of agar. Production and utilization of products from commercial seaweeds. FAO Fish. Tech. Pap 1987, 288, 1–57. [Google Scholar]
- Imeson, A. Agar. In Food Stabilisers, Thickeners and Gelling Agents; Wiley-Blackwell: New York, NY, USA, 2009; pp. 31–49. [Google Scholar]
- Hehemann, J.H.; Boraston, A.B.; Czjzek, M. A sweet new wave: Structures and mechanisms of enzymes that digest polysaccharides from marine algae. Curr. Opin. Struct. Biol. 2014, 28, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Koch, R. Die Atiologie der Tuberculose. Berl. Klin. Wochenschr. 1882, 19, 221–230. [Google Scholar]
- Armisén, R. Agar and agarose biotechnological applications. Hydrobiologia 1991, 221, 157–166. [Google Scholar] [CrossRef]
- Clark, A.H.; Ross-Murphy, S.B. Structural and mechanical properties of biopolymer gels. In Biopolymers; Springer: Berlin/Heidelberg, Germany, 1987; pp. 57–192. [Google Scholar]
- FAO (Food and Agriculture Organization of the United Nations). Training Manual on Gracilaria Culture and Seaweed Processing in China, Regional Seafarming Development and Demonstration Project. FAO Corporate Document Repository 1990. Available online: http://www.fao.org/docrep/field/003/AB730E/AB730E03.htm (accessed on 16 July 2016).
- McHugh, D.J. Worldwide distribution of commercial resources of seaweeds including Gelidium. Hydrobiologia 1991, 221, 19–29. [Google Scholar] [CrossRef]
- Araki, C. Structure of the agarose constituent of agar-agar. Bull. Chem. Soc. Jpn. 1956, 29, 543–544. [Google Scholar] [CrossRef]
- Ball, S.G.; Morell, M.K. From bacterial glycogen to starch: Understanding the biogenesis of the plant starch granule. Annu. Rev. Plant Biol. 2003, 54, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Venturi, F.; Andrich, G.; Quartacci, M.F.; Sanmartin, C.; Andrich, L.; Zinnai, A. A kinetic method to identify the optimum temperature for p-glucanase activity. S. Afr. J. Enol. Vitic. 2013, 34, 281–286. [Google Scholar] [CrossRef]
- Hashem, A.M.; Gamal, A.A.; Hassan, M.E.; Hassanein, N.M.; Esawy, M.A. Covalent immobilization of Enterococcus faecalis Esawy dextransucrase and dextran synthesis. Int. J. Biol. Macromol. 2016, 82, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Hamer, G.K.; Bhattacharjee, S.S.; Yaphe, W. Analysis of the enzymic hydrolysis products of agarose by 13 Cn. mr spectroscopy. Carbohydr. Res. 1977, 54, C7–C10. [Google Scholar] [CrossRef]
- Delattre, C.; Fenoradosoa, T.A.; Michaud, P. Galactans: An overview of their most important sourcing and applications as natural polysaccharides. Braz. Arch. Biol. Technol. 2011, 54, 1075–1092. [Google Scholar]
- Chi, W.J.; Chang, Y.K.; Hong, S.K. Agar degradation by microorganisms and agar-degrading enzymes. Appl. Microbiol. Biotechnol. 2012, 94, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Phillips, G.O. Introduction to food hydrocolloids. In Handbook of Hydrocolloids, 2nd ed.; Woodhead Publishing: Cambridge, UK, 2009; pp. 1–22. [Google Scholar]
- Lahaye, M.; Rochas, C. Chemical structure and physico-chemical properties of agar. Hydrobiologia 1991, 221, 137–148. [Google Scholar] [CrossRef]
- Pedroche, J.; del Mar Yust, M.; Mateo, C.; Fernández-Lafuente, R.; Girón-Calle, J.; Alaiz, M.; Vioque, J.; Guisán, J.M.; Millán, F. Effect of the support and experimental conditions in the intensity of the multipoint covalent attachment of proteins on glyoxyl-agarose supports: Correlation between enzyme-support linkages and thermal stability. Enzyme Microb. Technol. 2007, 40, 1160–1166. [Google Scholar] [CrossRef]
