
Synthesis and Determination of Physicochemical Properties of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Alkoxyethoxybenzoates

 and
and
Abstract
:1. Introduction
2. Results and Discussion
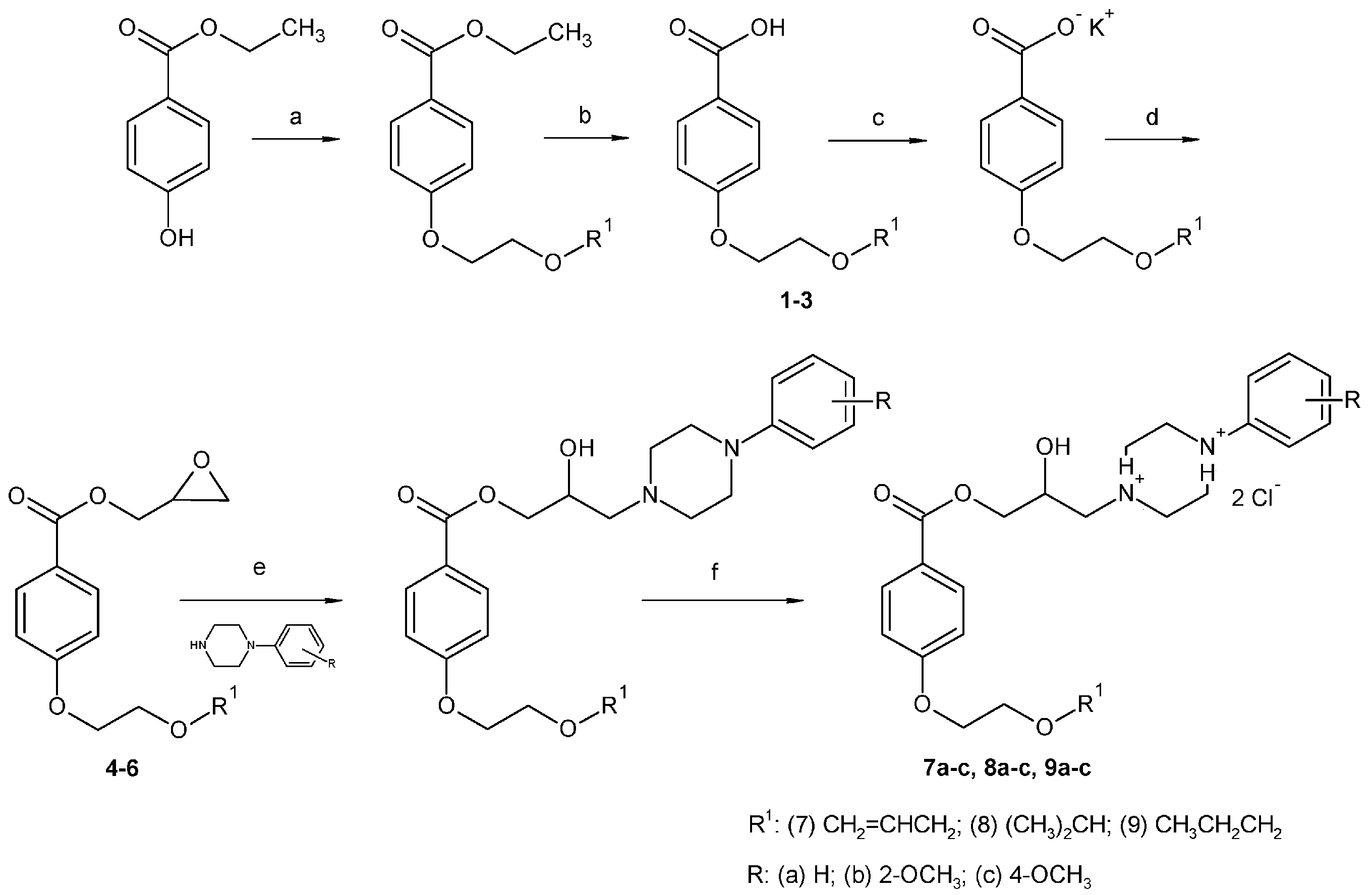
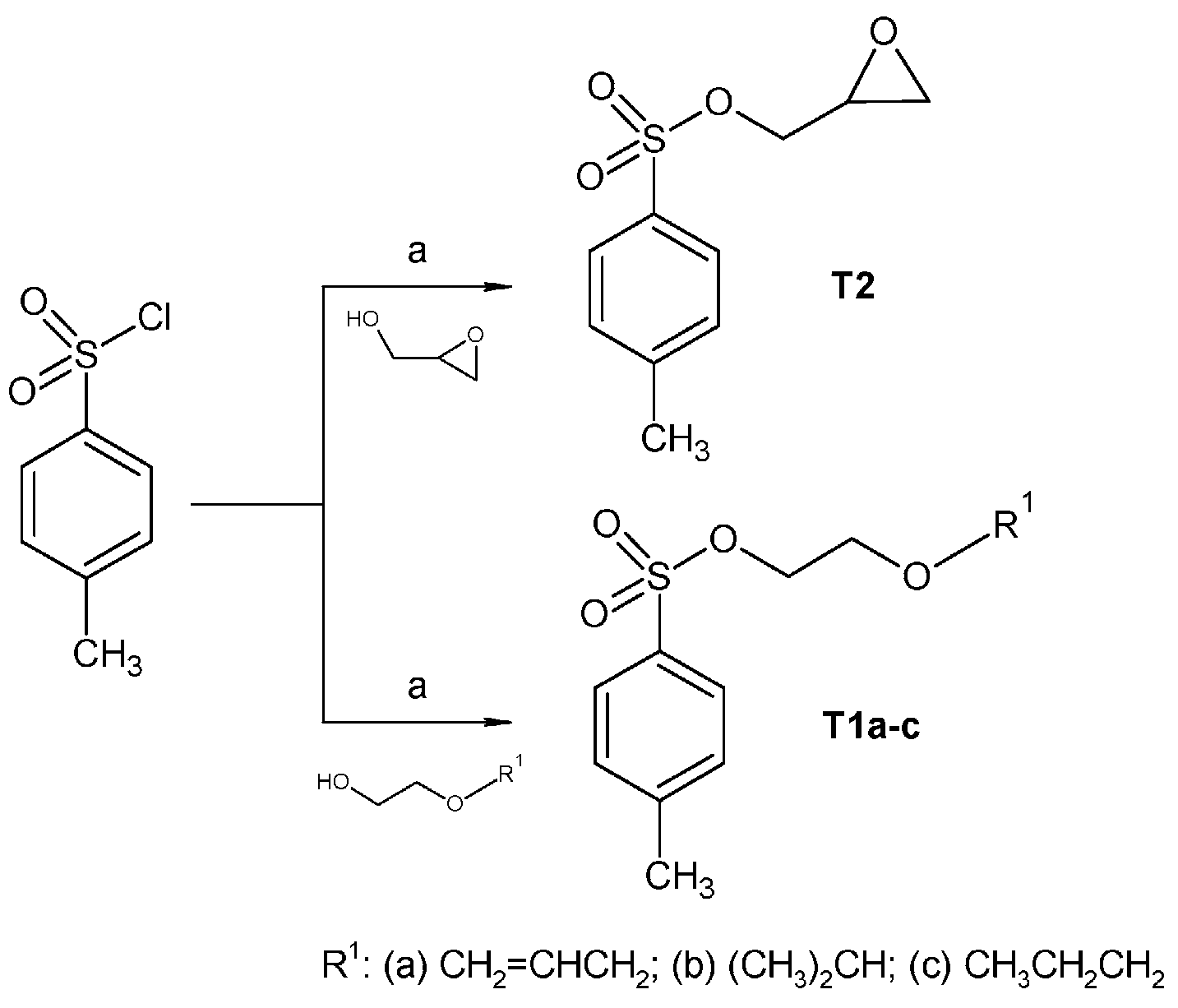
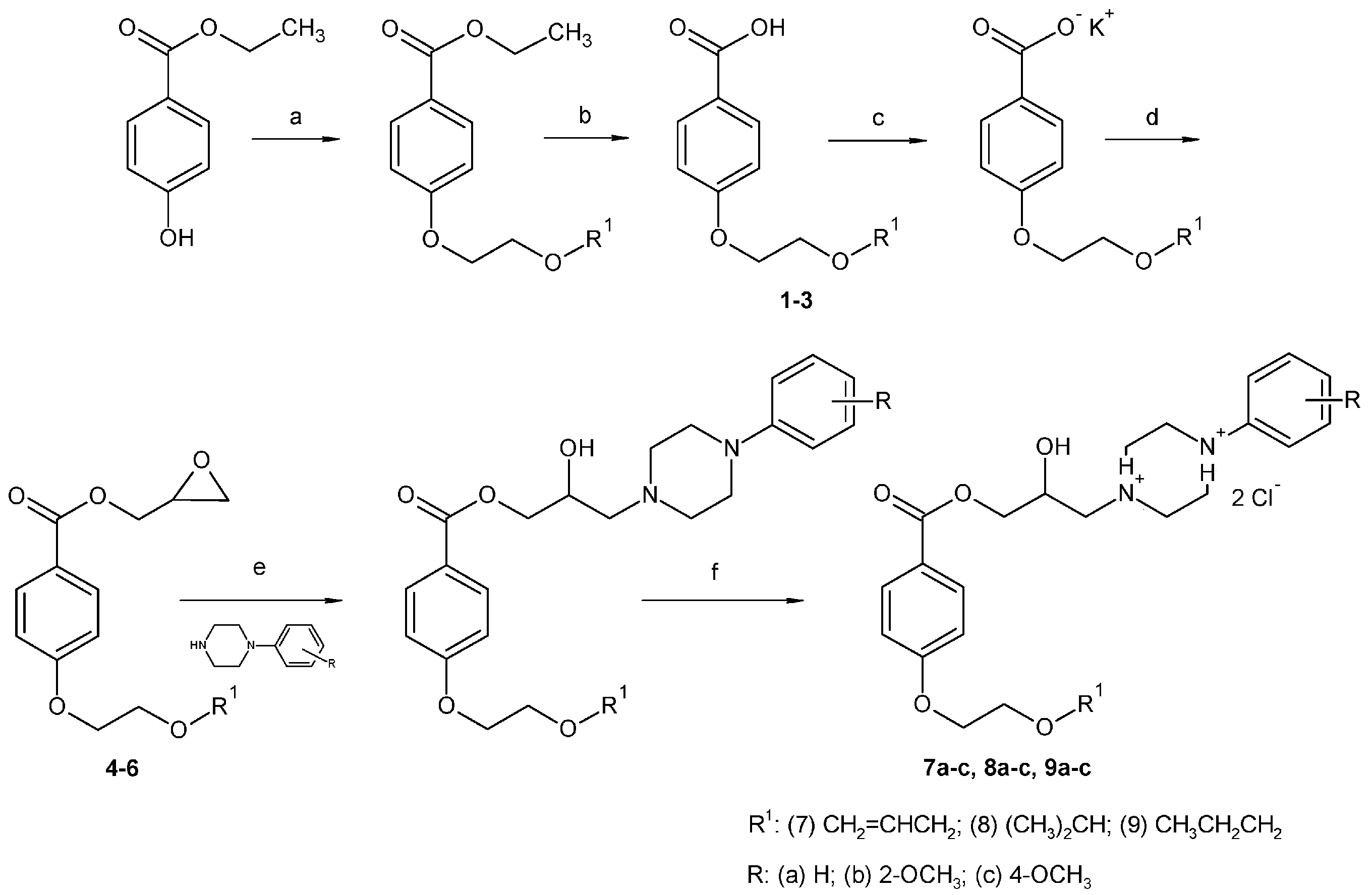
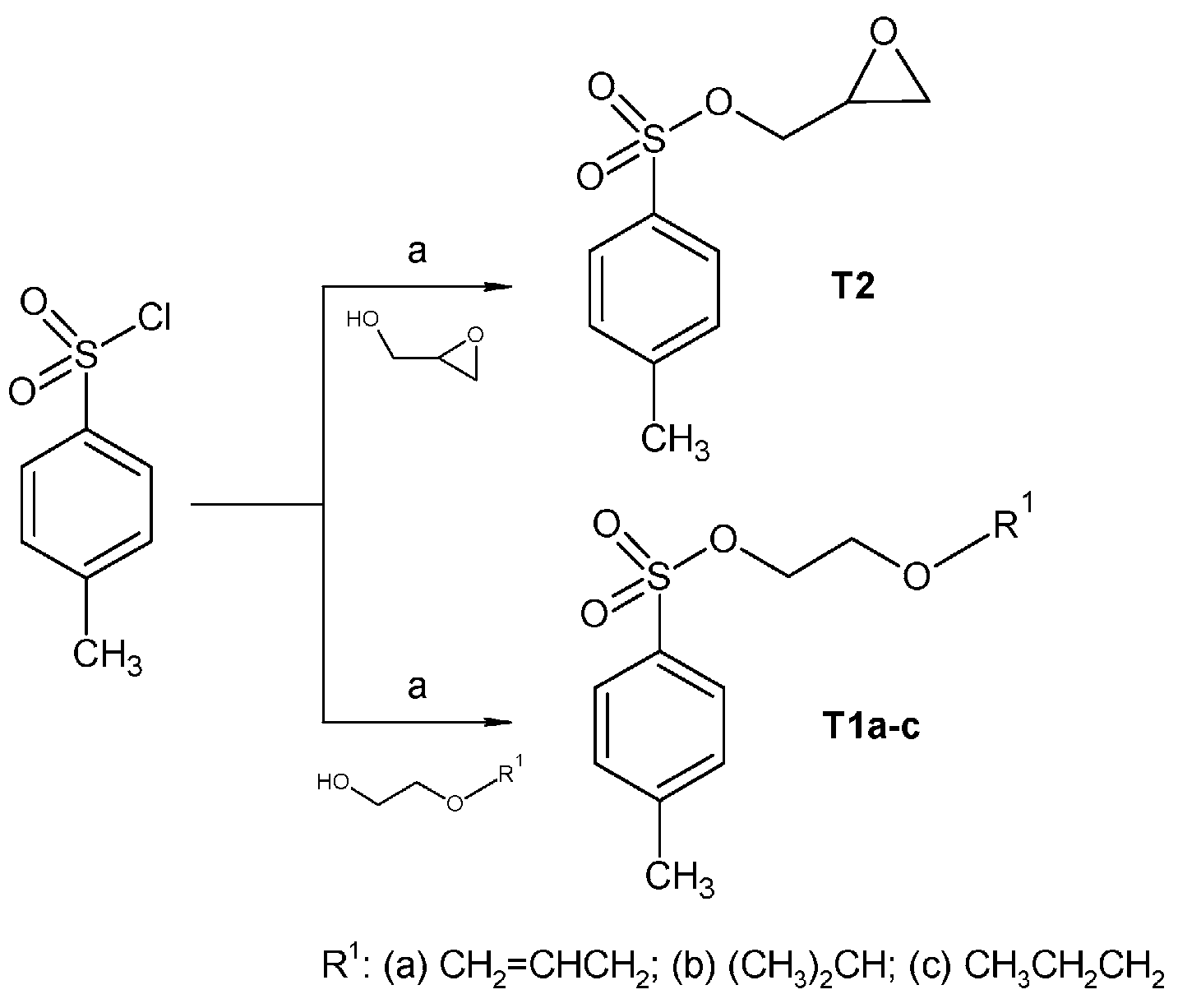
2.1. Chemistry
2.2. Physicochemical Properties
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. Synthesis of Tosylate Intermediates
3.2.2. Synthesis of 4-Alkoxyethoxybenzoic Acid
3.2.3. Synthesis of Oxirane-Intermediates
3.2.4. Synthesis of Final Compounds
3.3. Determination of Physicochemical Parameters
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pedersen, M.E.; Cockcroft, J.R. The vasodilatory Beta-blockers. Curr. Hypertens. Rep. 2007, 9, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Reiter, M.-J. Cardiovascular Drug Class Specifity: β-blockers. Prog. Cardiovasc. Dis. 2004, 47, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Wu, J.R.; Lee, C.H.; Liou, S.F.; Dai, Z.K.; Chen, I.J.; Yeh, J.L. Anti-Hypertension Effect of Vanylidilol: A Phenylaldehyde α/β-Adrenoceptor Blocker with Endothelium-Dependent and K+ Channels Opening-Associated