
Functionalization of a Triazine Dendrimer Presenting Four Maleimides on the Periphery and a DOTA Group at the Core

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
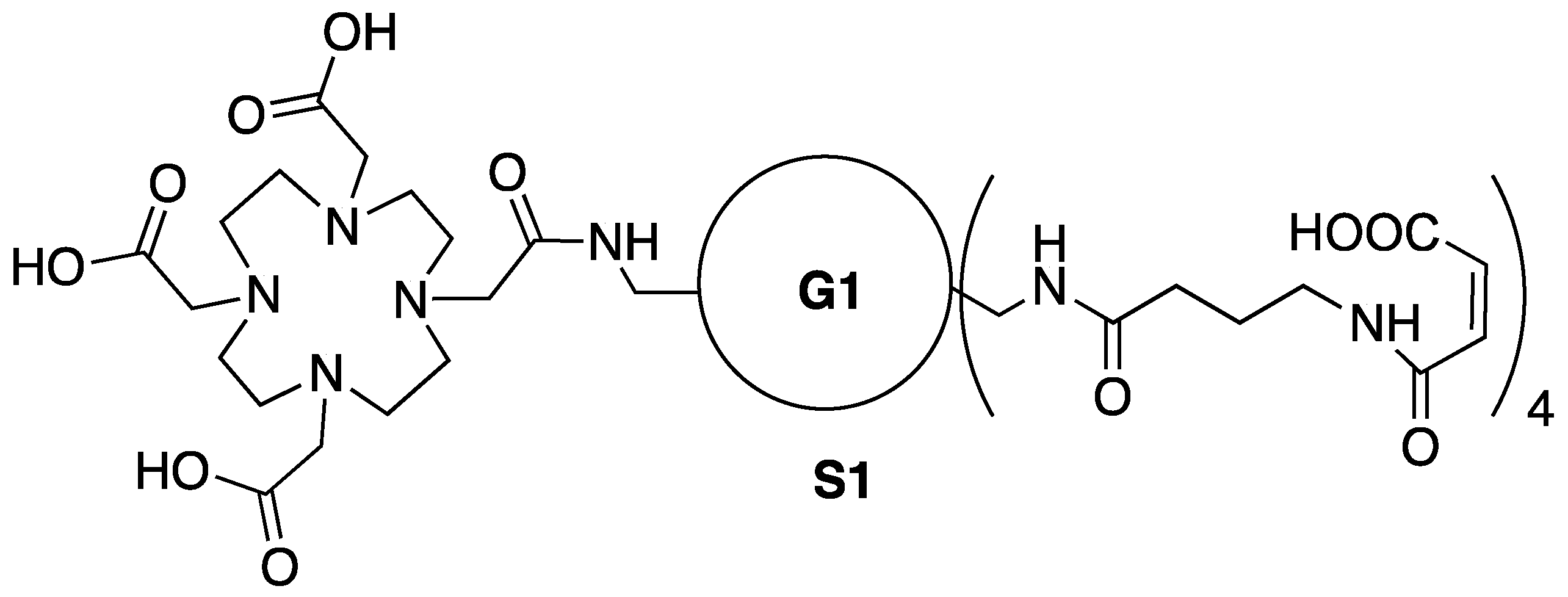
2.2. Hydrolytic Stability
2.3. Conjugation of Small Molecules
2.4. Conjugation of Pepties
3. Experimental Section
3.1. General Synthetic Procedures
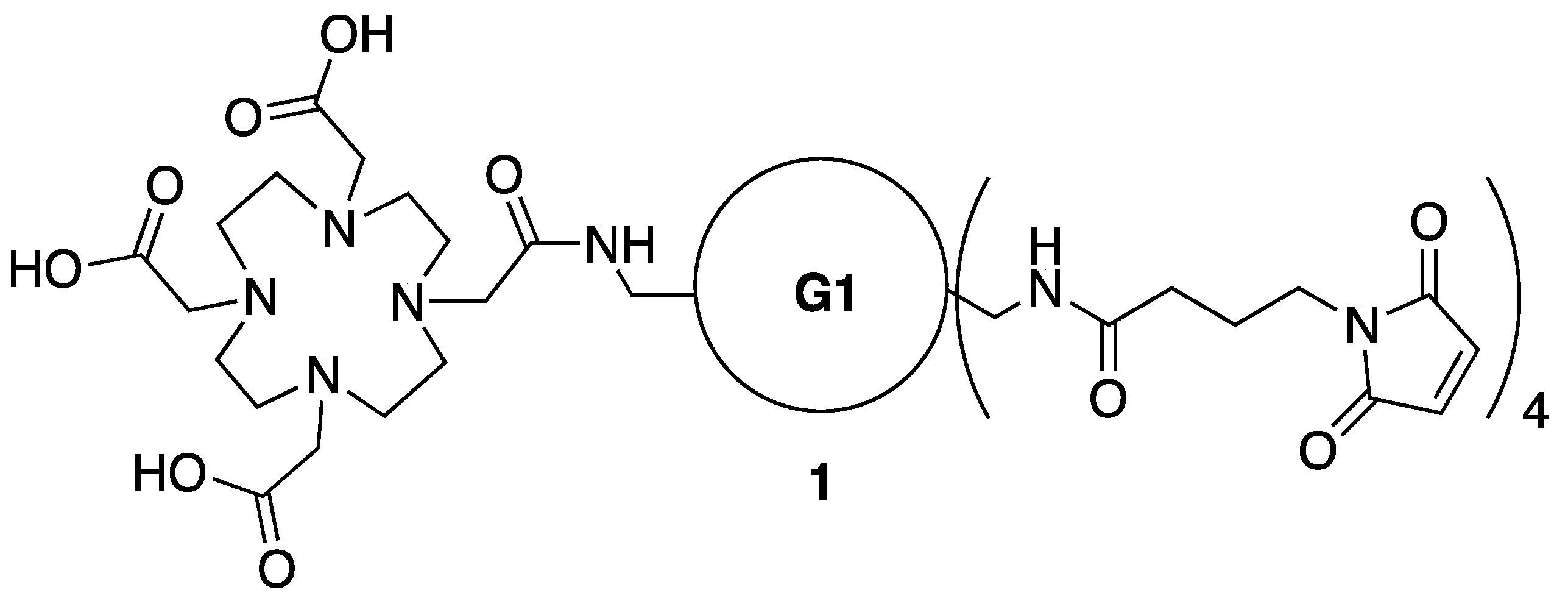
3.2. Synthesis of Dendrimer 1
3.3. Synthesis of Intermediate S1—Maleic Acid Amide of 1
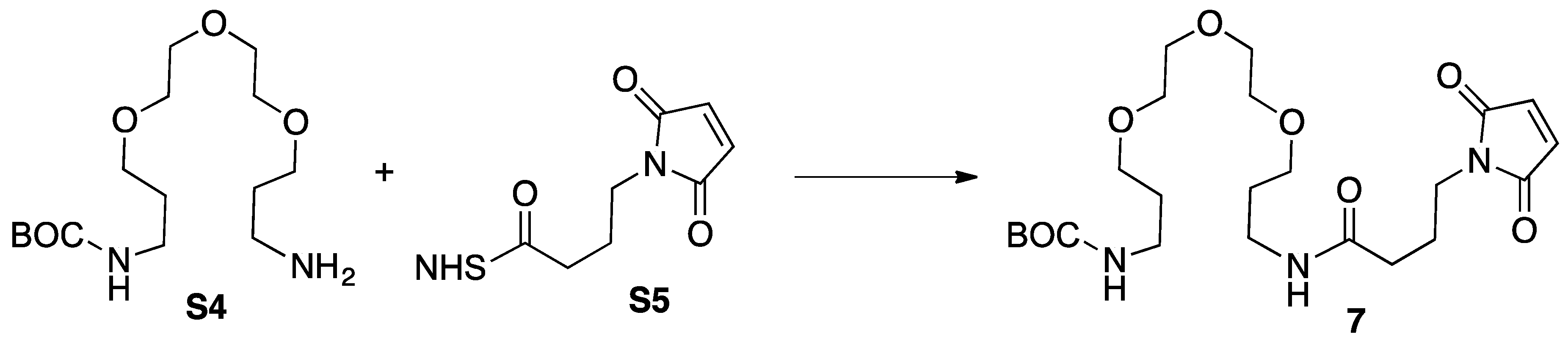
3.4. Synthesis of Intermediate 4
3.5. Synthesis of Intemediate 5
3.6. Synthesis of Intermediate—Generation 3 Dendrimer with DOTA
3.7. Synthesis of Intermediate 6
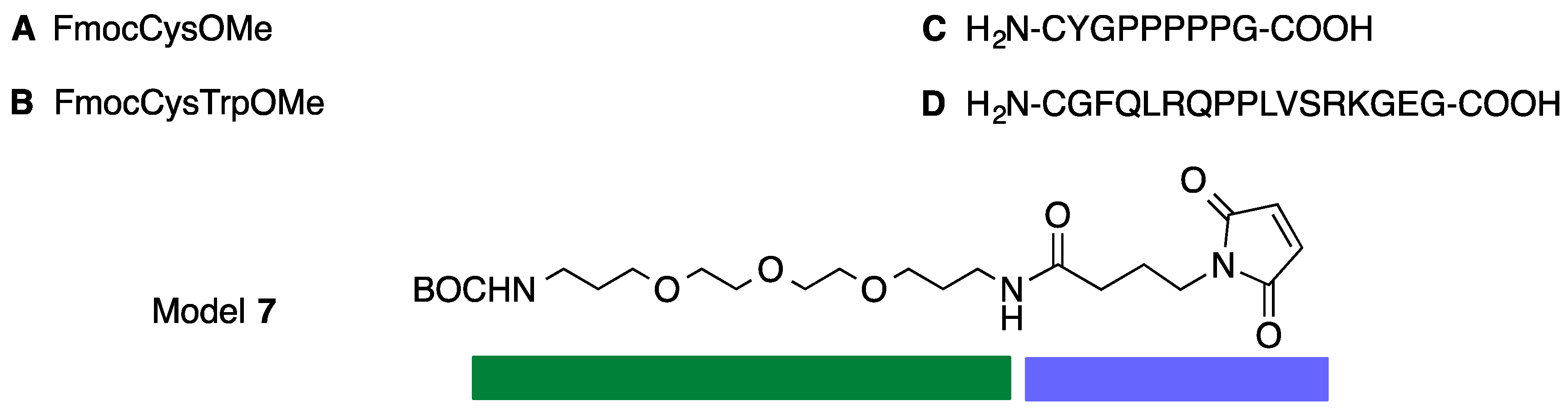
3.8. Synthesis of Model 7
3.9. Synthesis of Conjugate 7-A
3.10. Synthesis of Cystine (S6)
3.11. Synthesis of Conjugate 7-B
3.12. Synthesis of Conjugate 1-A4
3.13. Synthesis of Conjugate 1-B4
3.14. Synthesis of Conjugate 1-C4
3.15. Synthesis of Conjugate 1-D4
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCarthy, J.R.; Weissleder, R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv. Drug Deliv. Rev. 2008, 60, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Janib, S.M.; Moses, A.S.; MacKay, J.A. Imaging and drug delivery using theranostic nanoparticles. Adv. Drug Deliv. Rev. 2010, 62, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, S.S.; Reineke, T.M. Theranostics: Combining imaging and therapy. Bioconjug. Chem. 2011, 22, 1879–1903. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef] [PubMed]
- Jokerst, J.V.; Gambhir, S.S. Molecular imaging with theranostic nanoparticles. Acc. Chem. Res. 2011, 44, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.K.; Park, J.; Jon, S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics 2012, 2, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Lartigue, L.; Innocenti, C.; Kalaivani, T.; Awwad, A.; Sanchez Duque, M.D.M.; Guari, Y.; Larionova, J.; Guerin, C.; Montero, J.