- Polson, A. Fractionation of protein mixtures on columns of granulated agar. Biochim. Biophys. Acta 1961, 50, 565–567. [Google Scholar] [CrossRef]
- Porath, J.; Axén, R.; Ernback, S. Chemical coupling of proteins to agarose. Nature 1967, 215, 1491–1492. [Google Scholar] [CrossRef] [PubMed]
- Porath, J.; Janson, J.C.; Ls, T. Agar derivatives for chromatography, electrophoresis and gel-bound enzymes. I. Desulphated and reduced cross-linked agar and agarose in spherical bead form. J. Chromatogr. 1971, 60, 179–184. [Google Scholar] [CrossRef]
- Arnott, S.; Fulmer, A.; Scott, W.E.; Dea, I.C.M.; Moorhouse, R.; Rees, D.A. The agarose double helix and its function in agarose gel structure. J. Mol. Biol. 1974, 90, 269–284. [Google Scholar] [CrossRef]
- Nordqvist, D.; Vilgis, T.A. Rheological Study of the Gelation Process of Agarose-Based Solutions. Food Biophys. 2011, 6, 450–460. [Google Scholar] [CrossRef]
- Deszczynski, M.; Kasapis, S.; MacNaughton, W.; Mitchell, J.R. Effect of sugars on the mechanical and thermal properties of agarose gels. Food Hydrocoll. 2003, 17, 793–799. [Google Scholar] [CrossRef]
- Guenet, J.M.; Rochas, C. Agarose sols and gels revisited. Macromol. Symp. 2006, 242, 65–70. [Google Scholar] [CrossRef]
- Amsterdam, A.; Er-El, Z.; Shaltiel, S. Ultrastructure of beaded agarose. Arch. Biochem. Biophys. 1975, 171, 673–677. [Google Scholar] [CrossRef]
- Guiseley, K.B. Chemical and physical properties of algal polysaccharides used for cell immobilization. Enzyme Microb. Technol. 1989, 11, 706–716. [Google Scholar] [CrossRef]
- Normand, V.; Lootens, D.L.; Amici, E.; Plucknett, K.P.; Aymard, P. New insight into agarose gel mechanical properties. Biomacromolecules 2000, 1, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Normand, V.; Aymard, P.; Lootens, D.L.; Amici, E.; Plucknett, K.P.; Frith, W.J. Effect of sucrose on agarose gels mechanical behaviour. Carbohydr. Polym. 2003, 54, 83–95. [Google Scholar] [CrossRef]
- Hayashi, A.; Kanzaki, T. Swelling of agarose gel and its related changes. Food Hydrocoll. 1987, 1, 317–325. [Google Scholar] [CrossRef]
- Hjertén, S. The preparation of agarose spheres for chromatography of molecules and particles. Biochim. Biophys. Acta Biophys. 1964, 79, 393–398. [Google Scholar] [CrossRef]
- Egorov, A.M.; Vakhabov, A.K.; Chernyak, V.Y. Isolation of agarose and granulation of agar and agarose gel. J. Chromatogr. 1970, 46, 143–148. [Google Scholar] [CrossRef]
- Bengtsson, S.; Philipson, L. Chromatography of animal viruses on pearl-condensed agar. Biochim. Biophys. Acta Biophys. 1964, 79, 399–406. [Google Scholar] [CrossRef]
- Zhou, Q.-Z.; Wang, L.-Y.; Ma, G.-H.; Su, Z.-G. Preparation of uniform-sized agarose beads by microporous membrane emulsification technique. J. Colloid Interface Sci. 2007, 311, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Q.; Li, Q.; Gong, F.-L.; Lei, J.-D.; Zhao, X.; Ma, G.-H.; Su, Z.-G. Preparation of large-sized highly uniform agarose beads by novel rotating membrane emulsification. J. Membr. Sci. 2015, 476, 30–39. [Google Scholar] [CrossRef]
- Laas, T. Agar derivatives for chromatography, electrophoresis and gel-bound enzymes. J. Chromatogr. 1975, 111, 373–387. [Google Scholar] [CrossRef]
