
Design and Synthesis of Vandetanib Derivatives Containing Nitroimidazole Groups as Tyrosine Kinase Inhibitors in Normoxia and Hypoxia

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
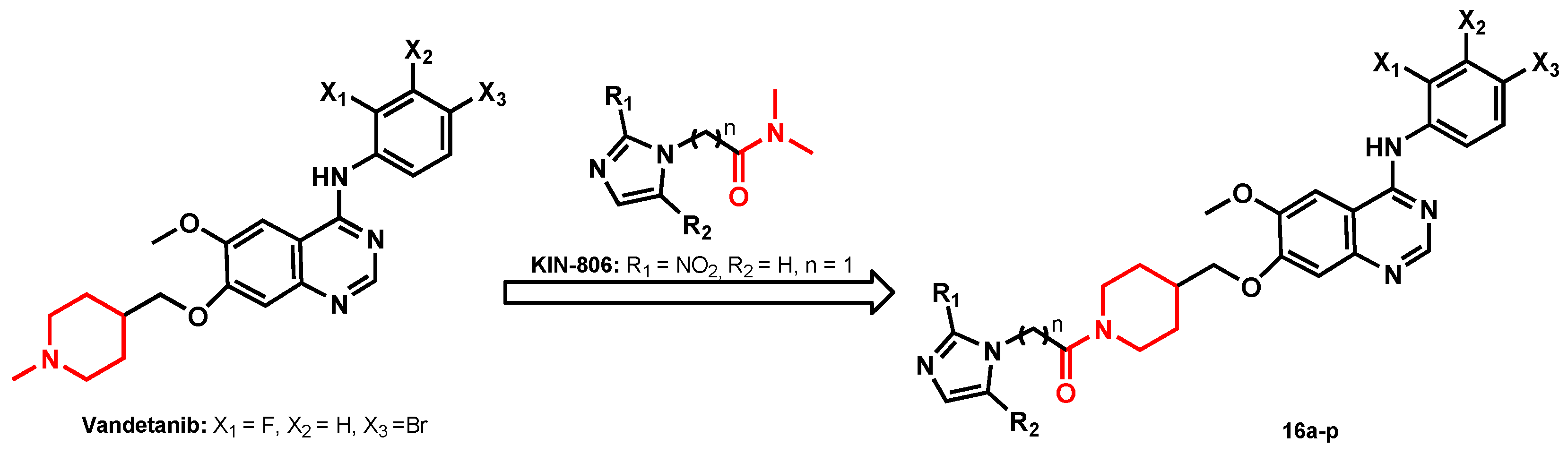
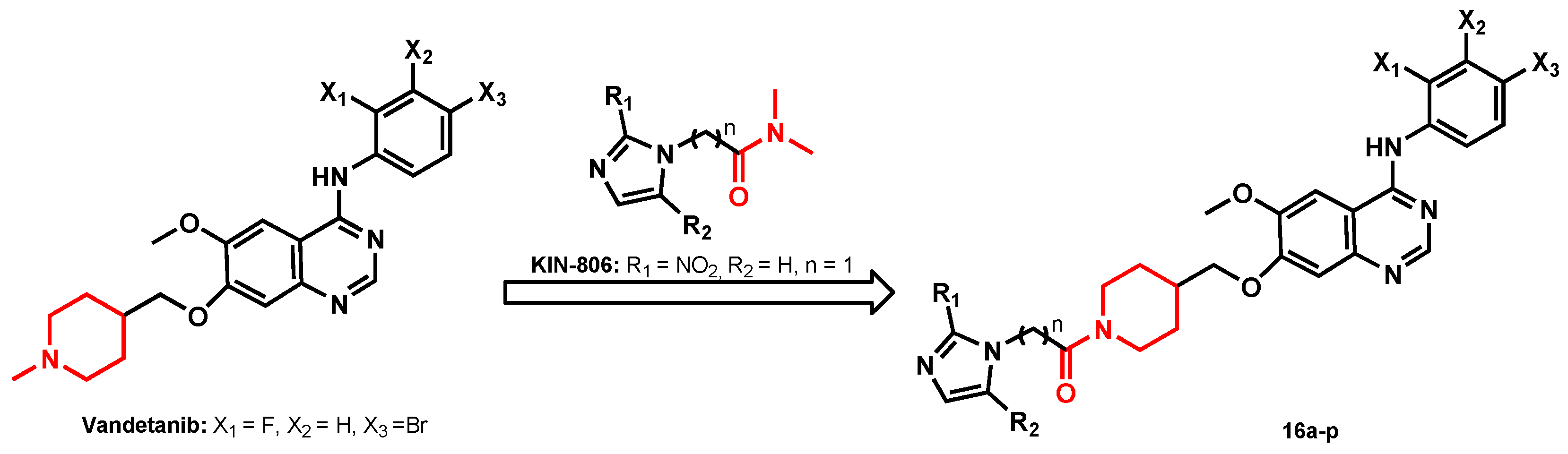
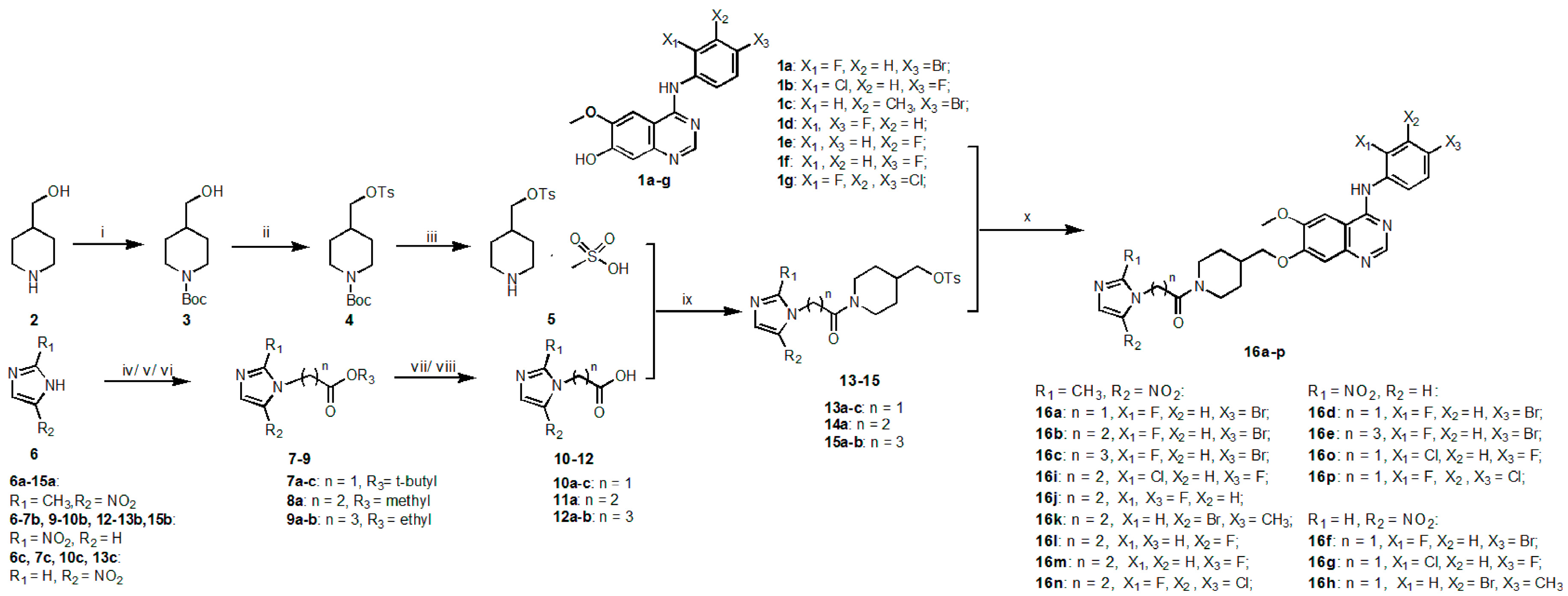
2.1. Chemistry
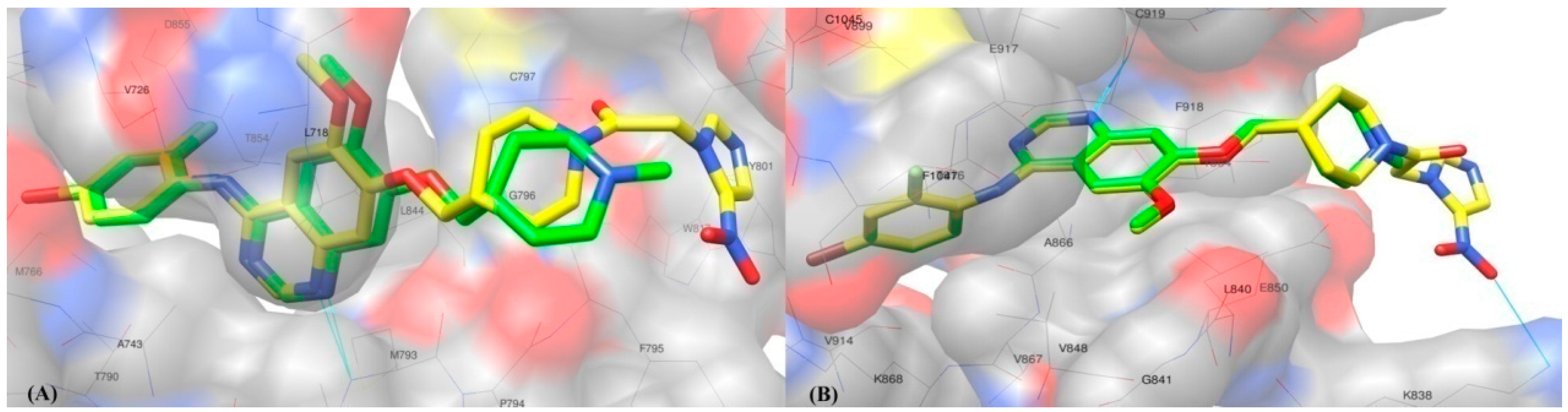
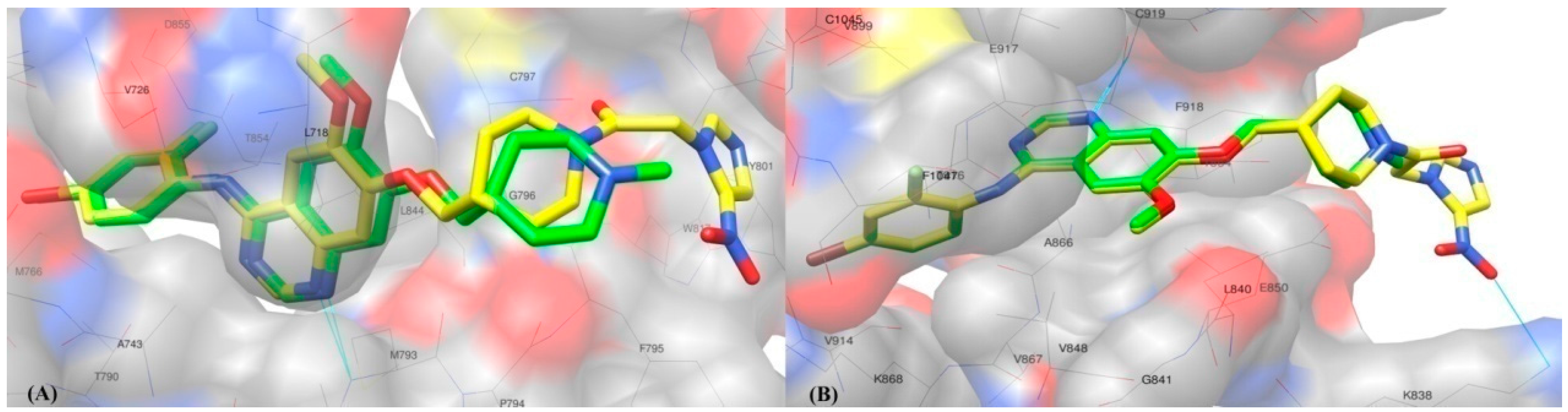
2.2. Molecular Docking Study
2.3. Biological Evaluation
2.3.1. In Vitro EGFR Inhibitory Activity
2.3.2. In Vitro Anti-Proliferative Activity Assays in Normoxia and Hypoxia
2.3.3. In Vitro VEGF/EGF Gene Expression Inhibitory Activity
3. Experimental Section
3.1. Materials and Methods
3.1.1. General Methods
3.1.2. Synthesis
General Synthetic Procedure of tert-Butyl 2-(Substituted-nitroimidazol-1-yl)acetate (7a–7c)
Methyl 3-(2-Methyl-5-nitroimidazol-1-yl)propionate (8a)
General Synthetic Procedure of Ethyl 4-(Substituted-nitroimidazol-1-yl)butyrate (9a–b)
General Synthetic Procedure of 2-(Substituted-nitroimidazol-1-yl)acetic acid (10a–c)
General Synthetic Procedure of 3-(Substituted-nitroimidazol-1-yl)propionic acid (11a) and 4-(Substituted-nitroimidazol-1-yl)butyric acid (12a–b)
General Synthetic Procedure of p-Toluenesulfonate Intermediates (13a–c, 14a and 15a–b)
General Synthetic Procedure of Target Compounds (16a–p)
3.2. Molecular Docking Study
3.3. Pharmacology
3.3.1. In Vitro EGFR Inhibitory Assay
3.3.2. In Vitro Anti-Proliferative Activity Assays in Normoxia and Hypoxia
3.3.3. In Vitro VEGF/EGF Gene Expression Inhibition Assays
4. Conclusions
Acknowledgment
Author Contributions
Conflicts of Interest
References
