
Recent Advances in Solid Catalysts Obtained by Metalloporphyrins Immobilization on Layered Anionic Exchangers: A Short Review and Some New Catalytic Results

 ,
,
Abstract
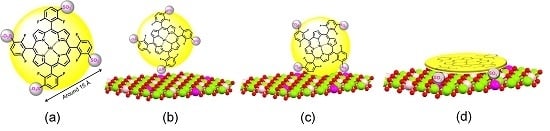
1. Introduction
1.1. Metalloporphyrins and Homogeneous Catalysis
1.2. Metalloporphyrins and Heterogeneous Catalysis
1.3. Layered Compounds
2. LDH and LHS as Support for Metalloporphyrin Immobilization
2.1. Layered Double Hydroxides (LDHs)
2.1.1. Other Macrocycles Immobilized in LDH
2.2. Layered Hydroxide Salts (LHSs)
2.2.1. Oxides Derived from LHSs
3. Influence of the LDH Composition on the MnP Immobilization Rates and Catalytic Performance: New Results
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Milgrom, L.R. The Colours of Life; Oxford University Press: New York, NY, USA, 1997; p. 93. [Google Scholar]
- Kadish, K.; Smith, K.; Guillard, R. Applications of Porphyrins and Metalloporphyrins to materials chemistry. In The Porphyrins Handbook; Chou, J.H., Nalwa, H.S., Kosal, M.E., Rakow, N.A., Suslick, K.S., Eds.; Academic Press: New York, NY, USA, 1999; Volume 6, p. 44. [Google Scholar]
- Singh, S.; Aggarwal, A.; Thompson, S.; Tomé, J.P.; Zhu, X.; Samaroo, D.; Vinodu, M.; Gao, R.; Drain, C.M. Synthesis and photophysical properties of thioglycosylated-chlorins, isobacteriochlorins and bacteriochlorins for bioimaging and diagnostics. Bioconj. Chem. 2010, 21, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Castro, K.A.D.F.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Wypych, F.; Cavaleiro, J.A.S.; Nakagaki, S. Glycol metalloporphyrin derivatives in solution or immobilized on LDH and silica: Synthesis, characterization and catalytic features in oxidation reactions. Catal. Sci. Technol. 2014, 4, 129–141. [Google Scholar] [CrossRef]
- Mansuy, D. A brief history of the contribution of metalloporphyrin models to cytochrome P450 chemistry and oxidation catalysis. C. R. Chim. 2007, 10, 392–413. [Google Scholar] [CrossRef]
- Groves, J.T.; Haushalter, R.C.; Nakamura, M.; Nemo, T.E.; Evans, B.J. High-valent iron-porphyrin complexes related to peroxidase and cytochrome P-450. J. Am. Chem. Soc. 1981, 103, 2884–2886. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.M.Q.; de Paula, R.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Metalloporphyrins in the biomimetic oxidative valorization of natural and other organic substrates. J. Porph. Phthal. 2009, 13, 589–596. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; Nemo, T.E.; Myers, R.S. Hydroxylation and epoxidation catalyzed by iron-porphine complexes. Oxygen transfer from iodosylbenzene. J. Am. Chem. Soc. 1979, 101, 1032–1033. [Google Scholar] [CrossRef]
- Bedioui, F. Zeolite-encapsulated and clay-intercalated metal porphyrin, phthalocyanine and Schiff-base complexes as models for biomimetic oxidation catalysts: An overview. Coord. Chem. Rev. 1995, 144, 39–68. [Google Scholar] [CrossRef]
- Suslick, K.S.; Bhyrappa, P.; Chou, J.H.; Kosal, M.E.; Nakagaki, S.; Smithenry, D.W.; Wilson, S.R. Microporous porphyrin solids. Acc. Chem. Res. 2005, 38, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Bolzon, L.B.; Airoldi, H.R.; Zanardi, F.B.; Granado, J.G.; Iamamoto, Y. Metalloporphyrin-functionalized hexagonal mesoporous silica: Synthesis, structural properties and catalytic activity as cytochrome P450 model. Microporours Mesoporous Mater. 2013, 168, 37–45. [Google Scholar] [CrossRef]
- Evans, S.; Smith, J.R.L. The oxidation of ethylbenzene by dioxygen catalysed by supported iron porphyrins derived from iron(III) tetrakis(pentafluoro-phenyl)porphyrin. J. Chem. Soc. Perkin Trans. 2001, 2, 174–180. [Google Scholar] [CrossRef]
- Made, A.W.; van der Smeets, J.W.H.; Nolte, R.J.M.; Drenth, W. Olefin epoxidation by a mono-oxygenase model: Effect of site isolation. J. Chem. Soc. Chem. Commun. 1983. [Google Scholar] [CrossRef]
- Machado, G.S.; de Lima, O.J.; Ciuffi, K.J.; Wypych, F.; Nakagaki, S. Iron(III) porphyrin supported on metahalloysite: An efficient and reusable catalyst for oxidation reactions. Catal. Sci. Technol. 2013, 3, 1094–1101. [Google Scholar] [CrossRef]
- Papacídero, A.T.; Rocha, L.A.; Caetano, B.L.; Molina, E.; Sacco, H.C.; Nassar, E.J.; Martinelli, Y.; Mello, C.; Nakagaki, S.; Ciuffi, K.J. Preparation and characterization of spherical silica-porphyrin catalysts obtained by the sol-gel methodology. Colloids Surf. A 2006, 275, 27–35. [Google Scholar] [CrossRef]
- Martinez-Lorente, M.A.; Battioni, P.; Kleemiss, W.; Bartoli, J.F.; Mansuy, D. Manganese porphyrins covalently bound to silica and montmorillonite Kl0 as efficient catalysts for alkene and alkane oxidation by hydrogen peroxide. J. Mol. Catal. A Chem. 1996, 113, 343–353. [Google Scholar] [CrossRef]
