
Bio-Guided Isolation of Methanol-Soluble Metabolites of Common Spruce (Picea abies) Bark by-Products and Investigation of Their Dermo-Cosmetic Properties

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
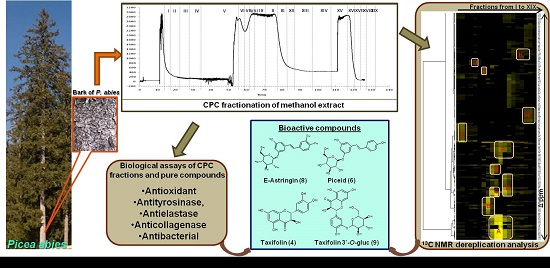
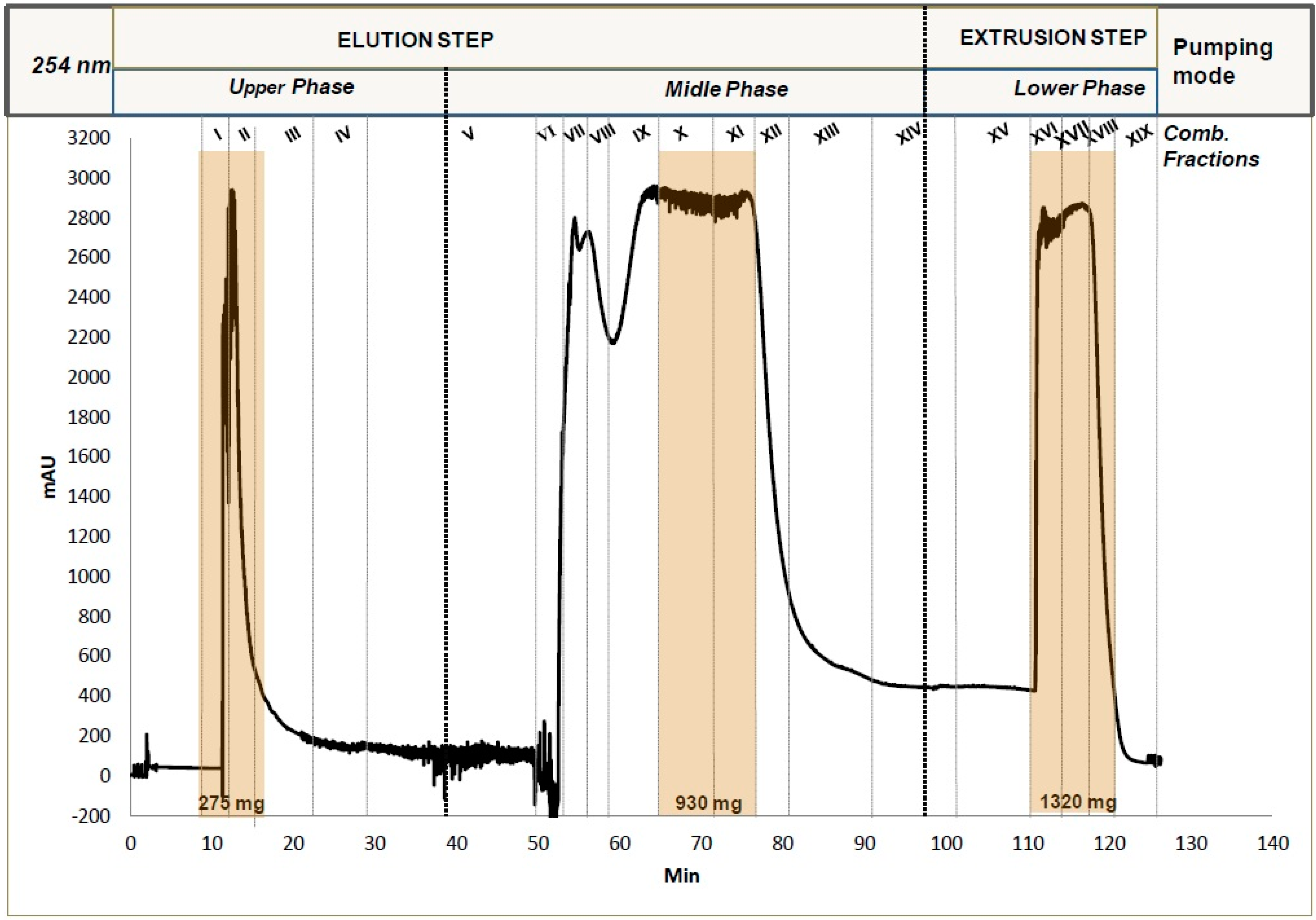
2.1. Fractionation of Crude Methanol Extract by CPC
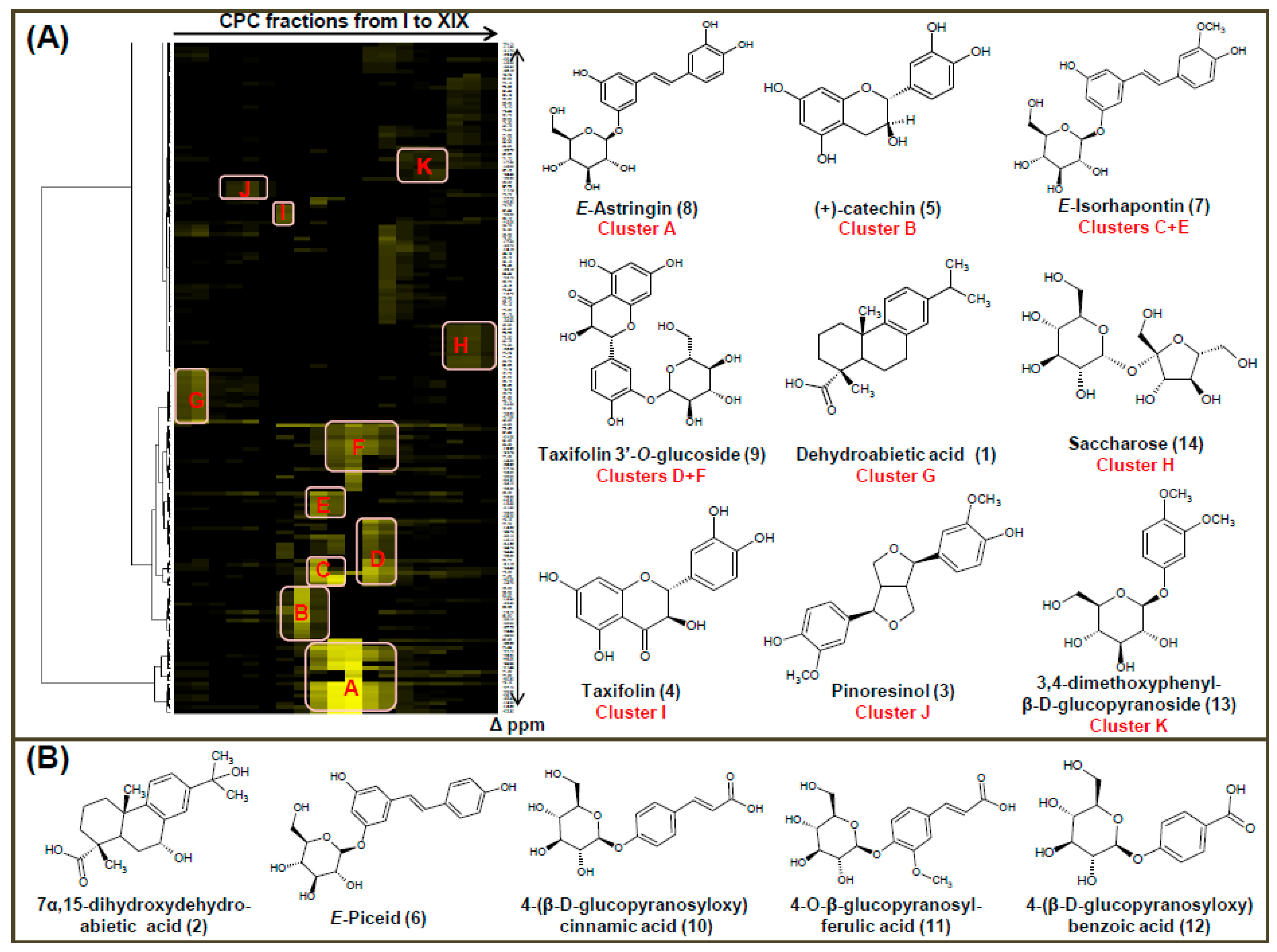
2.2. Chemical Characterization of CPC Fractions by 13C- and 2D-NMR Analysis
2.3. DPPH Radical Scavenging Activity of CPC Fractions
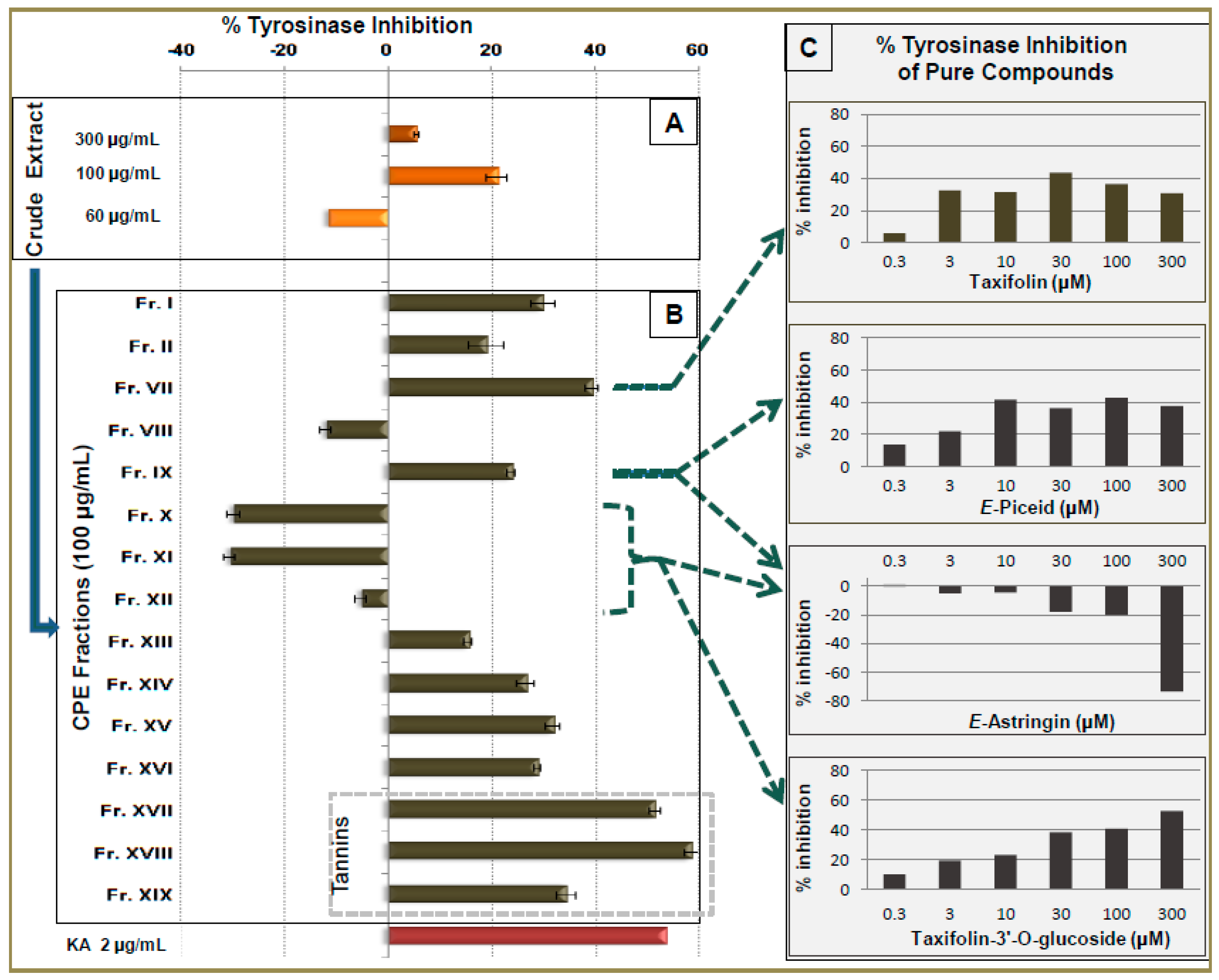
2.4. Tyrosinase Inhibitory Activity of CPC Fractions and Pure Compounds
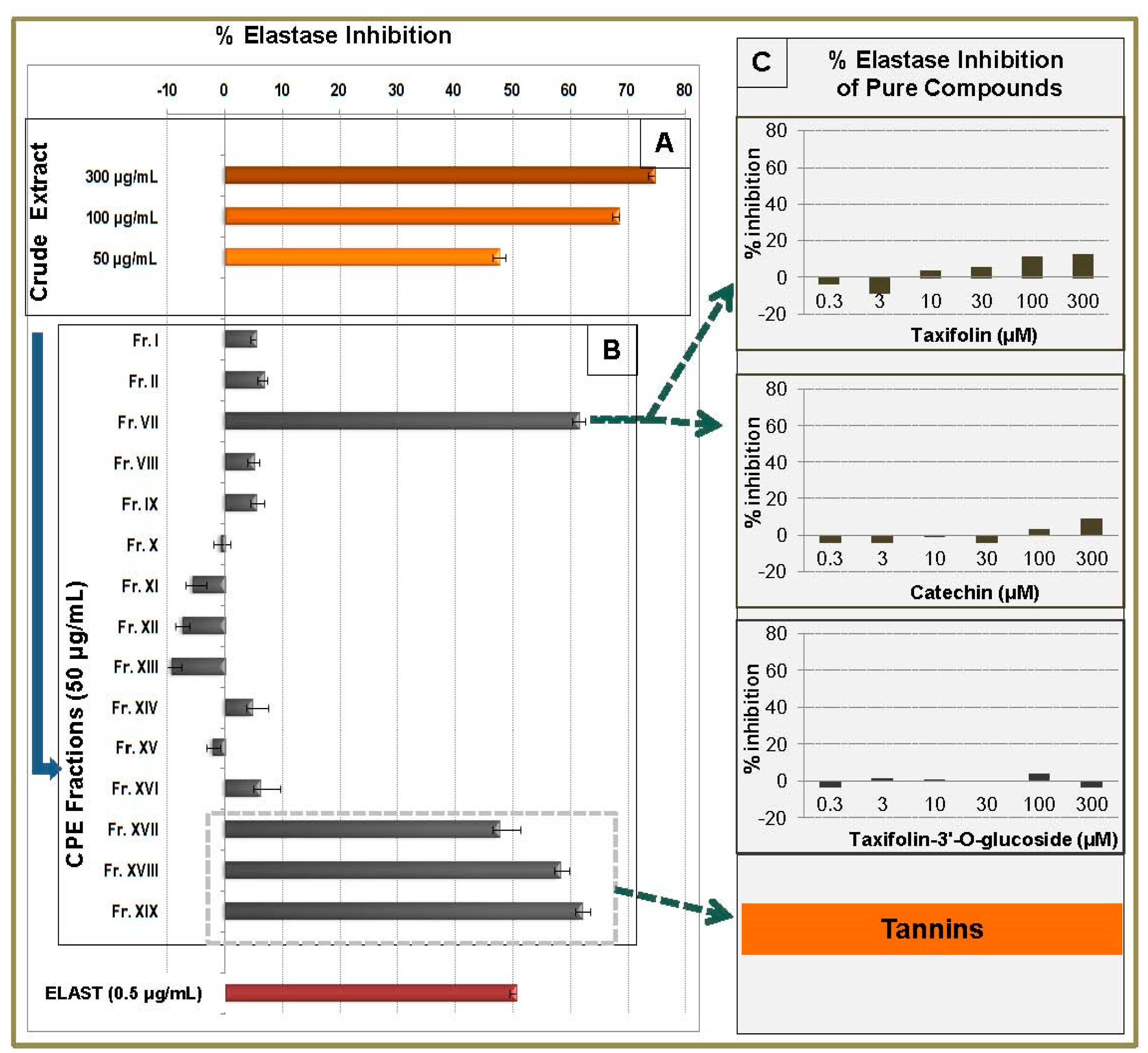
2.5. Elastase Inhibitory Activity of CPC Fractions and Pure Compounds
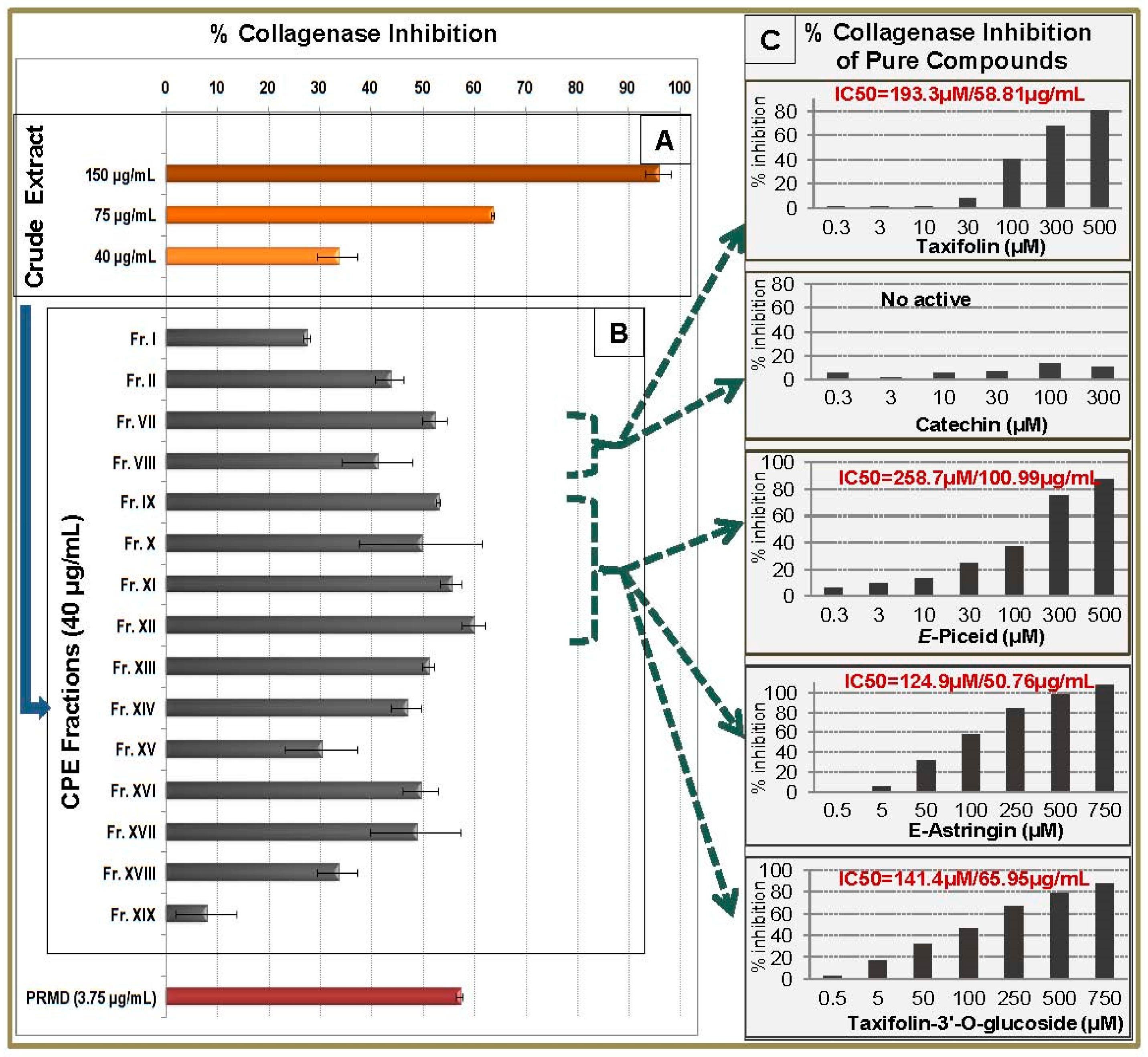
2.6. Collagenase Inhibitory Activity of CPC Fractions and Pure Compounds
2.7. In Vitro Antibacterial Activity of CPC Fractions and Pure Compounds
3. Materials and Methods
3.1. Reagents and Materials
3.2. Plant Material and Extraction Process
3.3. CPC Fractionation of the Methanol Extract
3.4. NMR Chemical Profiling of the CPE-Generated Fractions
3.5. Recovery of Pure E-Astringin from the CPE-Generated Fractions
3.6. Antioxidant Activity-DPPH Radical Scavenging Assay
3.7. Whitening Activity-Tyrosinase Inhibition Assay
3.8. Elastase Inhibition Assay
3.9. Collagenase Inhibition Assay
3.10. Antimicrobial Assay
3.10.1. Bioautography
3.10.2. MIC Determination