Vasorelaxant Activities. Pharmacology 2004, 70, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Deák, K.; Takács-Novák, K.; Kapás, M.; Vastag, M.; Tihanyi, K.; Noszál, B. Physico-chemical characterization of a novel group of dopamine D3/D2 receptor ligands, potential atypical antipsychotic agents. J. Pharm. Biomed. Anal. 2008, 48, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Tengler, J.; Kapustikova, I.; Stropnicky, O.; Mokry, P.; Oravec, M.; Csollei, J.; Jampilek, J. Synthesis of new (arylcarbonyloxy)aminopropanol derivatives and the determination of their physico-chemical properties. Cent. Eur. J. Chem. 2013, 11, 1757–1767. [Google Scholar] [CrossRef]
- Mälkiä, A.; Murtomäki, L.; Urtti, A.; Kontturi, K. Drug permeation in biomembranes in vitro and in silico prediction and influence of physicochemical properties. Eur. J. Pharm. Sci. 2004, 23, 13–47. [Google Scholar] [CrossRef] [PubMed]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Duchowicz, P.R.; Castro, E.A. QSPR studies on aqueous solubilities of drug-like compounds. Int. J. Mol. Sci. 2009, 10, 2558–2577. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, A.; Waterbeemd, H. Substructure and whole molecule approaches for calculating log P. J. Comput. Aided Mol. Des. 2001, 15, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Tsantili-Kakoulidou, A. Current state of the art in HPLC methodology for lipophilicity assessment of basic drugs. A review. J. Liq. Chromatogr. Relat. Technol. 2008, 31, 79–96. [Google Scholar] [CrossRef]
- Lombardo, F.; Shalaeva, M.Y.; Tupper, K.A.; Gao, F. ElogDoct: A tool for lipophilicity determination in drug discovery. 2. Basic and neutral compounds. J. Med. Chem. 2001, 44, 2490–2497. [Google Scholar] [CrossRef] [PubMed]
- Stella, C.; Galland, A.; Xiangli, L.; Testa, B.; Rudaz, S.; Veuthey, J.L.; Carrupt, P.A. Novel RPLC stationary phases for lipophilicity measurement: Solvatochromic analysis of retention mechanisms for neutral and basic compounds. J. Sep. Sci. 2004, 27, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Reijenga, J.; van Hoof, A.; van Loon, A.; Teunissen, B. Development of methods for the determination of pKa Values. Anal. Chem. Insights 2013, 8, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Kapustikova, I.; Tengler, J. Determination of Acid-Base Dissociation Constant of Selected β-Blockers Using Capillary Zone Electrophoresis, RP-HPLC and 1H-NMR. Chem. Listy 2014, 108, 1073–1079. [Google Scholar]