- Nakagaki, S.; Machado, G.S.; Halma, M.; Marangon, A.A.S.; Castro, K.A.D.F.; Mattoso, N.; Wypych, F. Immobilization of iron porphyrins in tubular kaolinite obtained by an intercalation/delamination procedure. J. Catal. 2006, 242, 110–117. [Google Scholar] [CrossRef]
- Machado, G.S.; Castro, K.A.D.F.; Wypych, F.; Nakagaki, S. Immobilization of metalloporphyrins into nanotubes of natural halloysite toward selective catalysts for oxidation reactions. J. Mol. Catal. A Chem. 2008, 283, 99–107. [Google Scholar] [CrossRef]
- Halma, M.; Bail, A.; Wypych, F.; Nakagaki, S. Catalytic activity of anionic iron(III) porphyrins immobilized on grafted disordered silica obtained from acidic leached chrysotile. J. Mol. Catal. A Chem. 2006, 243, 44–51. [Google Scholar] [CrossRef]
- Iamamoto, Y.; Idemori, Y.M.; Nakagaki, S. Cationic ironporphyrins as catalyst in comparative oxidation of hydrocarbons: Homogeneous and supported on inorganic matrices systems. J. Mol. Catal. A Chem. 1995, 99, 187–193. [Google Scholar] [CrossRef]
- Gandini, M.E.F.; Neri, C.R.; Vinhado, F.S.; Minorin, T.S.; Nascimento, O.R.; Serra, O.A.; Iamamoto, Y. Modified silicas covalently bounded to 5,10,15,20-tetrakis(2-hydroxy-5-nitrophenyl) porphyrinato iron(III). synthesis, spectroscopic and EPR characterization. Catalytic studies. J. Braz. Chem. Soc. 2008, 19, 344–351. [Google Scholar] [CrossRef]
- Huang, G.; Xiang, F.; Li, T.; Jiang, Y.; Guo, Y. Selective oxidation of toluene over the new catalyst cobalt tetra (4-hydroxyl) phenylporphyrin supported on zinc oxide. Catal. Commun. 2011, 12, 886–889. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, F.; Liu, P.; Hao, F.; Luo, H. Zinc oxide supported trans-CoD(p-Cl)PPCl-type metalloporphyrins catalyst for cyclohexane oxidation to cyclohexanol and cyclohexanone with high yield. Ind. Eng. Chem. Res. 2015, 54, 2425–2430. [Google Scholar] [CrossRef]
- Dos Santos, J.S.; Faria, A.L.; Amorin, P.M.S.; Luna, F.M.L.; Caiado, K.L.; Silva, D.O.C.; Sartoratto, P.P.C.; Assis, M.D. Iron(III) porphyrin covalently supported onto magnetic amino-functionalized nanospheres as catalyst for hydrocarbon and herbicide oxidations. J. Braz. Chem. Soc. 2012, 23, 1411–1420. [Google Scholar] [CrossRef]
- Zucca, P.; Sollai, F.; Garau, A.; Rescigno, A.; Sanjust, E. Fe(III)-5,10,15,20-Tetrakis(pentafluorophenyl)porphine supported on pyridyl-functionalized, crosslinked poly(vinyl alcohol) as a biomimetic versatile-peroxidase-like catalyst. J. Mol. Catal A Chem. 2009, 306, 89–96. [Google Scholar] [CrossRef]
- Naik, R.; Joshi, P.; Umbarkar, S.; Deshpande, R.K. Polystyrene encapsulation of manganese porphyrins: Highly efficient catalysts for oxidation of olefins. Catal. Commun. 2005, 6, 125–129. [Google Scholar] [CrossRef]
- Benedito, F.L.; Nakagaki, S.; Saczk, A.A.; Peralta-Zamora, P.G.; Costa, M.C.M. Study of metalloporphyrin covalently bound to silica as catalyst in the ortho-dianisidine oxidation. Appl. Catal. A Gen. 2003, 250, 1–11. [Google Scholar] [CrossRef]
- Demel, J.; Lang, K. Layered hydroxide-porphyrin hybrid materials: Synthesis, structure, and properties. Eur. J. Inorg. Chem. 2012, 2012, 5154–5164. [Google Scholar] [CrossRef]
- Halma, M.; Castro, K.A.D.F.; Taviot-Gueho, C.; Prévot, V.; Forano, C.; Wypych, F.; Nakagaki, S. Synthesis, characterization, and catalytic activity of anionic iron(III) porphyrins intercalated into layered double hydroxides. J. Catal. 2008, 257, 233–243. [Google Scholar] [CrossRef]
- Tonga, Z.; Shichia, T.; Takagi, K. Oxidation catalysis of a manganese(III)porphyrin intercalated in layered double hydroxide clays. Mater. Lett. 2003, 57, 2258–2261. [Google Scholar] [CrossRef]
- Machado, G.S.; Wypych, F.; Nakagaki, S. Anionic iron(III) porphyrins immobilized on zinc hydroxide chloride as catalysts for heterogeneous oxidation reactions. Appl. Catal. A 2012, 413–414, 94–102. [Google Scholar] [CrossRef]
- Machado, G.S.; Arízaga, G.G.C.; Wypych, F.; Nakagaki, S. Immobilization of anionic metalloporphyrins on zinc hydroxide nitrate and study of an unusual catalytic activity. J. Catal. 2010, 274, 130–141. [Google Scholar] [CrossRef]
- Warmuth, W.M.; Schöllhorn, R. Progress in Intercalation Reactions; Kluver Academic Press: Oxford, UK, 1994; p. 14. [Google Scholar]
- Mercury - Crystal Structure Visualisation, Exploration and Analysis Made Easy. Available online: http://www.ccdc.cam.ac.uk/Solutions/FreeSoftware/Pages/FreeMercury.aspx (accessed on 1 September 2015).
- Crystallography Open Database. Available online: http://www.crystallography.net/ (accessed on 15 August 2015).
