
Synthesis of 2-Alkenyl-2H-indazoles from 2-(2-Carbonylmethyl)-2H-indazoles

Abstract
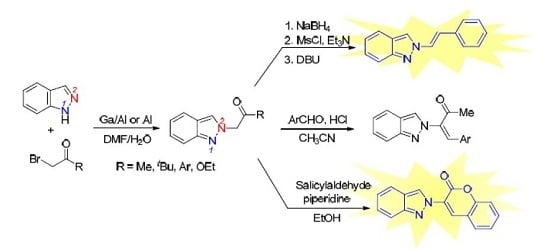
:
1. Introduction
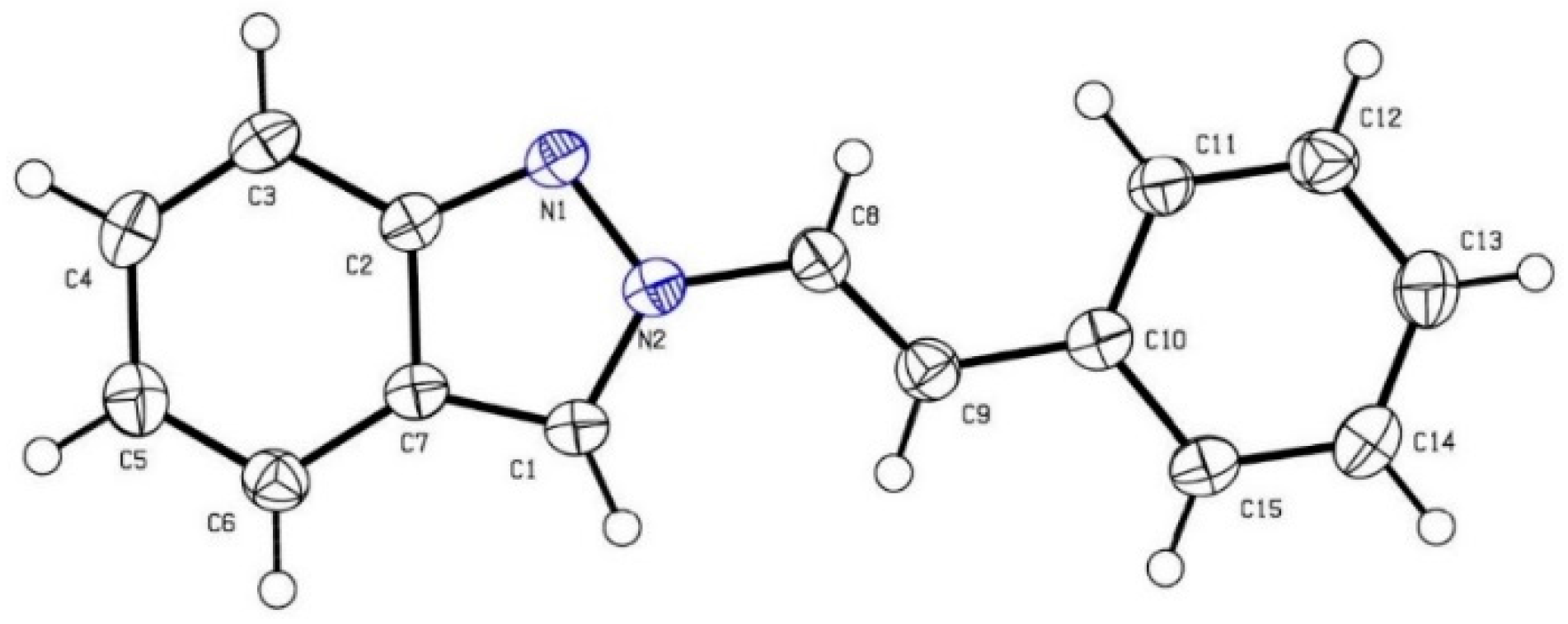
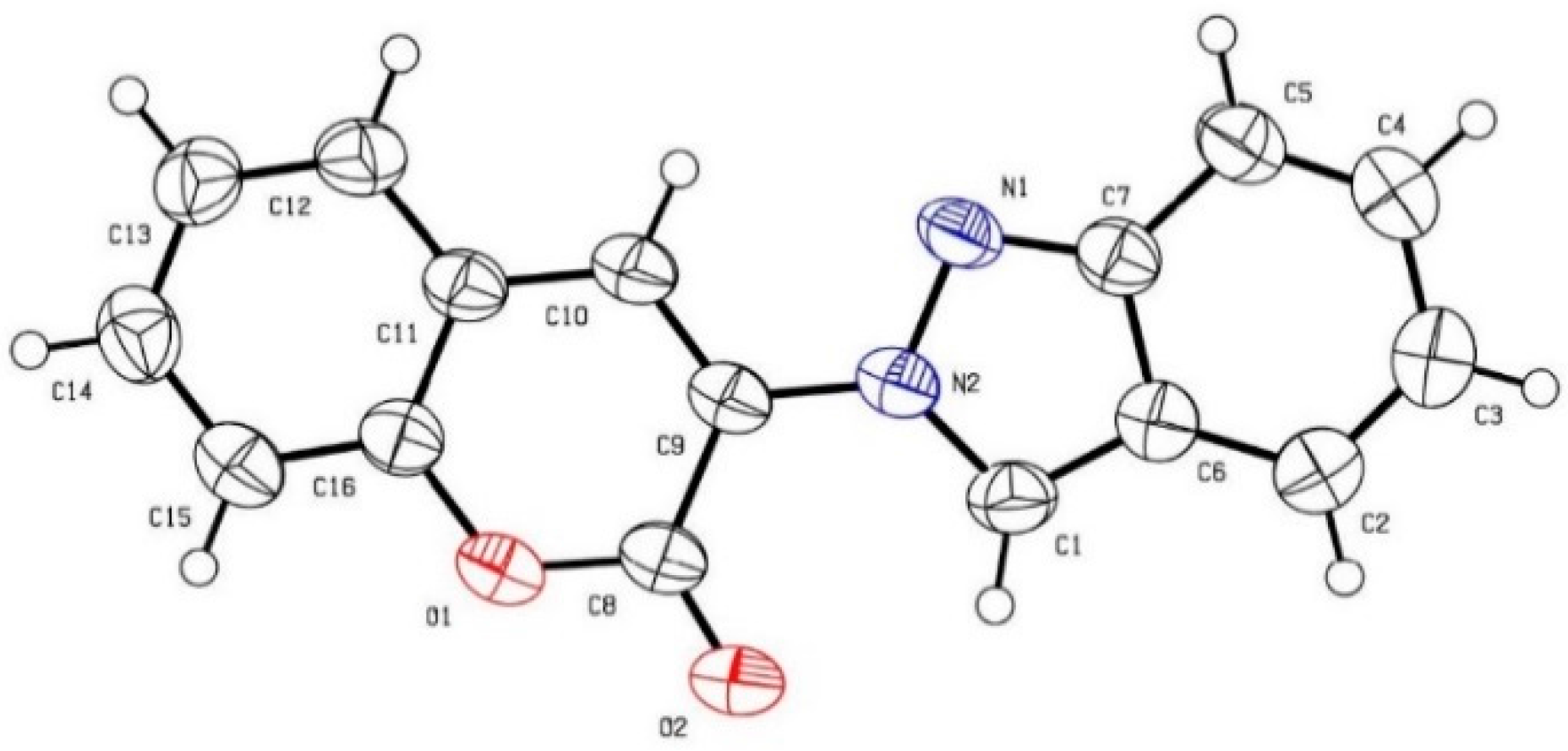
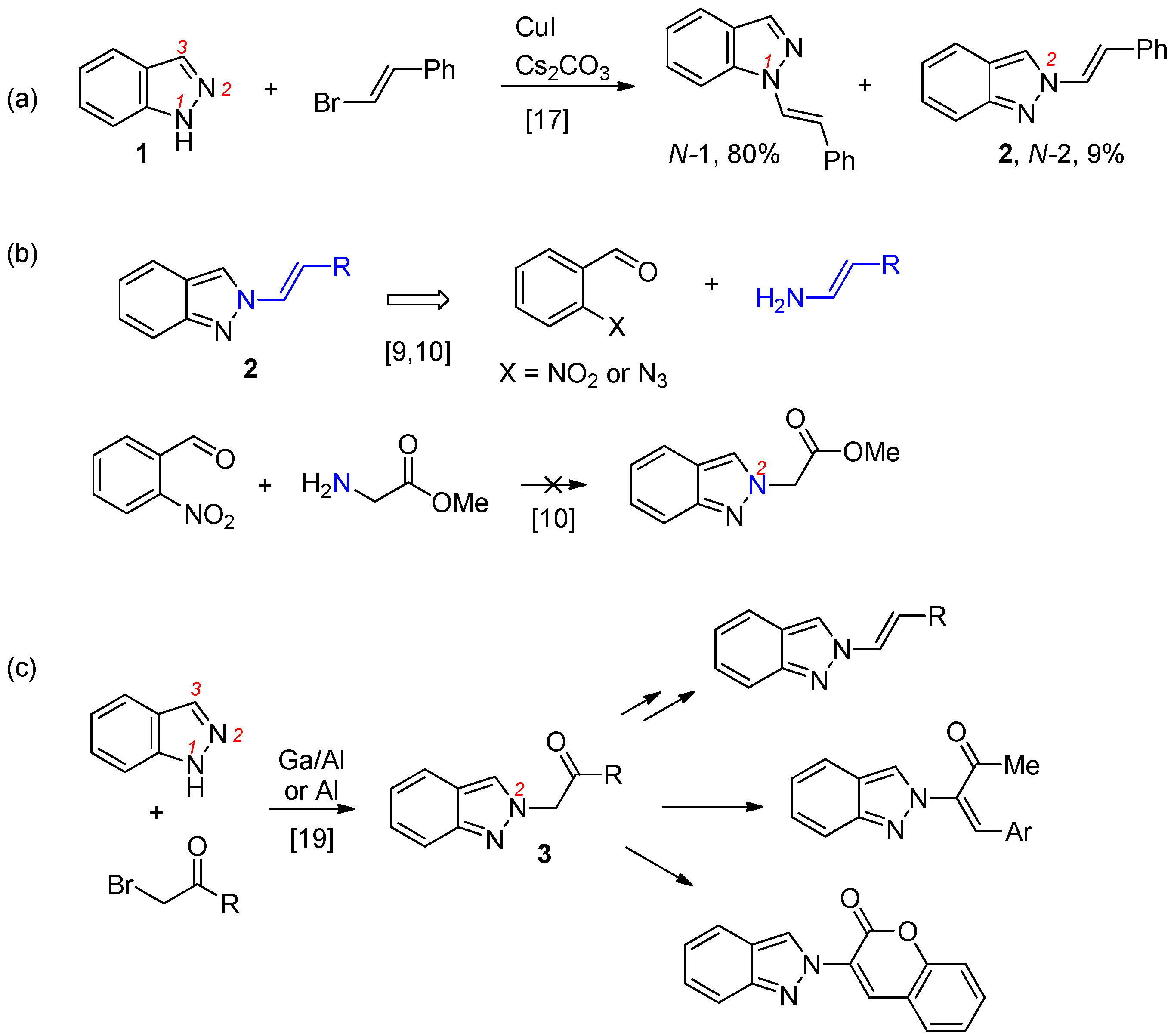
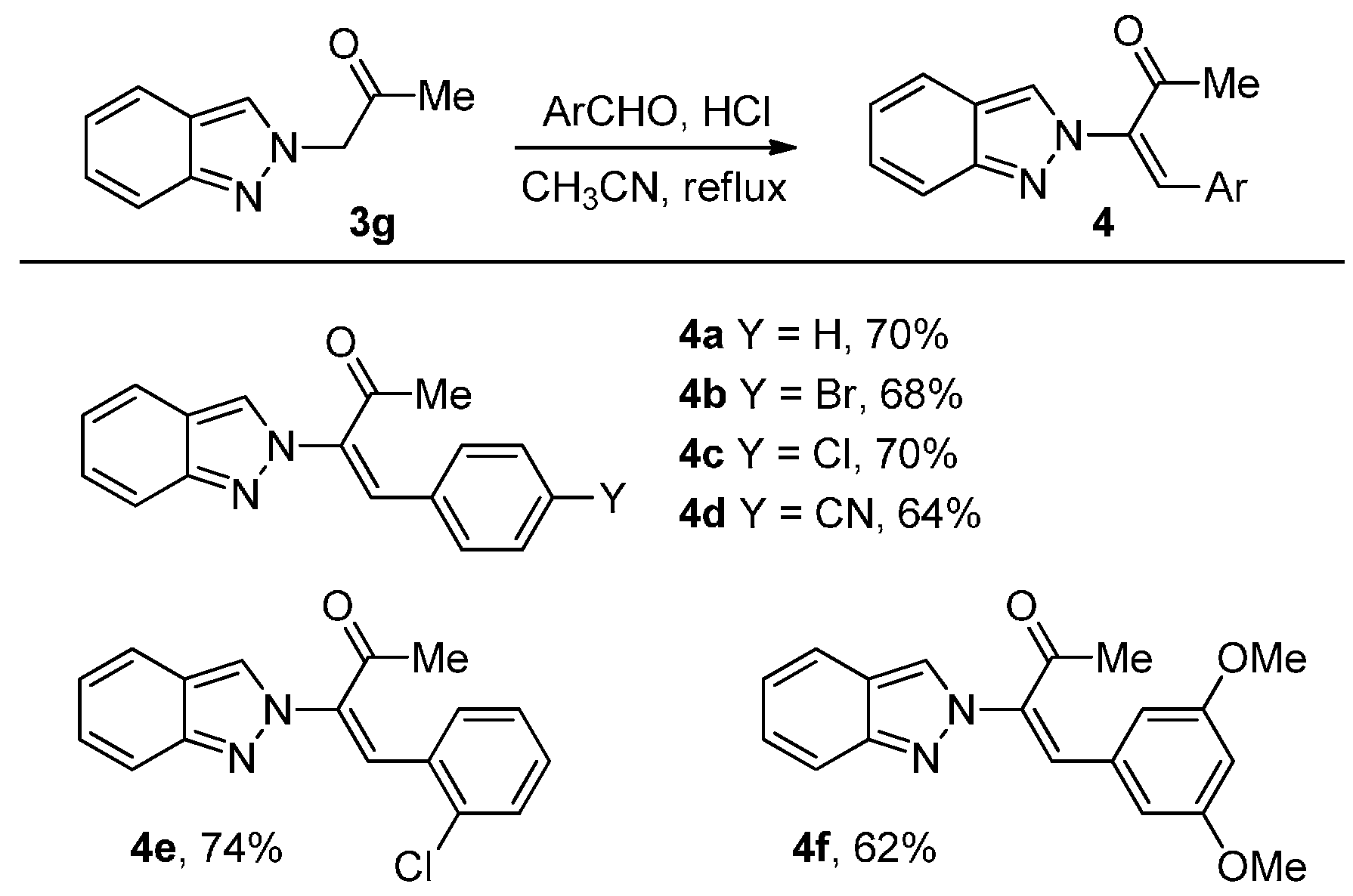
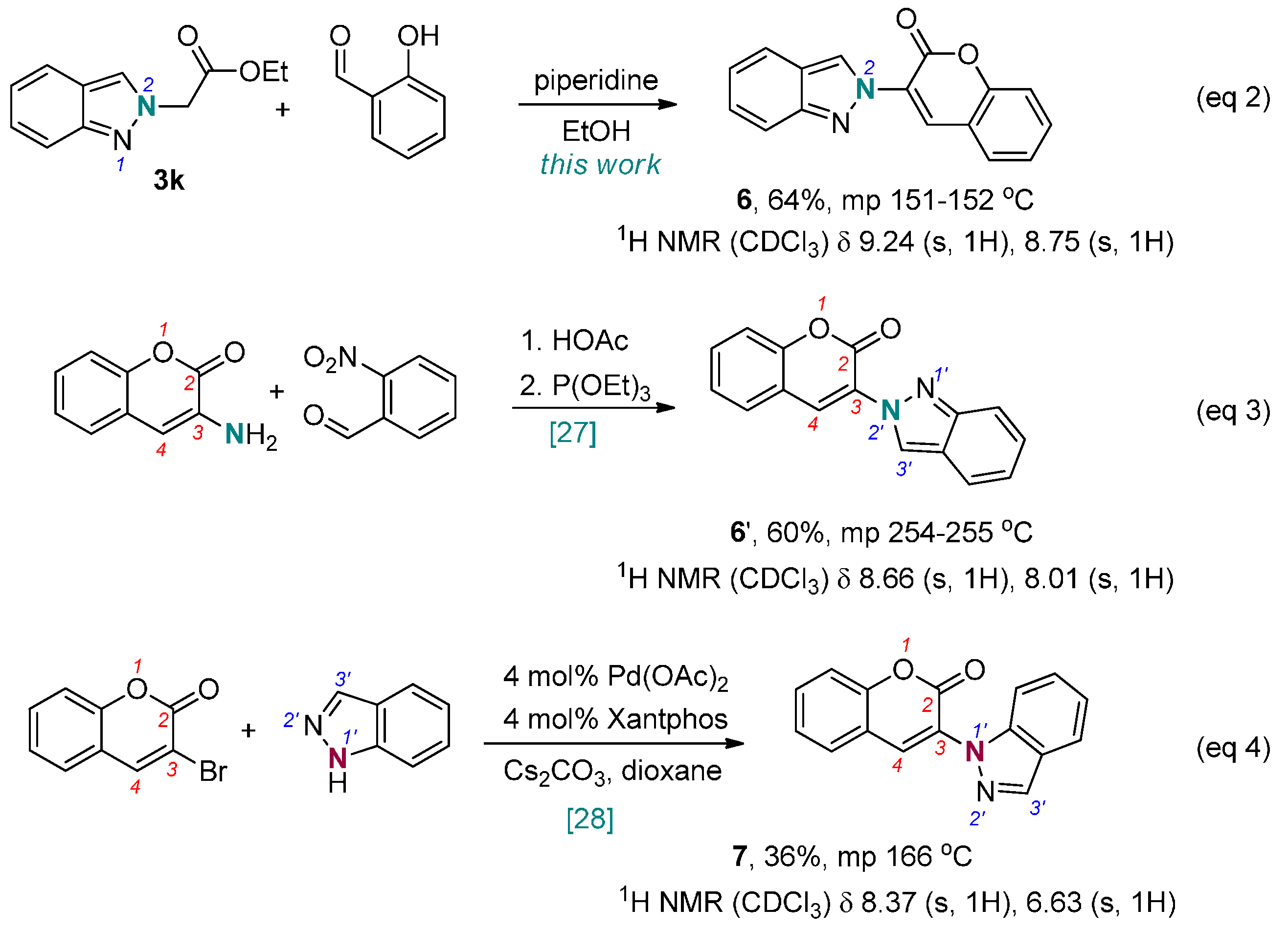
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of 2-Alkenyl-2H-indazoles 2 (Route A)
3.2.2. General Procedure for the Synthesis of 2-Alkenyl-2H-indazoles 2 (Route B)
3.2.3. General procedure for the synthesis of 2-alkenyl-2H-indazoles 4:
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Young, D.W. Heterocyclic Chemistry; Longman group Ltd.: London, UK, 1975. [Google Scholar]