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heikkilä, J.; Sirén, M.; Ahtikoski, A.; Hynynen, J.; Sauvula, T.; Lehtonen, M. Energy wood thinning as a part of stand management of scots pine and norway spruce. Silva Fennica 2009, 43, 129–146. [Google Scholar] [CrossRef]
- Brignolas, F.; Lieutier, F.; Sauvard, D.; Christiansen, E.; Berryman, A.A. Phenolic predictors for norway spruce resistance to the bark beetle ips typographus (coleoptera: Scolytidae) and an associated fungus, ceratocystis polonica. Can. J. Forest Res. 1998, 28, 720–728. [Google Scholar] [CrossRef]
- Willför, S.; Nisula, L.; Hemming, J.; Reunanen, M.; Holmbom, B. Bioactive phenolic substances in industrially important tree species. Part 2: Knots and stemwood of fir species. Holzforschung 2005, 58, 650–659. [Google Scholar] [CrossRef]
- Sultan, M.Z.; Jeon, Y.-M.; Moon, S.-S. Labdane-type diterpenes active against acne from pine cones (Pinus densiflora). Plant. Med. 2008, 74, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Mannila, E.; Talvitie, A. Stilbenes from Picea abies bark. Phytochemistry 1992, 31, 3288–3289. [Google Scholar]
- Pietarinen, S.; Willför, S.; Ahotupa, M.; Hemming, J.; Holmbom, B. Knotwood and bark extracts: Strong antioxidants from waste materials. J. Wood Sci. 2006, 52, 436–444. [Google Scholar] [CrossRef]