- Drits, V.A.; Bookin, A.S. Crystal struture and X-ray identification of layered double hydroxides. In Layered Double Hydroxides: Present and Future; Rives, V., Ed.; Nova Science Publishers: New York, NY, USA, 2006; pp. 41–100. [Google Scholar]
- Duan, X.; Evans, D.G. Layered Double Hydroxides; Springer-Verlag: Berlim, Germany, 2006; pp. 3–12. [Google Scholar]
- Fernandes, C.I.; Vaz, P.D.; Nunes, C.D. Catalytic applications of layered double hydroxide. In Layered Double Hydroxides (LDHs): Synthesis, Characterization and Applications; Sherman, I.T., Ed.; Nova Science Publisher: New York, NY, USA, 2015; pp. 1–32. [Google Scholar]
- Besserguenev, A.V.; Fogg, A.M.; Francis, R.J.; Price, S.J.; O’Hare, D.; Isupov, V.P.; Tolochko, B.P. Synthesis and structure of the Gibbsite intercalation compounds [LiAl2(OH)6]X {X = Cl, Br, NO3} and [LiAl2(OH)6]Cl·H2O using synchrotron X-ray and neutron powder diffraction. Chem. Mater. 1997, 9, 241–247. [Google Scholar] [CrossRef]
- Belskaya, O.B.; Baklanova, O.N.; Leont’eva, N.N.; Gulyaeva, T.I.; Likholobov, V.A. Mechanochemical synthesis of LiAl-layered hydroxides, precursors of oxidic supports and catalysts of the basic type. Procedia Eng. 2015, 113, 91–97. [Google Scholar] [CrossRef]
- Arízaga, G.G.C.; Satyanarayana, K.G.; Wypych, F. Layered hydroxide salts: Synthesis, properties and potential applications. Solid State Ion. 2007, 178, 1143–1162. [Google Scholar] [CrossRef]
- Satyanarayana, K.G.; Wypych, F. Clay Surfaces—Fundamentals and Applications; Academic Press: Oxford, UK, 2004; pp. 1–23. [Google Scholar]
- Aurichalcite: Aurichalcite Mineral Information and Data. Available online: http://www.mindat.org/min-422.html (accessed on 1 September 2015).
- Hill, R.J. The structure of loseyite. Acta Crystallogr. 1981, B37, 1323–1328. [Google Scholar] [CrossRef]
- Grice, J.D.; Dunn, P.J. Sclarite, a new mineral from Franklin, New Jersey, with essential octahedrally and tetrahedrally coordination zinc: Description and structure refinement. Am. Miner. 1989, 74, 1355–1359. [Google Scholar]
- Manju, G.N.; Gigi, M.C.; Anirudhan, T.S. Hydrotalcite as adsorbent for the removal of chromium (VI) from aqueous media: Equilibrium studies. Indian J. Chem. Technol. 1999, 6, 134–141. [Google Scholar]
- Centi, G.; Perathoner, S. Catalysis by layered materials: A review. Microporous Mesoporous Mater. 2008, 107, 3–15. [Google Scholar] [CrossRef]
- Choy, J.H.; Choi, S.J.; Oh, J.M.; Park, T. Clay minerals and layered double hydroxides for novel biological applications. Appl. Clay Sci. 2007, 36, 122–132. [Google Scholar] [CrossRef]
- Becker, C.M.; Gabbardo, A.D.; Wypych, F.; Amico, S.C. Mechanical and flame-retardant properties of epoxy/Mg-Al LDH composites. Composites Part A Appl. Sci. Manuf. 2011, 42, 196–202. [Google Scholar] [CrossRef]
- Wypych, F.; Bubniak, G.A.; Halma, M.; Nakagaki, S. Exfoliation and immobilization of anionic iron porphyrin in layered double hydroxides. J. Colloid Interface Sci. 2003, 264, 203–207. [Google Scholar] [CrossRef]
- Nakagaki, S.; Halma, M.; Bail, A.; Arízaga, G.G.C.; Wypych, F. First insight into catalytic activity of anionic iron porphyrins immobilized on exfoliated layered double hydroxides. J. Colloid Interface Sci. 2005, 281, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, Z.; Zhao, L.; Liu, H.; He, J. Enhanced catalytic efficiency in the epoxidation of alkenes for manganese complex encapsulated in the hydrophobic interlayer region of layered double hydroxides. Ind. Eng. Chem. Res. 2013, 52, 17821–17828. [Google Scholar] [CrossRef]
