
Improved Homology Model of the Human all-trans Retinoic Acid Metabolizing Enzyme CYP26A1

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
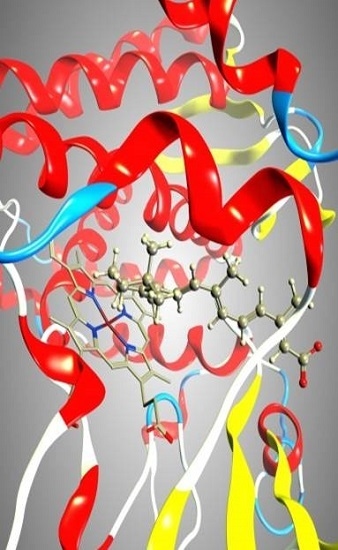
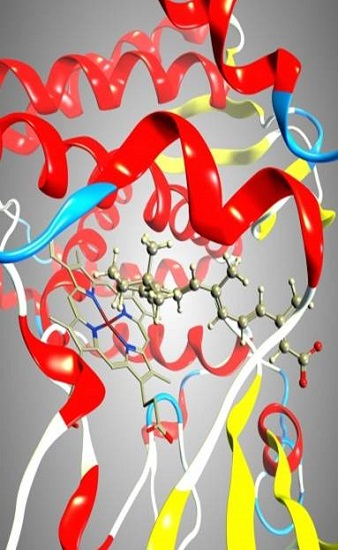
2.1. CYP26A1 Homology Model