- Cai, Z.; Rudie, A.W.; Stark, N.M.; Sabo, R.C.; Ralph, R.A. New products and product categories in the global forest sector. In The Global Forest Sector: Changes, Practices, and Prospects; Hansen, E., Panwar, R., Vlosky, R., Eds.; CRC Press—Taylor & Francis Group LLC: Boca Raton, FL, USA, 2013; pp. 129–150. [Google Scholar]
- Krogell, J.; Holmbom, B.; Pranovich, A.; Hemming, J.; Willför, S. Biorefinery: Extraction and chemical characterization of norway spruce inner and outer bark. Nordic. Pulp. Paper Res. J. 2012, 27, 6–17. [Google Scholar] [CrossRef]
- Co, M.; Fagerlund, A.; Engman, L.; Sunnerheim, K.; Sjöberg, P.J.R.; Turner, C. Extraction of antioxidants from spruce (Picea abies) bark using eco-friendly solvents. Chemistry 2012, 23. [Google Scholar] [CrossRef] [PubMed]
- Jyske, T.; Laakso, T.; Latva-Maenpaa, H.; Tapanila, T.; Saranpaa, P. Yield of stilbene glucosides from the bark of young and old Norway spruce stems. Biomass Bioenerg. 2014, 71, 216–227. [Google Scholar] [CrossRef]
- Latva-Mäenpää, H.; Laakso, T.; Sarjala, T.; Wähälä, K.; Saranpää, P. Variation of stilbene glucosides in bark extracts obtained from roots and stumps of Norway spruce (Picea abies [L.] karst.). Trees 2013, 27, 131–139. [Google Scholar] [CrossRef]
- Hubert, J.; Angelis, A.; Aligiannis, N.; Michalea, R.; Abedini, A.; Bakiri, A.; Reynaud, R.; Nuzillard, J.M.; Gangloff, S.C.; Skaltsounis, A.L.; Renault, J.H. In vitro dermo-cosmetic evaluation of bark extracts from common temperate trees. Plant. Med. 2016, 28. [Google Scholar] [CrossRef] [PubMed]