- Katritzky, A.R.; Rees, C.W. Comprehensive Heterocyclic Chemistry; Pergamon Press: New York, NY, USA, 1984. [Google Scholar]
- Nekrasov, D.D. Biological activity of 5- and 6-membered azaheterocycles and their synthesis from 5-aryl-2,3-dihydrofuran-2,3-diones. Chem. Heterocycl. Compd. 2001, 37, 263–275. [Google Scholar] [CrossRef]
- Balaban, A.T.; Oniciu, D.C.; Katritzky, A.R. Aromaticity as a cornerstone of heterocyclic chemistry. Chem. Rev. 2004, 104, 2777–2812. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Beutler, A.; Snovydovych, B. Recent advances in the chemistry of indazoles. Eur. J. Org. Chem. 2008, 24, 4073–4095. [Google Scholar] [CrossRef]
- Haddadin, M.J.; Conrad, W.E.; Kurth, M.J. The Davis-Beirut reaction: A novel entry into 2H-indazoles and indazolones. Recent biological activity of indazoles. Mini-Rev. Med. Chem. 2012, 12, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Thangadurai, A.; Minu, M.; Wakode, S.; Agrawal, S.; Narasimhan, B. Indazole: A medicinally important heterocyclic moiety. Med. Chem. Res. 2012, 21, 1509–1523. [Google Scholar] [CrossRef]
- Ali, N.A.S.; Dar, B.A.; Pradhan, V.; Farooqui, M. Chemistry and biology of indoles and indazoles: A mini-review. Mini-Rev. Med. Chem. 2013, 13, 1792–1800. [Google Scholar] [PubMed]
- Hu, J.; Cheng, Y.; Yang, Y.; Rao, Y. A general and efficient approach to 2H-indazoles and 1H-pyrazoles through copper-catalyzed intramolecular N-N bond formation under mild conditions. Chem. Commun. 2011, 47, 10133–10135. [Google Scholar] [CrossRef] [PubMed]