- Kumar, P.; Gill, K.; Kumar, S.; Ganguly, S.K.; Jaina, S.L. Magnetic Fe3O4@MgAl-LDH composite grafted with cobalt phthalocyanine as an efficient heterogeneous catalyst for the oxidation of mercaptans. J. Mol. Catal. A Chem. 2015, 401, 48–54. [Google Scholar] [CrossRef]
- Nakagaki, S.; Castro, K.A.D.F.; Ucoski, G.M.; Halma, M.; Prévot, V.; Forano, C.; Wypych, F. Anionic Iron(III) porphyrin immobilized on/into exfoliated macroporous layered double hydroxides as catalyst for oxidation reactions. J. Braz. Chem. Soc. 2014, 25, 2329–2338. [Google Scholar] [CrossRef]
- Ma, J.; Liu, L.; Chen, Y.; Zhuo, M.; Shao, F.; Gonga, J.; Tong, Z. Facile assembly for fast construction of intercalation hybrids of layered double hydroxides with anionic metalloporphyrin. Dalton Trans. 2014, 43, 9909–9915. [Google Scholar] [CrossRef] [PubMed]
- Teramura, K.; Tsuneoka, H.; Ogura, K.; Sugimoto, T.; Shishido, T.; Tanaka, T. Photoactivation of molecular oxygen by an Iron(III) porphyrin with a magnesium aluminum layered double hydroxide for the aerobic epoxidation of cyclohexene. ChemCatChem 2014, 6, 2276–2281. [Google Scholar] [CrossRef]
- Zhan, T.; Yang, Q.; Zhang, Y.; Wang, X.; Xu, J.; Hou, W. Structural characterization and electrocatalytic application of hemoglobin immobilized in layered double hydroxides modified with hydroxyl functionalized ionic liquid. J. Colloid Interface Sci. 2014, 433, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Prrez-Bernal, M.E.; Ruano-Casero, R.; Pinnavaia, T.J. Catalytic autoxidation of 1-decanethiol by cobalt(II) phthalocyaninetetrasulfonate intercalated in a layered double hydroxide. Catal. Lett. 1991, 11, 55–62. [Google Scholar] [CrossRef]
- Chibwe, M.; Pinnavaia, T.J. Stabilization of a cobalt(II) phthalocyanine oxidation catalyst by intercalation in a layered double hydroxide host. J. Chem. Soc. Chem. Commun. 1993. [Google Scholar] [CrossRef]
- Sun, Z.; Jin, L.; He, S.; Zhao, Y.; Wei, M.; Evans, D.G.; Duan, X. A structured catalyst based on cobalt phthalocyanine/calcined Mg-Al hydrotalcite film for the oxidation of mercaptan. Green Chem. 2012, 14, 1909–1916. [Google Scholar] [CrossRef]
- Parida, K.M.; Baliarsingh, N.; Patra, B.S.; Das, J. Copperphthalocyanine immobilized Zn/Al LDH as photocatalyst under solar radiation for decolorization of methylene blue. J. Mol. Catal. A Chem. 2007, 267, 202–208. [Google Scholar] [CrossRef]
- Carrado, K.A.; Forman, J.E.; Botto, R.E.; Winans, R.E. Incorporation of phthalocyanines by cationic and anionic clays via ion exchange and direct systhesis. Chem. Mater. 1993, 5, 472–478. [Google Scholar]
- Ukrainczyk, L.; Chibwe, M.; Pinnavaia, T.J.; Boydt, S.A. ESR study of Cobalt(II) tetrakis(N-methyl-4-pyridiniumy1)porphyrin and Cobalt(II) tetrasulfophthalocyanine intercalated in layered Aluminosilicates and a layered double hydroxide. J. Phys. Chem. 1994, 98, 2668–2676. [Google Scholar] [CrossRef]
- Chibwe, M.; Ukrainczyk, L.; Boydt, S.A.; Pinnavaia, T.J. Catalytic properties of biomimetic metallomacrocycles intercalated in layered double hydroxides and smectite clay: The importance of edge-site access. J. Mol. Catal. A Chem. 2004, 33, 790–791. [Google Scholar] [CrossRef]
- Ma, S.; Fan, C.; Du, L.; Huang, G.; Yang, X.; Tang, W.; Makita, Y.; Ooi, K. Intercalation of macrocyclic crown ether into well-crystallized LDH: Formation of staging structure and secondary host-guest reaction. Chem. Mater. 2009, 21, 3602–3610. [Google Scholar] [CrossRef]