- Hamzaoui, M.; Renault, J.H.; Nuzillard, J.M.; Reynaud, R.; Hubert, J. Stepwise elution of a three-phase solvent system in centrifugal partition extraction: A new strategy for the fractionation and phytochemical screening of a crude bark extract. Phytochem. Anal. 2013, 24, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Ekman, R.; Sjöholm, R. Hydroxylation of dehydroabietic acid by Fomes annosus. Acta Chem. Scand. 1979, B33, 76–78. [Google Scholar]
- Pan, H.; Lundgren, L.N. Phenolic extractives from root bark of Picea abies. Phytochemistry 1995, 39, 1423–1428. [Google Scholar] [CrossRef]
- Yang, X.-W.; Feng, L.; Li, S.-M.; Liu, X.-H.; Li, Y.-L.; Wu, L.; Shen, Y.-H.; Tian, J.-M.; Zhang, X.; Liu, X.-R.; et al. Isolation, structure, and bioactivities of abiesadines A–Y, 25 new diterpenes from abies georgei Orr. Bioorg. Med. Chem. 2010, 18, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Georges, P.; Legault, J.; Lavoie, S.; Grenon, C.; Pichette, A. Diterpenoids from the buds of pinus banksiana lamb. Molecules 2012, 17, 9716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-J.; Li, Y.-H.; Jiang, J.-D.; Yu, S.-S.; Wang, X.-J.; Zhuang, P.-Y.; Zhang, Y.; Qu, J.; Ma, S.-G.; Li, Y.; et al. Diterpenes and sesquiterpenes with anti-coxsackie virus b3 activity from the stems of illicium jiadifengpi. Tetrahedron 2014, 70, 4494–4499. [Google Scholar] [CrossRef]