- Genung, N.E.; Wei, L.; Aspnes, G.E. Regioselective synthesis of 2H-indazoles using a mild, one-pot condensation-Cadogan reductive cyclization. Org. Lett. 2014, 16, 3114–3117. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wu, C.; Larock, R.C.; Shi, F. Synthesis of 2H-indazoles by the [3+2] dipolar cycloaddition of sydnones with arynes. J. Org. Chem. 2011, 76, 8840–8851. [Google Scholar] [CrossRef] [PubMed]
- Haag, B.; Peng, Z.; Knochel, P. Preparation of polyfunctional indazoles and heteroarylazo compounds using highly functionalized zinc reagents. Org. Lett. 2009, 11, 4270–4273. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Yee, N.K. A novel synthesis of 2-aryl-2H-indazoles via a palladium-catalyzed intramolecular amination reaction. Org. Lett. 2000, 2, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, P.; Wang, L. Direct access to acylated azobenzenes via Pd-catalyzed C-H functionalization and further transformation into an indazole backbone. Org. Lett. 2013, 15, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Bergman, R.G.; Lavis, L.D.; Ellman, J.A. Rhodium(III)-catalyzed indazole synthesis by C-H bond functionalization and cyclative capture. J. Am. Chem. Soc. 2013, 135, 7122–7125. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, P.; Zhao, Q.; Wang, L. Unprecedented ortho-acylation of azoxybenzenes with α-oxocarboxylic acids by Pd-catalyzed C-H activation and decarboxylation. Chem. Commun. 2013, 49, 9170–9172. [Google Scholar] [CrossRef] [PubMed]
- Taillefer, M.; Ouali, A.; Renard, B.; Spindler, J.F. Mild copper-catalyzed vinylation reactions of azoles and phenols with vinyl bromides. Chem. Eur. J. 2006, 12, 5301–5313. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.W.; Moreno, D.A.; Suiter, N.; Clark, C.T.; Kim, G. Selective synthesis of 1-functionalized-alkyl-1H-indazoles. Org. Lett. 2009, 11, 5054–5057. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Liu, H.-J.; Lin, W.-C.; Kuo, C.-K.; Chuang, T.-H. Regioselective synthesis of 2H-indazoles through Ga/Al- and Al-mediated direct alkylation reactions of indazoles. Org. Biomol. Chem. 2015, 13, 11376–11381. [Google Scholar] [CrossRef] [PubMed]
- The Structure Has Been Deposited with the Cambridge Crystallographic Data Centre (2a: CCDC 1442986). These Data can be Obtained Free of Charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/ data_request/cif (accessed on 17 December 2015).