- Van Putten, L.M.; Kallman, R.F. Oxygenation status of a transplantable tumor during fractionated radiation therapy. J. Natl. Cancer Inst. 1968, 40, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.L.; Jin, Y.Z.; Shen, Y. Tumor Therapy Sensitizers, 1st ed.; Shanghai Science and Technical Literature Press: Shanghai, China, 2002; pp. 12–14. [Google Scholar]
- Toma-Daşu, I.; Daşu, A.; Karlsson, M. The relationship between temporal variation of hypoxia, polarographic measurements and predictions of tumour response to radiation. Phys. Med. Biol. 2004, 49, 4463–4475. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Dach, G.U.; Gleadle, J.M. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef] [PubMed]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Niki, T.; Goto, A.; Ota, S.; Morikawa, T.; Nakamura, Y.; Ohara, E.; Ishikawa, S.; Aburatani, H.; Nakajima, J.; Fukayama, M. Hypoxia increases the motility of lung adenocarcinoma cell line A549 via activation of the epidermal growth factor receptor pathway. Cancer Sci. 2007, 98, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Lancini, G.C.; Lazzari, E.; Arioli, V.; Bellani, P. Synthesis relationship between structure and activity of 2-nitroimidazole derivatives. J. Med. Chem. 1969, 12, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Chemical radiosensitizers for use in radiotherapy. Clin. Oncol. 2007, 19, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.E. Hypoxic cell sensitizers for radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1978, 4, 135–141. [Google Scholar] [CrossRef]
- Adams, G.E. Accomplishments, problems and prospects: A conference summary. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 805–808. [Google Scholar] [CrossRef]
- Adams, G.E.; Stratford, I.J.; Wallace, R.G.; Wardman, P.; Watts, M.E. Toxicity of nitro compounds toward hypoxic mammalian cells in vitro: Dependence on reduction potential. J. Natl. Cancer Inst. 1980, 64, 555–560. [Google Scholar] [PubMed]