- Okasaka, M.; Takaishi, Y.; Kashiwada, Y.; Kodzhimatov, O.K.; Ashurmetov, O.; Lin, A.J.; Consentino, L.M.; Lee, K.-H. Terpenoids from juniperus polycarpus var. Seravschanica. Phytochemistry 2006, 67, 2635–2640. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Spiteller, G. Comparison of phenolic compounds from galls and shoots of picea glauca. Phytochemistry 1997, 44, 59–67. [Google Scholar] [CrossRef]
- Cotoras, M.; Vivanco, H.; Melo, R.; Aguirre, M.; Silva, E.; Mendoza, L. In vitro and in vivo evaluation of the antioxidant and prooxidant activity of phenolic compounds obtained from grape (vitis vinifera) pomace. Molecules 2014, 19, 21154–21167. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Cordovés, C.; Bartolomé, B.; Vieira, W.; Virador, V.M. Effects of wine phenolics and sorghum tannins on tyrosinase activity and growth of melanoma cells. J. Agric. Food Chem. 2001, 49, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Tamura, N. Inhibitory compound of tyrosinase activity from the sprout of Polygonum hydropiper L. (Benitade). Biol. Pharm. Bull. 2007, 30, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Lamaison, J.; Carnat, A.; Petitjean-Freytet, C. Tannin content and inhibiting activity of elastase in Rosaceae. Ann. Pharm. Fr. 1990, 48, 335–340. [Google Scholar] [PubMed]
- Mrowietz, U.; Ternowitz, T.; Wiedow, O. Selective inactivation of human neutrophil elastase by synthetic tannin. J. Investig. Dermatol. 