- Poole, S.K.; Patel, S.; Dehring, K.; Workman, H.; Poole, C.F. Determination of acid dissociation constants by capillary electrophoresis. J. Chromatogr. A 2004, 1037, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.; Woźniakiewicz, M.; Kościelniak, P. Application of capillary electrophoresis in determination of acid dissociation constant values. J. Chromatogr. A 2015, 1377, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Andrasi, M.; Buglyo, P.; Zekany, L.; Gaspar, A. A comparative study of capillary zone electrophoresis and pH-potentiometry for determination of dissociation constants. J. Pharm. Biomed. Anal. 2007, 44, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Bodor, N.; Buchwald, P. Soft drug design: General principles and recent applications. Med. Res. Rev. 2000, 20, 58–101. [Google Scholar] [CrossRef]
- Handzlik, J.; Pertz, H.H.; Görnemann, T.; Jähnichen, S.; Kieć-Kononowicz, K. Search for influence of spatial properties on affinity at α1-adrenoreceptor subtypes for phenylpiperazine derivatives of phenytoin. Bioorg. Med. Chem. Lett. 2010, 20, 6152–6156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pei, Y.; Zhao, J.; Li, Z.; Chen, Y.; Zhuo, K. Formation of Ether-Functionalized Ionic-Liquid-Based Aqueous Two-Phase Systems and Their Application in Separation of Protein and Saccharides. J. Phys. Chem. B 2015, 119, 4471–4478. [Google Scholar] [CrossRef] [PubMed]