- Kumar, P.; Shustov, G.; Liang, H.; Khlebnikov, V.; Zheng, W.Z.; Yang, X.H.; Cheeseman, C.; Wiebe, L.I. Design, synthesis, and preliminary biological evaluation of 6-O-glucose-azomycin adducts for diagnosis and therapy of hypoxic tumors. J. Med. Chem. 2012, 55, 6033–6046. [Google Scholar] [CrossRef] [PubMed]
- Ziemer, L.S.; Evans, S.M.; Kachur, A.V.; Shuman, A.L.; Cardi, C.A.; Jenkins, W.T.; Karp, J.S.; Alavi, A.; Dolbier, W.R., Jr.; Koch, C.J. Noninvasive imaging of tumor hypoxia using the 2-nitroimidazole 18F-EF5 in rats. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Melo, T.; Duncan, J.; Ballinger, J.R. BRU59-21, a second-generation 99mTc-labled 2-nitroimidazole for imaging hypoxia in tumors. J. Nucl. Med. 2000, 41, 169–176. [Google Scholar]
- Hoigebazar, L.; Jeong, J.M.; Choi, S.Y.; Choi, J.Y.; Shetty, D.; Lee, Y.S.; Lee, D.S.; Chung, J.Y.; Lee, M.C.; Chung, Y.K. Synthesis and characterization of citroimidazole derivatives for 68Ga-labeling and testing in tumor xenografted mice. J. Med. Chem. 2010, 53, 6378–6385. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Jiao, H.; Kaizerman, J.; Stanton, T.; Evans, J.W.; Lan, L.; Lorente, G.; Banica, M.; Jung, D.; Wang, J.; et al. Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J. Med. Chem. 2008, 51, 2412–2420. [Google Scholar] [CrossRef] [PubMed]
- Inomata, T.; Ogawa, Y.; Itoh, S.; Kariya, S.; Hamada, N.; Nishioka, A.; Yoshida, S.; Nagasawa, H.; Hori, H.; Inayama, S. Lung metastasis suppression of the bifunctional new radiosensitizer KIN-806. Int. J. Mol. Med. 1999, 4, 257–317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.N. Modern Drug Design, 1st ed.; China Medical Science Press: Beijing, China, 2005; pp. 240–242, 586–587. [Google Scholar]