1991, 97, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Hrenn, A.; Steinbrecher, T.; Labahn, A.; Schwager, J.; Schempp, C.M.; Merfort, I. Plant phenolics inhibit neutrophil elastase. Plant. Med. 2006, 72, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Meghji, S.C.; Harvey, W.; Scutt, A.; Phillipson, D. Inhibition of collagenase activity by areca nut tannins—A mechanism of collagen accumulation in oral submucous fibrosis (OSF). J. Dent. Res. 1982, 61, 545. [Google Scholar]
- Kuspradini, H.; Mitsunaga, T.; Ohashi, H. Antimicrobial activity against streptococcus sobrinus and glucosyltransferase inhibitory activity of taxifolin and some flavanonol rhamnosides from kempas (koompassia malaccensis) extracts. J. Wood Sci. 2009, 55, 308–313. [Google Scholar] [CrossRef]
- Woodward, S.; Pearce, R.B. The role of stilbenes in resistance of sitka spruce (picea sitchensis (bong.) carr.) to entry of fungal pathogens. Physiol. Mol. Plant. Pathol. 1988, 33, 127–149. [Google Scholar] [CrossRef]
- Kolodziej, H.; Kayser, O.; Latté, K.P.; Ferreira, D. Evaluation of the antimicrobial potency of tannins and related compounds using the microdilution broth method. Plant. Med. 1999, 65, 444–446. [Google Scholar] [CrossRef]
- Lee, S.K.; Mbwambo, Z.H.; Chung, H.S.; Luyengi, L.; Gamez, E.J.C.; Mehta, R.G.; Kinghorn, A.D.; Pezzuto, J.M. Evaluation of the antioxidant potential of natural products. Comb. Chem. High. Throughput Screen. 1998, 1, 35–46. [Google Scholar] [PubMed]