- Ma, S.; Du, L.; Wang, J.; Chu, N.; Sun, Y.; Sun, G.; Yang, X.; Ooi, K. Structural adjustment during intercalation of macrocyclic crown ether into LDH via swelling/restoration reaction: Staging formation and mechanism insights. Dalton Trans. 2011, 40, 9835–9843. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wang, J.; Du, L.; Sun, Y.; Gu, Q.; Sun, G.; Yang, X. A new method for fast intercalation of bulk crown ether guest into LDH. J. Colloid Interface Sci. 2013, 393, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Aisawa, S.; Hirahara, H.; Sasaki, A.; Narita, E. Synthesis and adsorption property of calixarene-p-sulfonate-intercalated layered double hydroxides. Chem. Lett. 2004, 33, 790–791. [Google Scholar] [CrossRef]
- Sasaki, S.; Aisawa, S.; Hirahara, H.; Sasaki, A.; Nakayama, H.; Narita, E. Synthesis and adsorption properties of p-sulfonated calix[4 and 6]arene-intercalated layered double hydroxides. J. Solid State Chem. 2006, 179, 1129–1135. [Google Scholar] [CrossRef]
- Stählin, W.; Oswald, H.R. The crystal structure of zinc hydroxide nitrate, Zn5(OH)8(NO3)2·2H2O. Acta Crystallogr. 1970, B26, 860–863. [Google Scholar] [CrossRef]
- Stählin, W.; Oswald, H.R. The topotactic reaction of zinc hydroxide nitrate with aqueous metal chloride solutions. J. Solid State Chem. 1971, 3, 256–264. [Google Scholar] [CrossRef]
- Arízaga, G.G.C.; Mangrich, A.S.; Wypych, F. Cu2+ ions as a paramagnetic probe to study the surface chemical modification process of layered double hydroxides and hydroxide salts with nitrate and carboxylate anions. J. Colloid Interface Sci. 2008, 320, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Chitrakar, R.; Sakane, K.; Ooi, K.; Kobayashi, S.; Ohnishi, M.; Doi, A. Synthesis of ktenasite, a double hydroxide of zinc and copper, and its intercalation reaction. J. Solid State Chem. 2004, 177, 1624–1630. [Google Scholar] [CrossRef]
- Biswick, T.; Jones, W.; Pacuła, A.; Serwick, E. Synthesis, characterization and anion exchange properties of copper, magnesium, zinc and nickel hydroxy nitrates. J. Solid State Chem. 2006, 179, 49–55. [Google Scholar] [CrossRef]
- Wypych, F.; Arízaga, G.G.C.; Gardolinski, J.E.F.C. Intercalation and functionalization of zinc hydroxide nitrate with mono and dicarboxylic acids. J. Colloid Interface Sci. 2005, 283, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, R.; Ramos, L.P.; Wypych, F. New multifunctional materials obtained by the intercalation of anionic dyes into layered zinc hydroxide nitrate followed by dispersion into poly(vinyl alcohol) (PVA). J. Colloid Interface Sci. 2009, 330, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Demel, J.; Kubát, P.; Jirka, I.; Kovář, P.; Pospíšil, M.; Lang, K. Inorganic-organic hybrid materials: Layered zinc hydroxide salts with intercalated porphyrin sensitizers. J. Phys. Chem. C 2010, 114, 16321–16328. [Google Scholar] [CrossRef]
- Cordeiro, C.S.; Arizaga, G.G.C.; Ramos, L.P.; Wypych, F. A new zinc hydroxide nitrate heterogeneous catalyst for the esterification of free fatty acids and the transesterification of vegetable oils. Catal. Commun. 2008, 9, 2140–2143. [Google Scholar] [CrossRef]
- Dolphin, D.; Traylor, T.G.; Xie, L.Y. Polyhaloporphyrins: Unusual ligands for metals and metal-catalyzed oxidations. Acc. Chem. Res. 1997, 30, 251–259. [Google Scholar] [CrossRef]
- Groves, J.T. High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 2006, 100, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Nam, W. High-valent iron(IV)-oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc. Chem. Res. 2007, 40, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Machado, G.S.; Ucoski, G.M.; Lima, O.J.; Ciuffi, K.J.; Wypych, F.; Nakagaki, S. Cationic and anionic metalloporphyrins simultaneously immobilized onto raw halloysite nanoscrolls catalyze oxidation reactions. Appl. Catal. A 2013, 460–461, 124–131. [Google Scholar] [CrossRef]