- Hennequin, L.F.; Stokes, E.S.E.; Thomas, A.P.; Johnstone, C.; Ple, P.A.; Ogilvie, D.J.; Dukes, M.; Wedge, S.R.; Kendrew, J.; Curwen, J.O. Novel 4-anilinoquinazolines with C-7 basic side chains: Design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J. Med. Chem. 2002, 45, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, I.W.; Cox, B.G.; Meyrick, B. Kinetics and mechanism of N-Boc cleavage: Evidence of a second-order dependence upon acid concentration. J. Org. Chem. 2010, 75, 8117–8125. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hu, Q.Q.; Yu, B.; Li, Y.L.; Xiong, D.S. Anti-tumor activity of novel 4-aminobenzene quinazoline tyrosine kinase inhibitors. Drugs Clin. 2014, 29, 969–973. [Google Scholar]
- Chen, J.W.; Hu, X.J.; Lin, Q.; Mao, J.J.; Wang, L.; Bo, M.J.; Huang, M.S.; Shan, H. Effect of CoCl2 on expression of HIF-lα and VEGF, and tumor invasion in rat RH35 hepatoma cells. J. New. Med. 2014, 45, 790–795. [Google Scholar]
- Yun, C.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Zhu, S.J.; Ma, X.D.; Qiu, N.; Peng, P.; Sheng, R.; Hu, Y.Z. Design, synthesis and biological evaluation of 6-(nitroimidazole-1H-alkyloxyl)-4-anilinoquinazolines as efficient EGFR inhibitors exerting cytotoxic effects both under normoxia and hypoxia. Eur. J. Med. Chem. 2015, 89, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.


{kind=link}
{kind=link}
{kind=link}
| Compound | Substituent | IC50 (μmol/L) | ||||||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | n | X1 | X2 | X3 | A431 | H1975 | |
| 16a | CH3 | NO2 | 1 | F | H | Br | 1.82 | 58.46 |
| 16b | CH3 | NO2 | 2 | F | H | Br | 3.42 | 49.52 |
| 16c | CH3 | NO2 | 3 | F | H | Br | 2.03 | 53.42 |
| 16d | NO2 | H | 1 | F | H | Br | 2.10 | 60.12 |
| 16e | NO2 | H | 3 | F | H | Br | 4.81 | 40.15 |