- Masuda, T.; Yamashita, D.; Takeda, Y.; Yonemori, S. Screening for tyrosinase inhibitors among extracts of seashore plants and identification of potent inhibitors from garcinia subelliptica. Biosci. Biotechnol. Biochem. 2005, 69, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.C.; Taori, K.; Paul, V.J.; Luesch, H. Lyngbyastatins 8–10, elastase inhibitors with cyclic depsipeptide scaffolds isolated from the marine cyanobacterium lyngbya semiplena. Marine Drugs 2009, 7, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Gostner, J.; Fuchs, J.E.; Chaita, E.; Aligiannis, N.; Skaltsounis, L.; Ganzera, M. Inhibition of collagenase by mycosporine-like amino acids from marine sources. Plant. Med. 2015, 81, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Roumy, V.; Mahieux, S.; Biabiany, M.; Standaert-Vitse, A.; Riviere, C.; Sahpaz, S.; Bailleul, F.; Neut, C.; Hennebelle, T. Rosmarinic acid and its methyl ester as antimicrobial components of the hydromethanolic extract of Hyptis atrorubens Poit. (Lamiaceae). Evid.-Based Complement. Altern. Med. 2013, 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the crude bark extract and CPC fractions are available from the authors.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganisms | MIC (μg/mL) | ||||
|---|---|---|---|---|---|
| Taxifolin | Catechin | E-Piceid | E-Astringin | Tax-3’-O-gluc | |
| Gram positive bacteria | |||||
| Enterococcus faecalis ATCC 1034 | 125 | 125 | 125 | 31.2 | 125 |
| Staphylococcus aureus CIP 53.154 | 31.2 | 62.5 | 31.2 | 62.5 | 62.5 |