- Machado, G.S.; Wypych, F.; Nakagaki, S. Immobilization of anionic iron(III) porphyrins onto in situ obtained zinc oxide. J. Colloid Interface Sci. 2012, 377, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Sigoli, F.A.; Davolos, M.R.; Jafelicci, M., Jr. Morphological evolution of zinc oxide originating from zinc hydroxide carbonate. J. Alloys Compd. 1997, 262–263, 292–205. [Google Scholar] [CrossRef]
- Fah, C.P.; Xue, J.; Wang, J. Nanosized zinc-oxide particles derived from mechanical activation of Zn5(NO3)2(OH)8·2H2O in sodium chloride. J. Am. Ceram. Soc. 2002, 85, 273–275. [Google Scholar] [CrossRef]
- Machovsky, M.; Kuritka, I.; Sedlak, J.; Pastorek, M. Hexagonal ZnO porous plates prepared from microwave synthesized layered zinc hydroxide sulphate via thermal decomposition. Mater. Res. Bull. 2013, 48, 4002–4007. [Google Scholar] [CrossRef]
- Li, X.; He, G.; Xiao, G.; Liu, H.; Wang, M. Synthesis and morphology control of ZnO nanostructures in microemulsions. J. Colloid Interface Sci. 2009, 333, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Musić, S.; Dragčević, D.; Popović, S. Influence of synthesis route on the formation of ZnO particles and their morphologies. J. Alloys Compd. 2007, 429, 242–249. [Google Scholar] [CrossRef]
- Huang, G.; Mo, L.; Cai, J.; Cao, X.; Peng, Y.; Guo, Y.; Wei, S. Environmentally friendly and efficient catalysis of cyclohexane oxidation by iron meso-tetrakis(pentafluorophenyl)porphyrin immobilized on zinc oxide. Appl. Catal. B 2015, 162, 364–371. [Google Scholar] [CrossRef]
- Li, X.; Cheng, Y.; Kanga, S.; Mu, J. Preparation and enhanced visible light-driven catalytic activity of ZnO microrods sensitized by porphyrin heteroaggregate. Appl. Surf. Sci. 2010, 256, 6705–6709. [Google Scholar] [CrossRef]
- De Roy, A.; Forano, C.; Malki, K.E.; Besse, J.P. Anionic clays: Trends in pillaring chemistry. In Expanded Clays and Others Micropourous Solids, 2nd ed.; Occelli, M.L., Robson, H.E., Eds.; Springer: New York, NY, USA, 1992; Volume 2, pp. 108–167. [Google Scholar]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- De Roy, A.; Forano, C.; Besse, J.P. Layered double hydroxides: Systhesis and post-synthesis modification. In Layered Double Hydroxides: Present and Future; Rives, V., Ed.; Nova Science Publishers: New York, NY, USA, 2006; pp. 1–37. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds Part B, 6th ed.; John Wiley & Sons: New York, NY, USA, 2009; pp. 91–94. [Google Scholar]
- Conterosito, E.; Croce, G.; Palin, L.; Pagano, C.; Perioli, L.; Viterbo, D.; Boccaleri, E.; Paul, G.; Milanesio, M. Structural characterization and thermal and chemical stability of bioactive molecule-hydrotalcite (LDH) nanocomposites. Phys. Chem. Chem. Phys. 2013, 15, 13418–13433. [Google Scholar] [CrossRef] [PubMed]
- Conterosito, E.; Palin, L.; Antonioli, D.; Viterbo, D.; Mugnaioli, E.; Kolb, U.; Perioli, L.; Milanesio, M.; Gianotti, V. Structural characterisation of complex layered double hydroxides and TGA-GC-MS study on thermal response and carbonate contamination in nitrate- and organic-exchanged hydrotalcites. Chem. Eur. J. 2015, 21, 14975–14986. [Google Scholar] [CrossRef] [PubMed]
- Constantino, V.R.L.; Pinnavaia, T.J. Basic properties of Mg2+1−xAl3+x layered double hydroxides intercalated by carbonate, hydroxide, chloride, and sulfate anions. Inorg. Chem. 1995, 34, 883–892. [Google Scholar] [CrossRef]