| 16f | H | NO2 | 1 | F | H | Br | 1.64 | 60.01 |
| 16g | H | NO2 | 1 | Cl | H | F | 39.76 | 71.80 |
| 16h | H | NO2 | 1 | H | Br | CH3 | 12.34 | 78.25 |
| 16i | CH3 | NO2 | 2 | Cl | H | F | 15.14 | 78.82 |
| 16j | CH3 | NO2 | 2 | H | F | F | 65.42 | 87.90 |
| 16k | CH3 | NO2 | 2 | H | Br | CH3 | 17.95 | 74.84 |
| 16l | CH3 | NO2 | 2 | H | F | H | 78.94 | >100 |
| 16m | CH3 | NO2 | 2 | H | H | F | 66.75 | >100 |
| 16n | CH3 | NO2 | 2 | F | Cl | Cl | 6.75 | 46.22 |
| 16o | NO2 | H | 1 | Cl | H | F | 24.84 | 58.55 |
| 16p | NO2 | H | 1 | F | Cl | Cl | 9.45 | 37.85 |
| Vadetanib | - | - | - | F | H | Br | 0.85 | 4.81 |
| Compound | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vandet-anib | 16a | 16b | 16c | 16d | 16e | 16f | 16h | 16i | 16n | 16p | |||
| 5 μM inhibition ratio (%) | A549 | Normoxia | 37.30 | 40.43 | 37.32 | 43.78 | 46.74 | 66.09 | 12.02 | 33.17 | 38.76 | 42.23 | 55.84 |
| Hypoxia | 40.82 | 46.64 | 57.95 | 71.04 | 41.33 | 85.70 | 52.95 | 65.81 | 50.29 | 56.11 | 80.33 | ||
| H446 | Normoxia | 27.09 | 47.07 | 41.56 | 35.36 | 45.57 | 51.87 | 30.00 | 39.20 | 36.92 | 31.53 | 49.60 | |
| Hypoxia | 43.01 | 44.81 | 58.84 | 48.42 | 60.77 | 68.21 | 45.74 | 56.60 | 53.64 | 57.32 | 70.19 | ||
| Hela b | Hypoxia | 36.43 | 10.83 | 26.21 | 21.40 | 18.19 | 23.46 | 8.00 | 13.66 | 18.93 | 10.53 | 36.24 | |
| 0.5 μM inhibition ratio (%) | A549 | Normoxia | 11.22 | 4.02 | 6.81 | 15.24 | 11.20 | 24.43 | 0.52 | 15.42 | 7.04 | 12.75 | 22.67 |
| Hypoxia | 29.95 | 58.08 | 69.30 | 69.93 | 17.62 | 50.97 | 62.01 | 25.70 | 30.87 | 47.96 | 47.13 | ||
| H446 | Normoxia | 12.53 | 16.95 | 8.97 | 11.44 | 17.92 | 22.31 | 3.69 | 28.79 | 12.79 | 15.21 | 26.04 | |
| Hypoxia | 37.41 | 42.33 | 62.94 | 62.52 | 53.82 | 43.74 | 59.86 | 36.06 | 47.72 | 52.92 | 42.98 | ||