- León, L.G.; Carballo, R.M.; Vega-Hernández, M.C.; Miranda, P.O.; Martín, V.S.; Padrón, J.I.; Padrón, J.M. β’-Hydroxy-α,β-unsaturated ketones: A new pharmacophore for the design of anticancer drugs. Part 2. Chem. Med. Chem. 2008, 3, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Base promoted isomerization of the indene-linked 2H-indazole 5 was observed in a solution of 5 (0.2 mmol) in triethylamine (1 mL) at ambient temperature for 10 min to form 5′ in quantitative yield. A similar observation also see: Rosocha, G.; Batey, R.A. Synthesis of 2-bromo-1-aryl-1H-indenes via a Ag(I) promoted domino 2π-electrocyclic ring-opening/4π-electrocyclization reaction of 1,2-diaryl substituted gem-dibromocyclopropanes. Tetrahedron 2013, 69, 8758–8768. [Google Scholar]
![Molecules 21 00238 i012]()
- Finke, J.H.; Richter, C.; Gothsch, T.; Kwade, A.; Buttgenbach, S.; Müller-Goymann, C.C. Coumarin 6 as a fluorescent model drug: How to identify properties of lipid colloidal drug delivery systems via fluorescence spectroscopy? Eur. J. Lipid Sci. Technol. 2014, 116, 1234–1246. [Google Scholar] [CrossRef]
- Long, L.; Zhou, L.; Wang, L.; Meng, S.; Gong, A.; Du, F.; Zhang, C. A coumarin-based fluorescent probe for biological thiols and its application for living cell imaging. Org. Biomol. Chem. 2013, 11, 8214–8220. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Li, X.; Zhang, D.; Meng, S.; Zhang, J.; Sun, X.; Zhang, C.; Zhou, L.; Wang, L. Amino-coumarin based fluorescence ratiometric sensors for acidic pH and their application for living cells imaging. RSC Adv. 2013, 3, 12204–12209. [Google Scholar] [CrossRef]
- Jung, H.S.; Kwon, P.S.; Lee, J.W.; Kim, J.I.; Hong, C.S.; Kim, J.W.; Yan, S.; Lee, J.Y.; Lee, J.H.; Joo, T. Coumarin-derived Cu2+-selective fluorescence sensor: Synthesis, mechanisms, and applications in living cells. J. Am. Chem. Soc. 2009, 131, 2008–2012. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.R.; Darbarwar, M. Synthesis of 3-(2H-indazol-2-yl)-2H[1]-benzopyran-2-ones. Synth. Commun. 1992, 22, 1713–1722. [Google Scholar] [CrossRef]
- Soussi, M.A.; Audisio, D.; Messaoudi, S.; Provot, O.; Brion, J.-D.; Alami, M. Palladium-catalyzed coupling of 3-halo-substituted coumarins, chromenes, and quinolones with various nitrogen-containing nucleophiles. Eur. J. Org. Chem. 2011, 26, 5077–5088. [Google Scholar] [CrossRef]
- The Structure Has Been Deposited with the Cambridge Crystallographic Data Centre (6: CCDC 1442987). These Data can be Obtained Free of Charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/ data_request/cif (accessed on 17 December 2015).