- Porath, J.; Torgny, T.; Janson, J.-C. Agar derivatives for chromatography, electrophoresis and gel-bound enzymes. J. Chromatogr. 1975, 103, 49–62. [Google Scholar] [CrossRef]
- Jan-Christer, J. Microspheres for the Separation and Refolding of Proteins with an Emphasis on Particles Made of Agarose. In Microspheres and Microcapsules in Biotechnology; Pan Stanford Publishing: Singapore, 2013; pp. 123–152. [Google Scholar]
- Sundberg, L.; Porath, J. Preparation of adsorbents for biospecific affinity chromatography. J. Chromatogr. 1974, 90, 87–98. [Google Scholar] [CrossRef]
- De Koning, H.W.; Chamuleau, R.A.; Bantjes, A. Crosslinked agarose encapsulated sorbents resistant to steam sterilization. Preparation and mechanical properties. J. Biomed. Mat. Res. 1984, 18. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Palomo, J.M.; Fuentes, M.; Betancor, L.; Grazu, V.; López-Gallego, F.; Pessela, B.C.C.; Hidalgo, A.; Fernández-Lorente, G.; Fernández-Lafuente, R.; et al. Glyoxyl agarose: A fully inert and hydrophilic support for immobilization and high stabilization of proteins. Enzyme Microb. Technol. 2006, 39, 274–280. [Google Scholar] [CrossRef]
- López-Gallego, F.; Fernandez-Lorente, G.; Rocha-Martin, J.; Bolivar, J.M.; Mateo, C.; Guisan, J.M. Stabilization of enzymes by multipoint covalent immobilization on supports activated with glyoxyl groups. Methods Mol. Biol. 2013, 1051, 59–71. [Google Scholar] [PubMed]
- Mateo, C.; Abian, O.; Bernedo, M.; Cuenca, E.; Fuentes, M.; Fernandez-Lorente, G.; Palomo, J.M.; Grazu, V.; Pessela, B.C.C.; Giacomini, C.; et al. Some special features of glyoxyl supports to immobilize proteins. Enzyme Microb. Technol. 2005, 37, 456–462. [Google Scholar] [CrossRef]
- Bolivar, J.M.; López-Gallego, F.; Godoy, C.; Rodrigues, D.S.; Rodrigues, R.C.; Batalla, P.; Rocha-Martín, J.; Mateo, C.; Giordano, R.L.C.; Guisán, J.M. The presence of thiolated compounds allows the immobilization of enzymes on glyoxyl agarose at mild pH values: New strategies of stabilization by multipoint covalent attachment. Enzyme Microb. Technol. 2009, 45, 477–483. [Google Scholar] [CrossRef]
- Grazú, V.; López-Gallego, F.; Montes, T.; Abian, O.; González, R.; Hermoso, J.A.; García, J.L.; Mateo, C.; Guisán, J.M. Promotion of multipoint covalent immobilization through different regions of genetically modified penicillin G acylase from E. coli. Process Biochem. 2010, 45, 390–398. [Google Scholar] [CrossRef]
- Godoy, C.A.; Rivas, B.D.L.; Grazú, V.; Montes, T.; Guisàn, J.M.; López-Gallego, F. Glyoxyl-disulfide agarose: A tailor-made support for site-directed rigidification of proteins. Biomacromolecules 2011, 12, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Manoel, E.A.; dos Santos, J.C.S.; Freire, D.M.G.; Rueda, N.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports involves the open form of the enzyme. Enzyme Microb. Technol. 2015, 71, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.C.S.; Rueda, N.; Barbosa, O.; Millán-Linares, M.D.C.; Pedroche, J.; Del Mar Yuste, M.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Bovine trypsin immobilization on agarose activated with divinylsulfone: Improved activity and stability via multipoint covalent attachment. J. Mol. Catal. B Enzym. 2015, 117, 38–44. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R.; Rosell, C.M.; Rodriguez, V.; Santana, C.; Soler, G.; Bastida, A.; Guisán, J.M. Preparation of activated supports containing low pK amino groups. A new tool for protein immobilization via the carboxyl coupling method. Enzyme Microb. Technol. 1993, 15, 546–550. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernandez-Lafuente, R. Amination of enzymes to improve biocatalyst performance: Coupling genetic modification and physicochemical tools. RSC Adv. 2014, 4, 38350–38374. [Google Scholar] [CrossRef]
- Betancor, L.; López-Gallego, F.; Hidalgo, A.; Alonso-Morales, N.; Mateo, G.D.O.C.; Fernández-Lafuente, R.; Guisán, J.M. Different mechanisms of protein immobilization on glutaraldehyde activated supports: Effect of support activation and immobilization conditions. Enzyme Microb. Technol. 2006, 39, 877–882. [Google Scholar] [CrossRef]
- López-Gallego, F.; Betancor, L.; Mateo, C.; Hidalgo, A.; Alonso-Morales, N.; Dellamora-Ortiz, G.; Guisán, J.M.; Fernández-Lafuente, R. Enzyme stabilization by glutaraldehyde crosslinking of adsorbed proteins on aminated supports. J. Biotechnol. 2005, 119, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Turková, J.; Bláha, K.; Malaníková, M.; Vančurová, D.; Švec, F.; Kálal, J. Methacrylate gels with epoxide groups as supports for immobilization of enzymes in pH range 3–12. BBA Enzymol. 1978, 524, 162–169. [Google Scholar] [CrossRef]
- Grazu, V.; López-Gallego, F.; Guisán, J.M. Tailor-made design of penicillin G acylase surface enables its site-directed immobilization and stabilization onto commercial mono-functional epoxy supports. Process Biochem. 2012, 47, 2538–2541. [Google Scholar] [CrossRef]
- Mateo, C.; Fernández-Lorente, G.; Abian, O.; Fernández-Lafuente, R.; Guisán, J.M. Multifunctional epoxy supports: A new tool to improve the covalent immobilization of proteins. The promotion of physical adsorptions of proteins on the supports before their covalent linkage. Biomacromolecules 2000, 1, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Torres, R.; Fernández-Lorente, G.; Ortiz, C.; Fuentes, M.; Hidalgo, A.; López-Gallego, F.; Abian, O.; Palomo, J.M.; Betancor, L.; et al. Epoxy-amino groups: A new tool for improved immobilization of proteins by the epoxy method. Biomacromolecules 2003, 4, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Smalla, K.; Turkova, J.; Coupek, J.; Hermann, P. Influence of salts on the covalent immobilization of proteins to modified copolymers of 2-hydroxyethyl methacrylate with ethylene dimethacrylate. Biotechnol. Appl. Biochem. 1988, 10, 21–31. [Google Scholar] [PubMed]
- Barbosa, O.; Torres, R.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Heterofunctional supports in enzyme immobilization: From traditional immobilization protocols to opportunities in tuning enzyme properties. Biomacromolecules 2013, 14, 2433–2462. [Google Scholar] [CrossRef] [PubMed]