- Colley, H.E.; Muthana, M.; Danson, S.J.; Jackson, L.V.; Brett, M.L.; Harrison, J.; Coole, S.F.; Mason, D.P.; Jennings, L.R.; Wong, M.; et al. An Orally Bioavailable, Indole-3-glyoxylamide Based Series of Tubulin Polymerization Inhibitors Showing Tumor Growth Inhibition in a Mouse Xenograft Model of Head and Neck Cancer. J. Med. Chem. 2015, 58, 9309–9333. [Google Scholar] [CrossRef] [PubMed]
- Ayme, J.-F.; Lux, J.; Sauvage, J.-P.; Sour, A. Catenanes Built Around Octahedral Transition-Metal Complexes that Contain Two Intertwined Endocyclic but Non-sterically Hindering Tridentate Ligands. Chem. Eur. J. 2012, 18, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; Amoroso, R.; Bettoni, G.; Fantacuzzi, M.; De Filippis, B.; Giampietro, L.; Maccallini, C.; Paludi, D.; Tricca, M.L. Synthesis and antibacterial evaluation of oxazolidin-2-ones structurally related to linezolid. Farmaco 2004, 59, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Marvanova, P.; Padrtova, T.; Pekarek, T.; Brus, J.; Czernek, J.; Mokry, P.; Humpa, O.; Oravec, M.; Jampilek, J. Synthesis and Characterization of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Propoxybenzoates and Their Hydrochloride Salts. Molecules 2016, 21, 707. [Google Scholar] [CrossRef] [PubMed]
- McAinsh, J.; Cruickshank, J.M. Beta-blockers and central nervous system side effects. Pharmacol. Ther. 1990, 46, 163–197. [Google Scholar] [CrossRef]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Vilar, S.; Chakrabarti, M.; Costanzi, S. Prediciton of passive blood-brain partitioning: Straightforward and effective classification models based on in silico derived physicochemical descriptors. J. Mol. Graph. Model. 2010, 28, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M.; Alavijeh, M.S. Translation CNS medicines research. Drug Discov. Today 2012, 17, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidate. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood-brain barrier penetration. J. Pharm. Sci. 1999, 88, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Hutter, M.C. Prediction of blood–brain barrier permeation using quantum chemically derived information. J. Comput. Aided Mol. Des. 2003, 17, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of compounds are available from authors P. Marvanova, O. Hosik and P. Mokry.