| Staphylococcus epidermidis | 250 | 125 | 125 | 31.2 | 125 |
| Gram negative bacteria | |||||
| Escherichia coli CIP 54.127 | 250 | 250 | 250 | 125 | 250 |
| Pseudomonas aeruginosa ATCC 9027 | 125 | 125 | 125 | 62.5 | 125 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelis, A.; Hubert, J.; Aligiannis, N.; Michalea, R.; Abedini, A.; Nuzillard, J.-M.; Gangloff, S.C.; Skaltsounis, A.-L.; Renault, J.-H. Bio-Guided Isolation of Methanol-Soluble Metabolites of Common Spruce (Picea abies) Bark by-Products and Investigation of Their Dermo-Cosmetic Properties. Molecules 2016, 21, 1586. https://doi.org/10.3390/molecules21111586
Angelis A, Hubert J, Aligiannis N, Michalea R, Abedini A, Nuzillard J-M, Gangloff SC, Skaltsounis A-L, Renault J-H. Bio-Guided Isolation of Methanol-Soluble Metabolites of Common Spruce (Picea abies) Bark by-Products and Investigation of Their Dermo-Cosmetic Properties. Molecules. 2016; 21(11):1586. https://doi.org/10.3390/molecules21111586
Chicago/Turabian StyleAngelis, Apostolis, Jane Hubert, Nektarios Aligiannis, Rozalia Michalea, Amin Abedini, Jean-Marc Nuzillard, Sophie C. Gangloff, Alexios-Leandros Skaltsounis, and Jean-Hugues Renault. 2016. "Bio-Guided Isolation of Methanol-Soluble Metabolites of Common Spruce (Picea abies) Bark by-Products and Investigation of Their Dermo-Cosmetic Properties" Molecules 21, no. 11: 1586. https://doi.org/10.3390/molecules21111586