- Brady, L.; Brzozowski, A.M.; Derewenda, Z.S.; Dodson, E.; Dodson, G.; Tolley, S.; Turkenburg, J.P.; Christiansen, L.; Huge-Jensen, B.; Norskov, L.; et al. A serine protease triad forms the catalytic centre of a triacylglycerol lipase. Nature 1990, 343, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Derewenda, U.; Derewenda, Z.S.; Dodson, G.G.; Lawson, D.M.; Turkenburg, J.P.; Bjorkling, F.; Huge-Jensen, B.; Patkar, S.A.; Thim, L. A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature 1991, 351, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, U.; Brzozowski, A.M.; Lawson, D.M.; Derewenda, Z.S. Catalysis at the interface: The anatomy of a conformational change in a triglyceride lipase. Biochemistry 1992, 31, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S. A twist in the tale of lipolytic enzymes. Nat. Struct. Biol. 1995, 2, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S.; Derewenda, U.; Dodson, G.G. The crystal and molecular structure of the Rhizomucor miehei triacylglyceride lipase at 1.9 Å resolution. J. Mol. Biol. 1992, 227, 818–839. [Google Scholar] [CrossRef]
- Bastida, A.; Sabuquillo, P.; Armisen, P.; Fernández-Lafuente, R.; Huguet, J.; Guisán, J.M. A single step purification, immobilization, and hyperactivation of lipases via interfacial adsorption on strongly hydrophobic supports. Biotechnol. Bioeng. 1998, 58, 486–493. [Google Scholar] [CrossRef]
- Rueda, N.; Dos Santos, J.C.S.; Torres, R.; Ortiz, C.; Barbosa, O.; Fernandez-Lafuente, R. Improved performance of lipases immobilized on heterofunctional octyl-glyoxyl agarose beads. RSC Adv. 2015, 5, 11212–11222. [Google Scholar] [CrossRef]
- Suescun, A.; Rueda, N.; Dos Santos, J.C.S.; Castillo, J.J.; Ortiz, C.; Torres, R.; Barbosa, O.; Fernandez-Lafuente, R. Immobilization of lipases on glyoxyl-octyl supports: Improved stability and reactivation strategies. Process Biochem. 2015, 50, 1211–1217. [Google Scholar] [CrossRef]
- Rueda, N.; Albuquerque, T.L.; Bartolome-Cabrero, R.; Fernandez-Lopez, L.; Torres, R.; Ortiz, C.; Dos Santos, J.C.S.; Barbosa, O.; Fernandez-Lafuente, R. Reversible immobilization of lipases on heterofunctional octyl-Amino agarose beads prevents enzyme desorption. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; Dos Santos, C.S.; Rodriguez, M.D.; Albuquerque, T.L.; Barbosa, O.; Torres, R.; Ortiz, C.; Fernandez-Lafuente, R. Reversible immobilization of lipases on octyl-glutamic agarose beads: A mixed adsorption that reinforces enzyme immobilization. J. Mol. Catal. B Enzym. 2016, 128, 10–18. [Google Scholar] [CrossRef]
- Hirata, D.B.; Albuquerque, T.L.; Rueda, N.; Sánchez-Montero, J.M.; Garcia-Verdugo, E.; Porcar, R.; Fernandez-Lafuente, R. Advantages of Heterofunctional Octyl Supports: Production of 1, 2-Dibutyrin by Specific and Selective Hydrolysis of Tributyrin Catalyzed by Immobilized Lipases. Chem. Select 2016, 1, 3259–3270. [Google Scholar] [CrossRef]
- Hirata, D.B.; Albuquerque, T.L.; Rueda, N.; Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Fernandez-Lafuente, R. Evaluation of different immobilized lipases in transesterification reactions using tributyrin: Advantages of the heterofunctional octyl agarose beads. J. Mol. Catal. B Enzym. 2016, 133, 117–123. [Google Scholar] [CrossRef]
- Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Hirata, D.B.; Torrestiana-Sanchez, B.; Rosales-Quintero, A.; Fernandez-Lafuente, R. Relevance of substrates and products on the desorption of lipases physically adsorbed on hydrophobic supports. Enzyme Microb. Technol. 2016, 96, 30–35. [Google Scholar] [CrossRef]
- Dos Santos, J.C.S.; Rueda, N.; Barbosa, O.; Fernández-Sánchez, J.F.; Medina-Castillo, A.L.; Ramón-Márquez, T.; Arias-Martos, M.C.; Millán-Linares, M.C.; Pedroche, J.; Yust, M.D.M.; et al. Characterization of supports activated with divinyl sulfone as a tool to immobilize and stabilize enzymes via multipoint covalent attachment. Application to chymotrypsin. RSC Adv. 2015, 5, 20639–20649. [Google Scholar] [CrossRef]