| Comp. | R1 | R | pKa a (exp.) | pKa b (calc.) | pKa c (calc.) |
|---|---|---|---|---|---|
| 7a | allyl | H | 6.47 ± 0.02 | 6.60 | 7.70 |
| 7b | allyl | 2-MeO | 6.88 ± 0.02 | 6.70 | 7.40 |
| 7c | allyl | 4-MeO | 6.49 ± 0.02 | 6.70 | 7.63 |
| 8a | isopropyl | H | 6.49 ± 0.01 | 6.60 | 7.70 |
| 8b | isopropyl | 2-MeO | 6.92 ± 0.01 | 6.70 | 7.40 |
| 8c | isopropyl | 4-MeO | 6.35 ± 0.02 | 6.70 | 7.63 |
| 9a | propyl | H | 6.50 ± 0.02 | 6.60 | 7.70 |
| 9b | propyl | 2-MeO | 6.79 ± 0.02 | 6.70 | 7.40 |
| 9c | propyl | 4-MeO | 6.39 ± 0.01 | 6.70 | 7.63 |

{kind=link}
{kind=link}
| Comp. | R1 | R | log kw7.4 a | log D7.4 b | log D7.4 c | log kw | log P b | log P c |
|---|---|---|---|---|---|---|---|---|
| 7a | allyl | H | 3.97 ± 0.03 | 3.97 | 3.15 | 4.01 ± 0.09 | 4.03 | 3.63 |
| 7b | allyl | 2-MeO | 3.78 ± 0.02 | 3.47 | 3.17 | 3.89 ± 0.08 | 3.65 | 3.47 |
| 7c | allyl | 4-MeO | 3.67 ± 0.02 | 3.40 | 3.04 | 3.71 ± 0.08 | 3.55 | 3.47 |
| 8a | isopropyl | H | 4.21 ± 0.01 | 4.03 | 3.19 | 4.25 ± 0.05 | 4.09 | 3.67 |
| 8b | isopropyl | 2-MeO | 4.03 ± 0.02 | 3.38 | 3.21 | 4.15 ± 0.07 | 3.56 | 3.51 |
| 8c | isopropyl | 4-MeO | 3.90 ± 0.02 | 3.35 | 3.08 | 3.93 ± 0.06 | 3.50 | 3.51 |
| 9a | propyl | H | 4.29 ± 0.03 | 4.01 | 3.30 | 4.34 ± 0.10 | 4.07 | 3.78 |
| 9b | propyl | 2-MeO | 4.11 ± 0.02 | 3.49 | 3.32 | 4.20 ± 0.09 | 3.67 | 3.62 |
| 9c | propyl | 4-MeO | 3.99 ± 0.02 | 3.41 | 3.19 | 4.03 ± 0.09 | 3.56 | 3.62 |
| Comp. | MW a (<500) | nOHNH (<5) | nON (<10) | nRTB (≤10) | PSApH 7.0 b (≤140) | Log BB Class c (Model 1) | Log BB Class c (Model 2) |
|---|---|---|---|---|---|---|---|
| 7a | 440.53 | 3 | 7 | 13 | 72.67 | −0.29 | 0.32 |
| 7b | 470.56 | 3 | 8 | 14 | 81.90 | −0.61 | −0.01 |
| 7c | 470.56 | 3 | 8 | 14 | 81.90 | −0.70 | −0.05 |
| 8a | 442.56 | 3 | 7 | 12 | 72.67 | −0.17 | 0.38 |
| 8b | 472.58 | 3 | 8 | 13 | 81.90 | −0.48 | 0.06 |
| 8c | 472.58 | 3 | 8 | 13 | 81.90 | −0.59 | 0.01 |
| 9a | 442.56 | 3 | 7 | 13 | 72.67 | −0.12 | 0.40 |
| 9b | 472.58 | 3 | 8 | 14 | 81.90 | −0.45 | 0.07 |
| 9c | 472.58 | 3 | 8 | 14 | 81.90 | −0.54 | 0.03 |
| esmolol | 295.37 | 2 | 5 | 10 | 72.37 | −1.37 d | −0.15 d |
| flestolol | 327.35 | 4 | 7 | 9 | 118.26 | −3.52 d | −3.17 d |
| landiolol | 510.60 | 3 | 11 | 14 | 132.40 | −3.96 d | −3.65 d |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marvanova, P.; Padrtova, T.; Odehnalova, K.; Hosik, O.; Oravec, M.; Mokry, P. Synthesis and Determination of Physicochemical Properties of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Alkoxyethoxybenzoates. Molecules 2016, 21, 1682. https://doi.org/10.3390/molecules21121682
Marvanova P, Padrtova T, Odehnalova K, Hosik O, Oravec M, Mokry P. Synthesis and Determination of Physicochemical Properties of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Alkoxyethoxybenzoates. Molecules. 2016; 21(12):1682. https://doi.org/10.3390/molecules21121682
Chicago/Turabian StyleMarvanova, Pavlina, Tereza Padrtova, Klara Odehnalova, Ondrej Hosik, Michal Oravec, and Petr Mokry. 2016. "Synthesis and Determination of Physicochemical Properties of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Alkoxyethoxybenzoates" Molecules 21, no. 12: 1682. https://doi.org/10.3390/molecules21121682