-L.G.; Barragan-Montero, V.; et al. Water-dispersible sugar-coated iron oxide nanoparticles. An evaluation of their relaxometric and magnetic hyperthermia properties. J. Am. Chem. Soc. 2011, 133, 10459–10472. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Lee, N.; Kim, T.; Kim, J.; Hyeon, T. Multifunctional mesoporous silica nanocomposite nanoparticles for theranostic applications. Acc. Chem. Res. 2011, 44, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-H.; Lee, C.-H.; Chen, M.-C.; Souris, J.S.; Tseng, F.-G.; Yang, C.-S.; Mou, C.-Y.; Chen, C.-T.; Lo, L.-W.J. Tri-functionalization of mesoporous silica nanoparticles for comprehensive cancer theranostics-the trio of imaging, targeting and therapy. Mater. Chem. 2010, 20, 6149–6157. [Google Scholar] [CrossRef]
- Jain, T.K.; Foy, S.P.; Erokwu, B.; Dimitrijevic, S.; Flask, C.A.; Labhasetwar, V. Magnetic resonance imaging of multifunctional pluronic stabilized iron-oxide nanoparticles in tumor-bearing mice. Biomaterials 2009, 30, 6748–6756. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.D.; Choi, S.-W.; Cai, X.; Li, W.; Cho, E.C.; Jeong, U.; Wang, L.V.; Xia, Y. A new theranostic system based on gold nanocages and phase-change materials with unique features for photoacoustic imaging and controlled release. J. Am. Chem. Soc. 2011, 133, 4762–4765. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-P.; Leong, K.W. Quantum dot-based theranostics. Nanoscale 2010, 2, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Wang, J.; Dai, Z.; Jin, Y.; Qu, E.; Xing, Z.; Guo, C.; Yue, X.; Liu, J. Gold-nanoshelled microcapsules: A theranostic agent for ultrasound contrast imaging and photothermal therapy. Angew. Chem. Int. Ed. Eng. 2011, 50, 3017–3021. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Feng, L.; Shi, X.; Liu, Z. Nano-graphene in biomedicine: Theranostic applications. Chem. Soc. Rev. 2013, 42, 530–547. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, W.T.; Al-Jamal, K.T.; Bomans, P.H.; Frederik, P.M.; Kostarelos, K. Functionalized-quantum-dot-liposome hybrids as multimodal nanoparticles for cancer. Small 2008, 4, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, W.T.; Kostarelos, K. Liposomes: From a clinically established drug delivery system to a nanoparticle platform for theranostic nanomedicine. Acc. Chem. Res. 2011, 44, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, Y.; Liu, T.; Hu, X.; Zhang, G.; You, Y.; Liu, S. Amphiphilic multiarm star block copolymer-based multifunctional unimolecular micelles for cancer targeted drug delivery and magnetic resonance imaging. Biomaterials 2011, 32, 6595–6805. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Xiao, Z.; Kamaly, N.; Farokhzad, O.C. Self-assembled targeted nanoparticles: Evolution of technologies and bench to bedside translation. Acc. Chem. Res. 2011, 44, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Thomas, T.P.; Peters, J.; Kotlyar, A.; Myc, A.; Bakers, J.R., Jr. Tumor angiogenic vasculature targeting with PAMAM dendrimer-RGD conjugates. Chem. Comm. 2005, 5739–5741. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, H.; Jia, X.; Lu, W.-L.; Lou, J.; Wei, Y. A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials 2012, 33, 3899–3908. [Google Scholar] [CrossRef] [PubMed]
- Tomalia, D.A. In quest of a systematic framework for unifying and defining nanoscience. J. Nanopart. Res. 2009, 11, 1251–1310. [Google Scholar] [CrossRef] [PubMed]