- Dos Santos, J.C.S.; Rueda, N.; Sanchez, A.; Villalonga, R.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Versatility of divinylsulfone supports permits the tuning of CALB properties during its immobilization. RSC Adv. 2015, 5, 35801–35810. [Google Scholar] [CrossRef]
- Dos Santos, J.C.S.; Rueda, N.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Tuning the catalytic properties of lipases immobilized on divinylsulfone activated agarose by altering its nanoenvironment. Enzyme Microb. Technol. 2015, 77, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Andersson, T.; Carlsson, M.; Hagel, L.; Pernemalm, P.-Å.; Jansson, J.C. Agarose-based media for high-resolution gel filtration of biopolymers. J. Chromatogr. 1985, 326, 33–44. [Google Scholar] [CrossRef]
- Margolis, S. Separation and size determination of human serum lipoproteins by agarose gel filtration. J. Lipid Res. 1967, 8, 501–507. [Google Scholar] [PubMed]
- Prakash, O.; Jaiswal, N. Immobilization of a Thermostable α-Amylase on Agarose and Agar Matrices and its Application in Starch Stain Removal. World Appl. Sci. J. 2011, 13, 572–577. [Google Scholar]
- Krzek, M.; van Beek, H.L.; Permentier, H.P.; Bischoff, R.; Fraaije, M.W. Covalent immobilization of a flavoprotein monooxygenase via its flavin cofactor. Enzyme Microb. Technol. 2016, 82, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Gioia, L.; Rodríguez-Couto, S.; Menéndez, M.d.P.; Manta, C.; Ovsejevi, K. Reversible covalent immobilization of Trametes villosa laccase onto thiolsulfinate-agarose: An insoluble biocatalyst with potential for decoloring recalcitrant dyes. Biotechnol. Appl. Biochem. 2015, 62, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Ricca, E.; Brucher, B.; Schrittwieser, J.H. Multi-enzymatic cascade reactions: Overview and perspectives. Adv. Synth. Catal. 2011, 353, 2239–2262. [Google Scholar] [CrossRef]
- Lopez-Gallego, F.; Schmidt-Dannert, C. Multi-enzymatic synthesis. Curr. Opin. Chem. Biol. 2010, 14, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Kazenwadel, F.; Franzreb, M.; Rapp, B. Synthetic enzyme supercomplexes: Co-immobilization of enzyme cascades. Anal. Methods 2015, 7, 4030–4037. [Google Scholar] [CrossRef]
- Mateo, C.; Chmura, A.; Rustler, S.; van Rantwijk, F.; Stolz, A.; Sheldon, R.A. Synthesis of enantiomerically pure (S)-mandelic acid using an oxynitrilase–nitrilase bienzymatic cascade: A nitrilase surprisingly shows nitrile hydratase activity. Tetrahedron Asymmetry 2006, 17, 320–323. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R.; Rodriguez, V.; Guisán, J.M. The coimmobilization of d-amino acid oxidase and catalase enables the quantitative transformation of d-amino acids (d-phenylalanine) into α-keto acids (phenylpyruvic acid). Enzyme Microb. Technol. 1998, 23, 28–33. [Google Scholar] [CrossRef]
- Rocha-Martín, J.; Rivas, B.L.; Muñoz, R.; Guisán, J.M.; López-Gallego, F. Rational co-immobilization of bi-enzyme cascades on porous supports and their applications in bio-redox reactions with insitu recycling of soluble cofactors. ChemCatChem 2012, 4, 1279–1288. [Google Scholar] [CrossRef]