- Li, L.; Dou, L.; Zhang, H. Layered double hydroxide supported gold nanoclusters by glutathione-capped Au nanoclusters precursor method for highly efficient aerobic oxidation of alcohols. Nanoscale 2014, 6, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Jin, L.; Zhao, Y.; He, S.; Li, S.; Wei, M.; Wang, L. A structured catalyst toward mercaptan sweetening with largely enhanced synergistic effect. Ind. Eng. Chem. Res. 2014, 53, 4595–4603. [Google Scholar] [CrossRef]
- Schiavon, M.A.; Iamamoto, Y.; Nascimento, O.R.; Assis, M.D. Catalytic activity of nitro- and carboxy-substituted iron porphyrins in hydrocarbon oxidation Homogeneous solution and supported systems. J. Mol. Catal. A Chem. 2001, 174, 213–222. [Google Scholar] [CrossRef]
- Machado, G.S.; Groszewicz, P.B.; Castro, K.A.D.F.; Wypych, F.; Nakagaki, S. Catalysts for heterogeneous oxidation reaction based on metalloporphyrins immobilized on kaolinite modified with triethanolamine. J. Colloid Interface Sci. 2012, 374, 278–286. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MP | LDH | Use | Reference |
|---|---|---|---|
| Fe or Mn glycol metalloporphyrin | Mg/Al-LDH (nitrate anions) | Oxidation of cyclooctene and cyclohexane | [4] |
| [Fe(TDFSPP)] and [Fe(TCFSPP)] | Mg/Al-LDH (glycinate anions) | Oxidation of cyclooctene and cyclohexane | [53] |
| Mn(TPP)OAc | Zn/Al-LDH (dodecyl sulfonate anions) | Epoxidation of alkenes | [54] |
| CoPcS (phthalocyanine) | Mg/Al-LDH@Fe3O4 (magnetic) | Oxidation of mercaptans | [55] |
| [Fe(TDCSPP)] | Mg/Al-LDH (macroporous) | Oxidation of cyclooctene and cyclohexane | [56] |
| MnTSPP | Mg or Ni/Al-LDH (intercalated porphyrin) | Epoxidation of cyclohexene | [57] |
| [Fe(TPFPP)Cl] | Mg/Al-LDH (nitrate anions) | Epoxidation of cyclohexene | [58] |
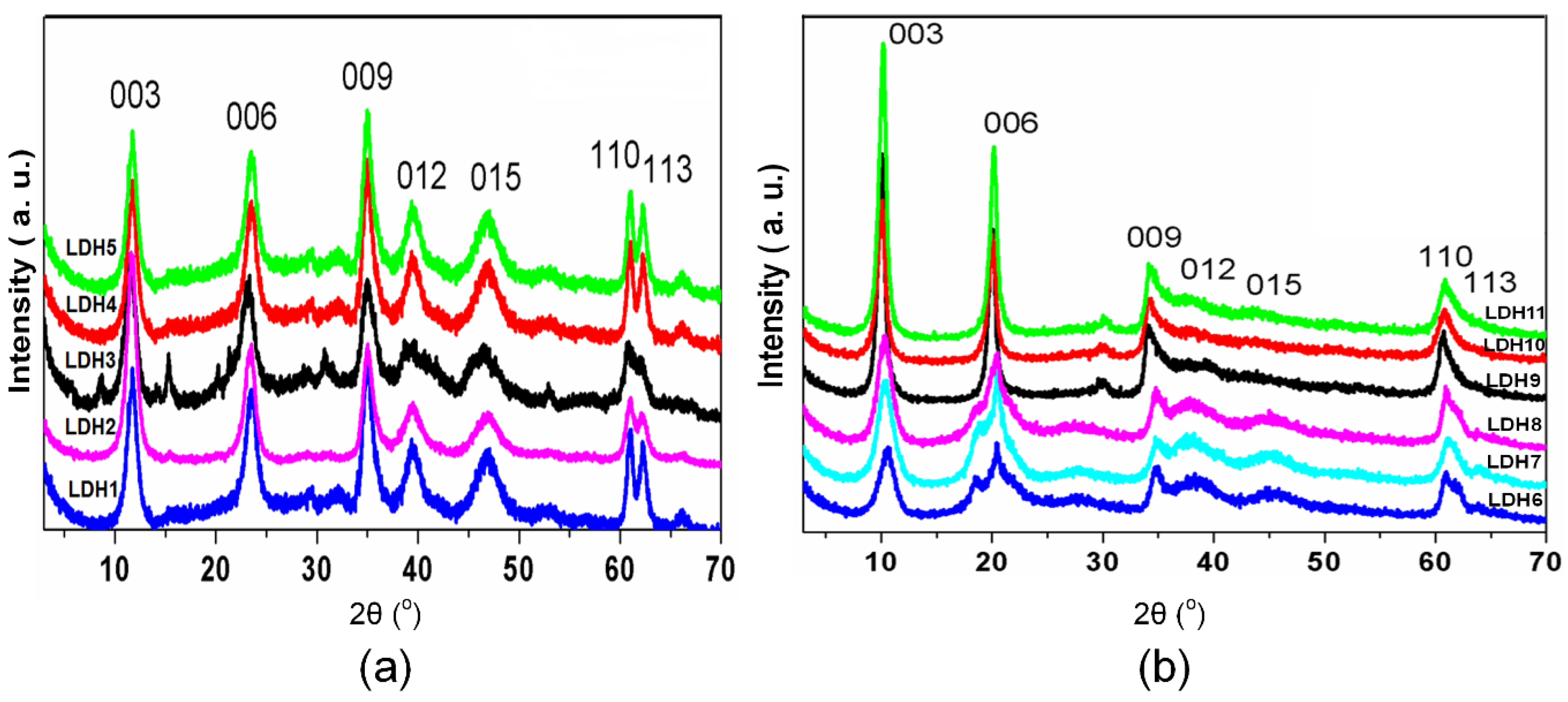
| Mg/Al-Anion | Mg:Al (Molar Ratio) | Basal Distance/(Å) | “a” Parameter/(Å) c | Zn/Al-Anion * | Zn:Al (Molar Ratio) | Basal Distance/(Å) | “a” Parameter/(Ǻ) c |
|---|---|---|---|---|---|---|---|
| LDH1 a | 2:1 | 7.57 | 3.036 | LDH4 a | 2:1 | 7.38 | 3.041 |
| LDH2 a | 3:1 | 7.60 | 3.037 | LDH5 a | 3:1 | 7.22 | 3.052 |
| LDH3 a | 4:1 | 7.67 | 3.042 | * | - | - | - |
| LDH6 b | 2:1 | 8.66 | 3.037 | LDH9 b | 2:1 | 8.87 | 3.042 |
| LDH7 b | 3:1 | 8.66 | 3.038 | LDH10 b | 3:1 | 8.87 | 3.047 |
| LDH8 b | 4:1 | 8.66 | 3.041 | LDH11 b | 4:1 | 8.80 | 3.054 |
| LDH-CO3 | %H2O | M2+/M3+ Molar Ratio | LDH-NO3 | %H2O | M2+/M3+ Molar Ratio | ||
|---|---|---|---|---|---|---|---|
| Calculated | Theoretical | Calculated | Theoretical | ||||
| LDH1 | 19.9 | 2.3 | 2 | LDH6 | 7.08 | 1.8 | 2 |
| LDH2 | 7.41 | 3.5 | 3 | LDH7 | 3.84 | 3.3 | 3 |
| LDH3 | 9.44 | 4.6 | 4 | LDH8 | 6.33 | 4.4 | 4 |
| LDH4 | 3.5 | 2.2 | 2 | LDH9 | 4.38 | 2.0 | 2 |
| LDH5 | 8.82 | 3.5 | 3 | LDH10 | 2.84 | 3.4 | 3 |
| LDH11 | 1.16 | 4.5 | 4 | ||||
| LDH-CO3 | Loading b/10−6 mol·g−1 | LDH-NO3 | Loading b/10−6 mol·g−1 |
|---|---|---|---|
| LDH1 (Mg/Al—2:1) | 5.7 | LDH6 (Mg/Al—2:1) | 4.0 |
| LDH2 (Mg/Al—3:1) | 4.6 | LDH7 (Mg/Al—3:1) | 4.0 |
| LDH3 (Mg/Al—4:1) | 6.4 | LDH8 (Mg/Al—4:1) | 4.1 |
| LDH4 (Zn/Al—2:1) | 5.9 | LDH9 (Zn/Al—2:1) | 4.4 |
| LDH5 (Zn/Al—3:1) | 5.3 | LDH10 (Zn/Al—3:1) | 4.0 |
| LDH11 (Zn/Al—4:1) | 4.1 |
| LDHX Solid | d 1,2 (Å) | d 2,3 (Å) | d 3,4 (Å) | d 4,1 (Å) | d 1,3 (Å) | d 2,4 (Å) | As Shown in |
|---|---|---|---|---|---|---|---|
| LDH1 (Mg/Al–2:1) | 10.52 | 10.52 | 10.52 | 10.52 | 10.52 | 10.52 | Figure 10b |
| LDH2 (Mg/Al—3:1) | 12.15 | 12.15 | 12.15 | 12.15 | 12.15 | 21.04 | Figure 10c |