| Hela b | Hypoxia | 14.78 | 8.09 | 15.91 | 21.01 | 15.48 | 18.47 | 11.59 | <0 | 10.54 | 12.44 | 18.55 | |
| Compound | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Control b,c | Vandetanib | 16a | 16b | 16c | 16d | 16e | 16f | ||
| VEGF gene expression level | Normoxia | 1.0000 | 0.6457 | 0.1070 | 0.1387 | 0.0949 | 0.0606 | 0.1866 | 0.0706 |
| Hypoxia a | 1.0000 | 0.2457 | 0.0009 | 0.0009 | 0.0007 | 0.0008 | 0.0006 | 0.0002 | |
| EGF gene expression level | Normoxia | 1.0000 | 0.3440 | 0.5260 | 0.9732 | 0.5471 | 0.4366 | 1.1758 | 0.5614 |
| Hypoxia a | 1.0000 | 5.4907 | 2.5398 | 2.8108 | 1.7447 | 1.9433 | 2.4923 | 0.3199 | |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, H.; Li, D.; Yang, X.; Shang, H.; Fan, S.; Li, Y.; Song, D. Design and Synthesis of Vandetanib Derivatives Containing Nitroimidazole Groups as Tyrosine Kinase Inhibitors in Normoxia and Hypoxia. Molecules 2016, 21, 1693. https://doi.org/10.3390/molecules21121693
Wei H, Li D, Yang X, Shang H, Fan S, Li Y, Song D. Design and Synthesis of Vandetanib Derivatives Containing Nitroimidazole Groups as Tyrosine Kinase Inhibitors in Normoxia and Hypoxia. Molecules. 2016; 21(12):1693. https://doi.org/10.3390/molecules21121693
Chicago/Turabian StyleWei, Huiqiang, Deguan Li, Xiangbo Yang, Haihua Shang, Saijun Fan, Yiliang Li, and Dan Song. 2016. "Design and Synthesis of Vandetanib Derivatives Containing Nitroimidazole Groups as Tyrosine Kinase Inhibitors in Normoxia and Hypoxia" Molecules 21, no. 12: 1693. https://doi.org/10.3390/molecules21121693
APA StyleWei, H., Li, D., Yang, X., Shang, H., Fan, S., Li, Y., & Song, D. (2016). Design and Synthesis of Vandetanib Derivatives Containing Nitroimidazole Groups as Tyrosine Kinase Inhibitors in Normoxia and Hypoxia. Molecules, 21(12), 1693. https://doi.org/10.3390/molecules21121693