- Sample Availability: Samples of the compounds. 2a–j, 4a–f, 5 and 6 are available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
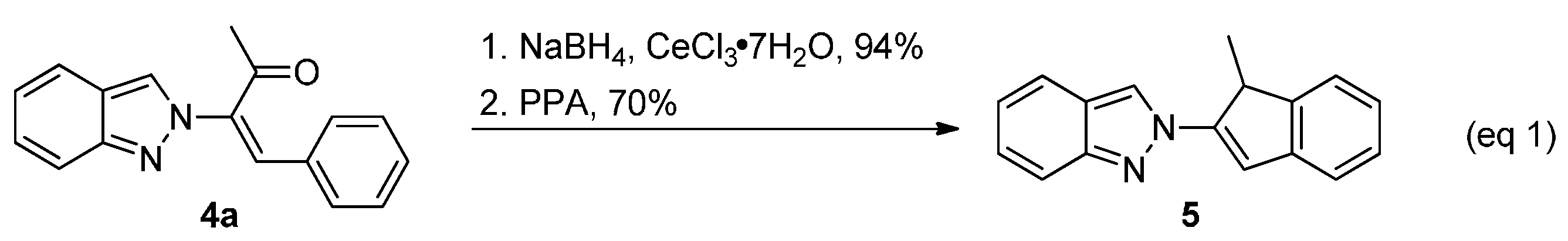{kind=link}
{kind=link}
| Entry | Product | 2; Yield(%) a from Route A | 2; Yield (%) a from Route B |
|---|---|---|---|
| 1 |  | 65 | 90 |
| 2 |  | 52 (10:1) c | 98 (8:1) c |
| 3 |  | 48 | 75 |
| 4 |  | 63 | 79 |
| 5 |  | 81 b | 87 |
| 6 |  | 63 | 85 |
| 7 |  | 72 (10:1) c | 84 (6.5:1) c |
| 8 |  | 24 | 70 |
| 9 |  | decomposed | 80 (20:1) c |
| 10 |  | 75 | 80 |
| Compd. | λabs (nm) | λem (nm) | ε(M−1cm−1) | φ |
|---|---|---|---|---|
| 2a | 322 | 398, 415 | 35,400 | 0.65 |
| 5 | 336 | 402 | 29,949 | 0.71 |
| 6 | 345 | 517 | 24,012 | 0.01 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.-H.; Liang, K.-Y.; Tsai, C.-H.; Chen, Y.-C.; Hsiao, H.-C.; Li, Y.-S.; Chen, C.-H.; Wu, H.-C. Synthesis of 2-Alkenyl-2H-indazoles from 2-(2-Carbonylmethyl)-2H-indazoles. Molecules 2016, 21, 238. https://doi.org/10.3390/molecules21020238
Lin M-H, Liang K-Y, Tsai C-H, Chen Y-C, Hsiao H-C, Li Y-S, Chen C-H, Wu H-C. Synthesis of 2-Alkenyl-2H-indazoles from 2-(2-Carbonylmethyl)-2H-indazoles. Molecules. 2016; 21(2):238. https://doi.org/10.3390/molecules21020238
Chicago/Turabian StyleLin, Mei-Huey, Kung-Yu Liang, Chang-Hsien Tsai, Yu-Chun Chen, Hung-Chang Hsiao, Yi-Syuan Li, Chung-Hao Chen, and Hau-Chun Wu. 2016. "Synthesis of 2-Alkenyl-2H-indazoles from 2-(2-Carbonylmethyl)-2H-indazoles" Molecules 21, no. 2: 238. https://doi.org/10.3390/molecules21020238
APA StyleLin, M. -H., Liang, K. -Y., Tsai, C. -H., Chen, Y. -C., Hsiao, H. -C., Li, Y. -S., Chen, C. -H., & Wu, H. -C. (2016). Synthesis of 2-Alkenyl-2H-indazoles from 2-(2-Carbonylmethyl)-2H-indazoles. Molecules, 21(2), 238. https://doi.org/10.3390/molecules21020238