| LDH3 (Mg/Al—4:1) | 16.10 * | 10.54 | 16.10 * | 10.54 | 16.10 * | 27.88 | Figure 10d |
| LDH4 (Zn/Al—2:1) | 10.53 | 10.53 | 10.53 | 10.53 | 10.53 | 10.53 | Figure 10b |
| LDH5 (Zn/Al—3:1) | 12.21 | 12.21 | 12.21 | 12.21 | 12.21 | 21.14 | Figure 10c |
| LDH6 (Mg/Al—2:1) | 10.52 | 10.52 | 10.52 | 10.52 | 10.52 | 10.52 | Figure 10b |
| LDH7 (Mg/Al—3:1) | 12.15 | 12.15 | 12.15 | 12.15 | 12.15 | 21.05 | Figure 10c |
| LDH8 (Mg/Al—4:1) | 16.09 * | 10.53 | 16.09 * | 10.53 | 16.09 * | 27.87 | Figure 10d |
| LDH9 (Zn/Al—2:1) | 10.54 | 10.54 | 10.54 | 10.54 | 10.54 | 10.54 | Figure 10b |
| LDH10 (Zn/Al—3:1) | 12.19 | 12.19 | 12.19 | 12.19 | 12.19 | 21.11 | Figure 10c |
| LDH11 (Zn/Al—4:1) | 16.16 * | 10.58 | 16.16 * | 10.58 | 16.16 * | 27.99 | Figure 10d |
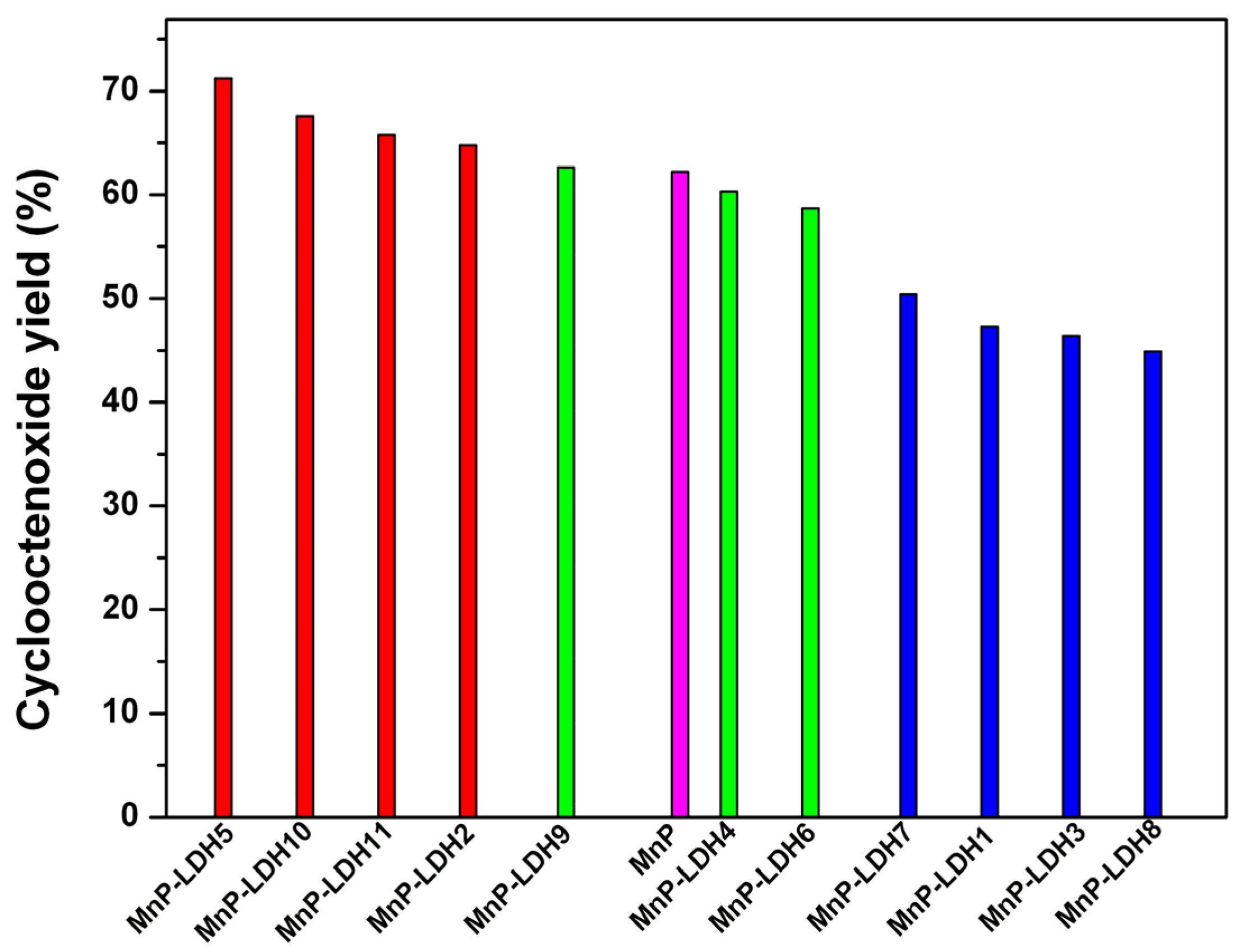
| Catalyst | Run | LDH (M2+/M3+) | LDH Anion | Epoxide Yield (%) a | Epoxide Yield, Corrected d |
|---|---|---|---|---|---|
| MnP b | 1 | 76.3 ± 3.8 | 62.2 | ||
| MnP-LDH1 | 2 | 2:1 | Mg/Al-CO3 | 51.3 ± 3.2 | 47.3 |
| MnP-LDH2 | 3 | 3:1 | Mg/Al-CO3 | 71.2 ± 2.9 | 64.8 |
| MnP-LDH3 | 4 | 4:1 | Mg/Al-CO3 | 49.9 ± 5.7 | 46.4 |
| MnP-LDH4 | 5 | 2:1 | Zn/Al-CO3 | 68.2 ± 4.4 | 60.3 |
| MnP-LDH5 | 6 | 3:1 | Zn/Al-CO3 | 77.1 ± 5.4 | 71.2 |
| MnP-LDH6 | 7 | 2:1 | Mg/Al-NO3 | 61.2 ± 2.6 | 55.3 |
| MnP-LDH7 | 8 | 3:1 | Mg/Al-NO3 | 57.6 ± 3.7 | 50.4 |
| MnP-LDH8 | 9 | 4:1 | Mg/Al-NO3 | 50.2 ± 4.1 | 44.9 |
| MnP-LDH9 | 10 | 2:1 | Zn/Al-NO3 | 71.7 ± 5.2 | 62.6 |
| MnP-LDH10 | 11 | 3:1 | Zn/Al-NO3 | 75.0 ± 3.6 | 67.6 |
| MnP-LDH11 | 12 | 4:1 | Zn/Al-NO3 | 73.1 ± 4.1 | 65.8 |
| LDH1 | 13 | 2:1 | Mg/Al-CO3 | 4.0 ± 3.8 | - |
| LDH2 | 14 | 3:1 | Mg/Al-CO3 | 6.4 ± 2.3 | - |
| LDH3 | 15 | 4:1 | Mg/Al-CO3 | 3.5 ± 4.8 | - |
| LDH4 | 16 | 2:1 | Zn/Al-CO3 | 7.9 ± 6.3 | - |
| LDH5 | 17 | 3:1 | Zn/Al-CO3 | 5.9 ± 4.4 | - |
| LDH6 | 18 | 2:1 | Mg/Al-NO3 | 2.5 ± 5.6 | - |
| LDH7 | 19 | 3:1 | Mg/Al-NO3 | 7.2 ± 2.2 | - |
| LDH8 | 20 | 4:1 | Mg/Al-NO3 | 5.3 ± 3.7 | - |
| LDH9 | 21 | 2:1 | Zn/Al-NO3 | 9.1 ± 5.3 | - |
| LDH10 | 22 | 3:1 | Zn/Al-NO3 | 7.4 ± 6.1 | - |
| LDH11 | 23 | 4:1 | Zn/Al-NO3 | 7.3 ± 4.3 | - |
| Control c | 24 | 14.1 ± 1.3 | - |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagaki, S.; Mantovani, K.M.; Sippel Machado, G.; Dias de Freitas Castro, K.A.; Wypych, F. Recent Advances in Solid Catalysts Obtained by Metalloporphyrins Immobilization on Layered Anionic Exchangers: A Short Review and Some New Catalytic Results. Molecules 2016, 21, 291. https://doi.org/10.3390/molecules21030291
Nakagaki S, Mantovani KM, Sippel Machado G, Dias de Freitas Castro KA, Wypych F. Recent Advances in Solid Catalysts Obtained by Metalloporphyrins Immobilization on Layered Anionic Exchangers: A Short Review and Some New Catalytic Results. Molecules. 2016; 21(3):291. https://doi.org/10.3390/molecules21030291
Chicago/Turabian StyleNakagaki, Shirley, Karen Mary Mantovani, Guilherme Sippel Machado, Kelly Aparecida Dias de Freitas Castro, and Fernando Wypych. 2016. "Recent Advances in Solid Catalysts Obtained by Metalloporphyrins Immobilization on Layered Anionic Exchangers: A Short Review and Some New Catalytic Results" Molecules 21, no. 3: 291. https://doi.org/10.3390/molecules21030291
APA StyleNakagaki, S., Mantovani, K. M., Sippel Machado, G., Dias de Freitas Castro, K. A., & Wypych, F. (2016). Recent Advances in Solid Catalysts Obtained by Metalloporphyrins Immobilization on Layered Anionic Exchangers: A Short Review and Some New Catalytic Results. Molecules, 21(3), 291. https://doi.org/10.3390/molecules21030291