- Peirce, S.; Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Rueda, N.; Bartolome-Cabrero, R.; Fernandez-Lopez, L.; Russo, M.E.; Marzocchella, A.; Fernandez-Lafuente, R. Development of simple protocols to solve the problems of enzyme coimmobilization. Application to coimmobilize a lipase and a β-galactosidase. RSC Adv. 2016, 6, 61707–61715. [Google Scholar] [CrossRef]
- Peirce, S.; Tacias-Pascacio, V.G.; Russo, M.E.; Marzocchella, A.; Virgen-Ortíz, J.J.; Fernandez-Lafuente, R. Stabilization of Candida antarctica Lipase B (CALB) Immobilized on Octyl Agarose by Treatment with Polyethyleneimine (PEI). Molecules 2016, 21, 751. [Google Scholar] [CrossRef] [PubMed]
- Virgen-Ortíz, J.J.; Peirce, S.; Tacias-Pascacio, V.G.; Cortes-Corberan, V.; Marzocchella, A.; Russo, M.E.; Fernandez-Lafuente, R. Reuse of anion exchangers as supports for enzyme immobilization: Reinforcement of the enzyme-support multiinteraction after enzyme inactivation. Process Biochem. 2016, 51, 1391–1396. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Fernández-Lorente, G.; Pedroche, J.; Fernández-Lafuente, R.; Guisan, J.M.; Tam, A.; Daminati, M. Epoxy Sepabeads: A novel epoxy support for stabilization of industrial enzymes via very intense multipoint covalent attachment. Biotechnol. Prog. 2002, 18, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Hidalgo, A.; Sánchez-Ruiloba, L.; Berenguer, J.; Guisán, J.M.; López-Gallego, F. Modulation of the distribution of small proteins within porous matrixes by smart-control of the immobilization rate. J. Biotechnol. 2011, 155, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Kong, J.; Dong, A.; Yu, S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Eisl, I.; Nidetzky, B. Advanced characterization of immobilized enzymes as heterogeneous biocatalysts. Catal. Today 2016, 259, 66–80. [Google Scholar] [CrossRef]
- Orrego, A.H.; García, C.; Mancheno, J.M.; Guisán, J.M.; Lillo, M.P.; López-Gallego, F. Two-Photon Fluorescence Anisotropy Imaging to Elucidate the Dynamics and the Stability of Immobilized Proteins. J. Phys. Chem. B 2016, 120, 485–491. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Location |
|---|---|
| Gelidium amansii | Japan, China |
| Gelidium cartilagineum | USA, Mexico, South Africa |
| Gelidium corneum | South Africa, Portugal, Spain, Morocco |
| Gelidium liatulum | Japan |
| Gelidium lingulatam | Chile |
| Gelidium pacificum | Japan |
| Gelidium sesquipedale | Portugal, Morocco |
| Gelidiella acerosa | Japan, India, China |
| Gracilaria verrucosa | Turkey |
| Gracilaria dura | France |
| Gracilaria tenuistipitata | Philippines |
| Pterocladia lucida | New Zealand, Azores |
| Pterocladia capilacea | Egypt, Japan, New Zealand |
| Ahnfeltia plicata | Russia |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules 2016, 21, 1577. https://doi.org/10.3390/molecules21111577
Zucca P, Fernandez-Lafuente R, Sanjust E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules. 2016; 21(11):1577. https://doi.org/10.3390/molecules21111577
Chicago/Turabian StyleZucca, Paolo, Roberto Fernandez-Lafuente, and Enrico Sanjust. 2016. "Agarose and Its Derivatives as Supports for Enzyme Immobilization" Molecules 21, no. 11: 1577. https://doi.org/10.3390/molecules21111577
APA StyleZucca, P., Fernandez-Lafuente, R., & Sanjust, E. (2016). Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules, 21(11), 1577. https://doi.org/10.3390/molecules21111577