- Tomalia, D.A.; Khanna, S.N. A systematic framework and nanoperiodic concept for unifying nanoscience: Hard/Soft nanoelements, superatoms, meta-atoms, new emerging properties, periodic property patterns, and predictive mendeleev-like nanoperiodic tables. Chem. Rev. 2016, 116, 2705–2774. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.M.; Nance, E.; Kannan, S.; Tomalia, D.A. Emerging concepts in dendrimer-based nanomedicine: From design principles to clinical applications. J. Internal Med. 2014, 276, 579–617. [Google Scholar] [CrossRef] [PubMed]
- Enciso, A.E.; Abid, Z.M.; Simanek, E.E. Rapid, semi-automated convergent synthesis of low generation triazine dendrimers using microwave assisted reactions. Polym. Chem. 2014, 5, 4635–4640. [Google Scholar] [CrossRef]
- Gray, W.D.; Wu, R.J.; Yin, X.; Zhou, J.; Davis, M.E.; Luo, Y. Dendrimeric bowties featuring hemispheric-selective decoration of ligands for microRNA-based therapy. Biomacromolecules 2013, 14, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wängler, C.; Mashauer, S.; Prante, O.; Schafer, M.; Schmirrmacher, R.; Bartenstein, P.; Eisenhut, M.; Wängler, B. Multimerization of cRGD peptides by click chemistry: Synthetic strategies, chemical limitations, and influence on biological properties. ChemBioChem 2010, 11, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.



















© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Ji, K.; Simanek, E.E. Functionalization of a Triazine Dendrimer Presenting Four Maleimides on the Periphery and a DOTA Group at the Core. Molecules 2016, 21, 335. https://doi.org/10.3390/molecules21030335
Lee C, Ji K, Simanek EE. Functionalization of a Triazine Dendrimer Presenting Four Maleimides on the Periphery and a DOTA Group at the Core. Molecules. 2016; 21(3):335. https://doi.org/10.3390/molecules21030335
Chicago/Turabian StyleLee, Changsuk, Kun Ji, and Eric E. Simanek. 2016. "Functionalization of a Triazine Dendrimer Presenting Four Maleimides on the Periphery and a DOTA Group at the Core" Molecules 21, no. 3: 335. https://doi.org/10.3390/molecules21030335