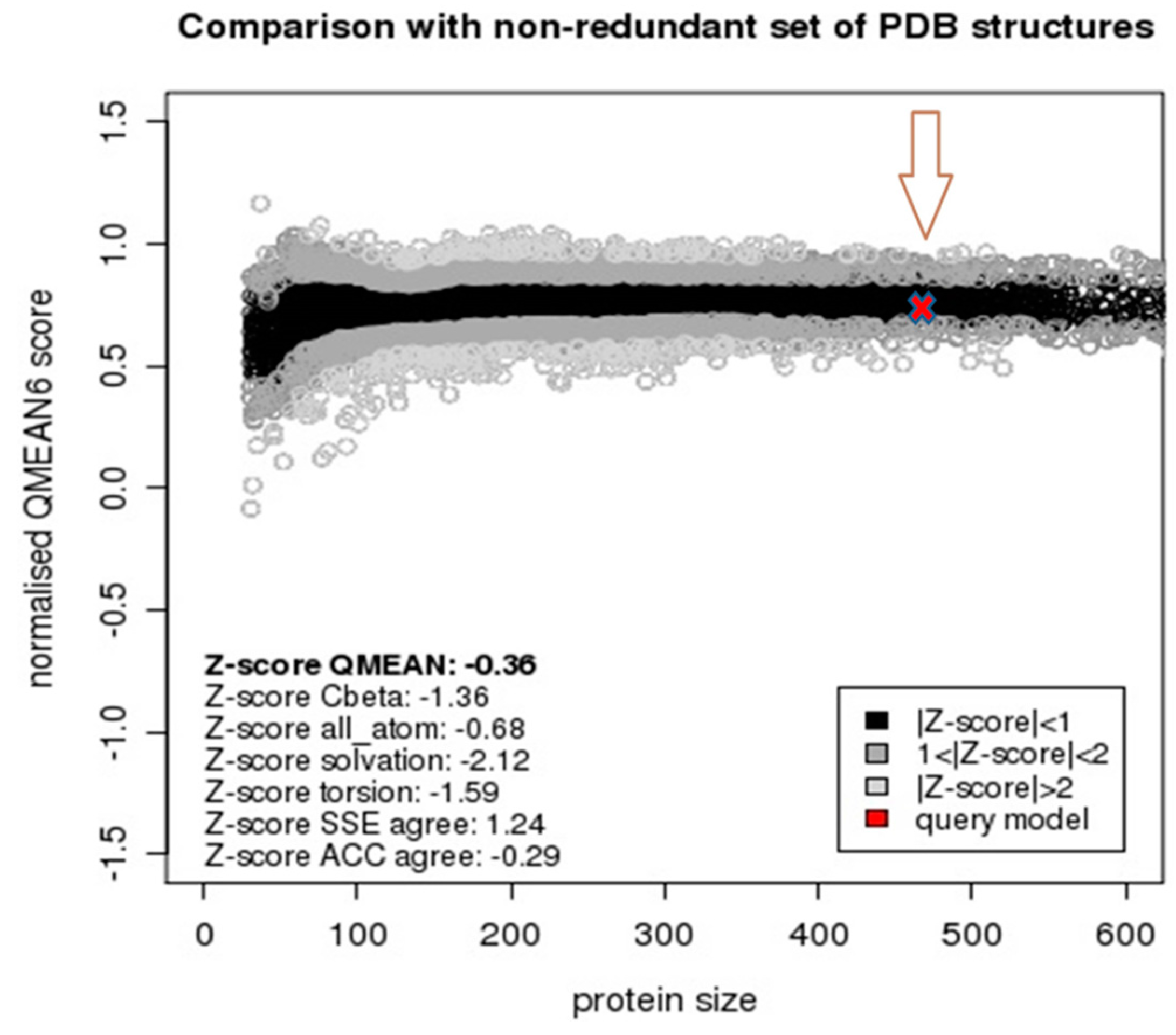
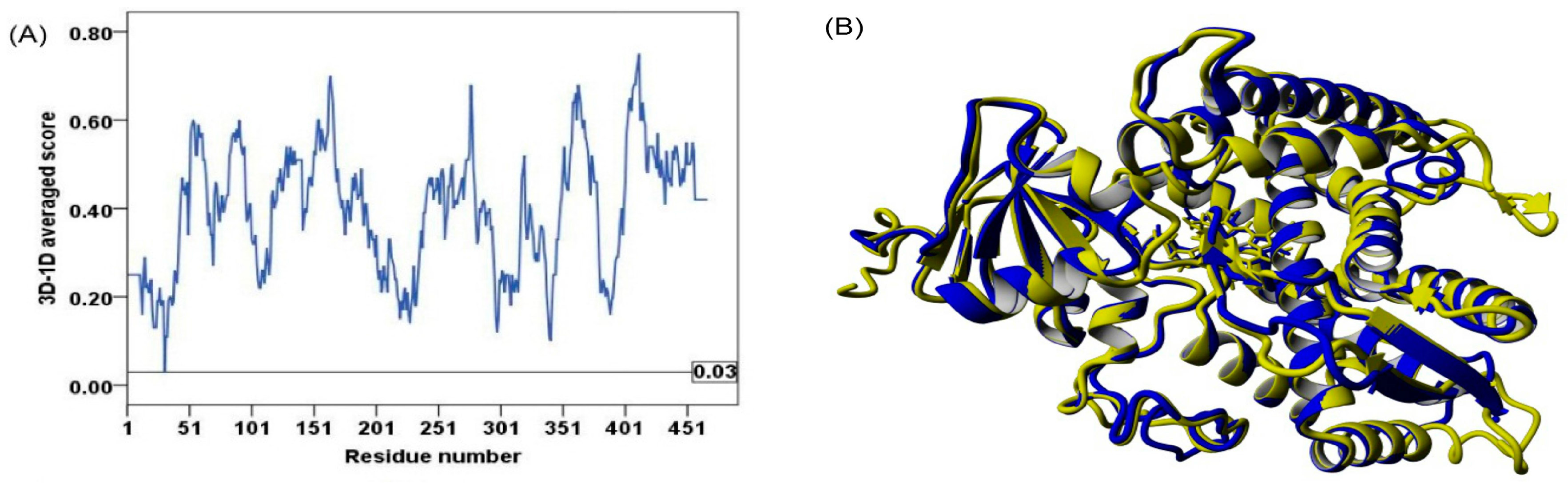
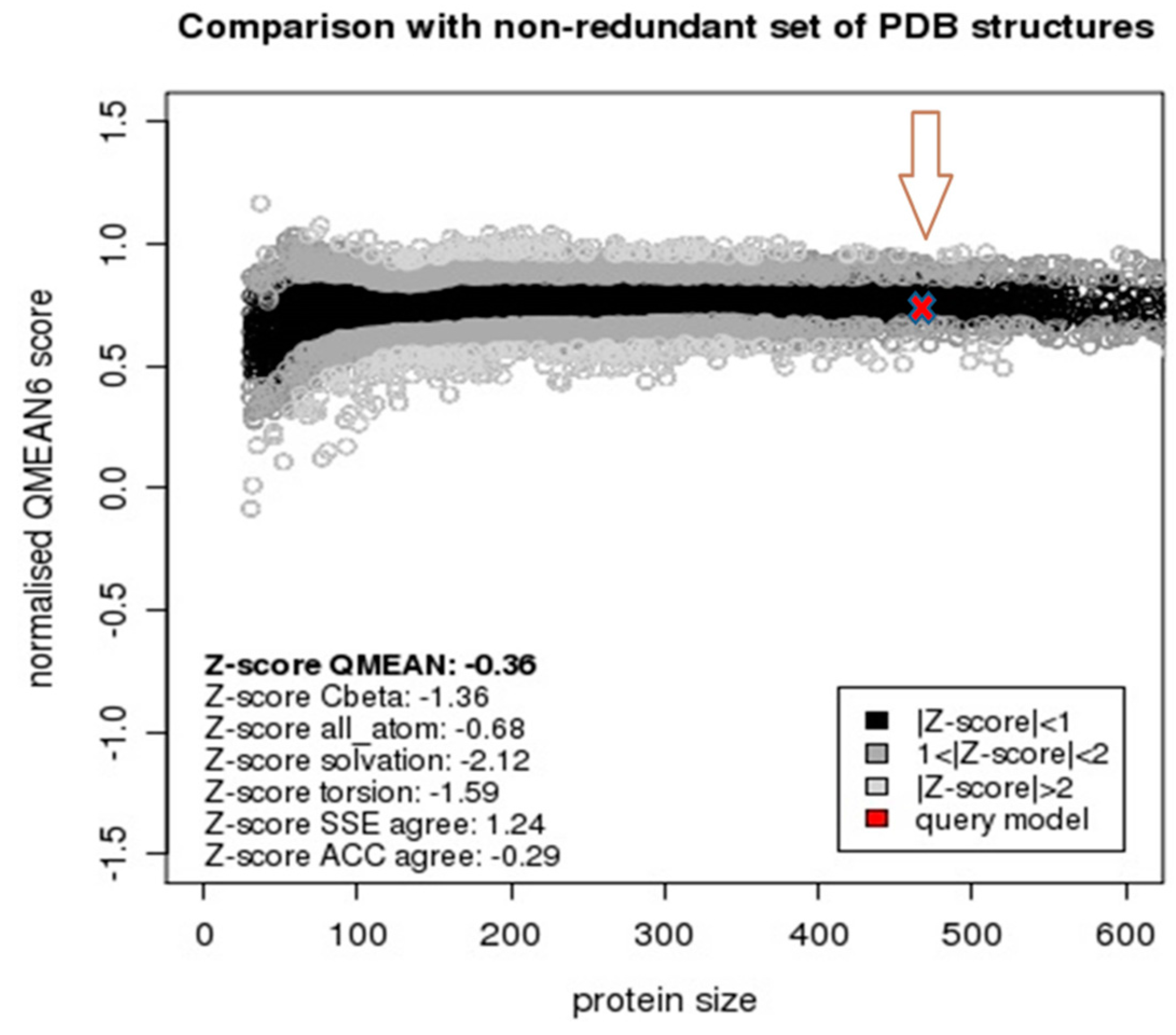
2.2. Model Quality
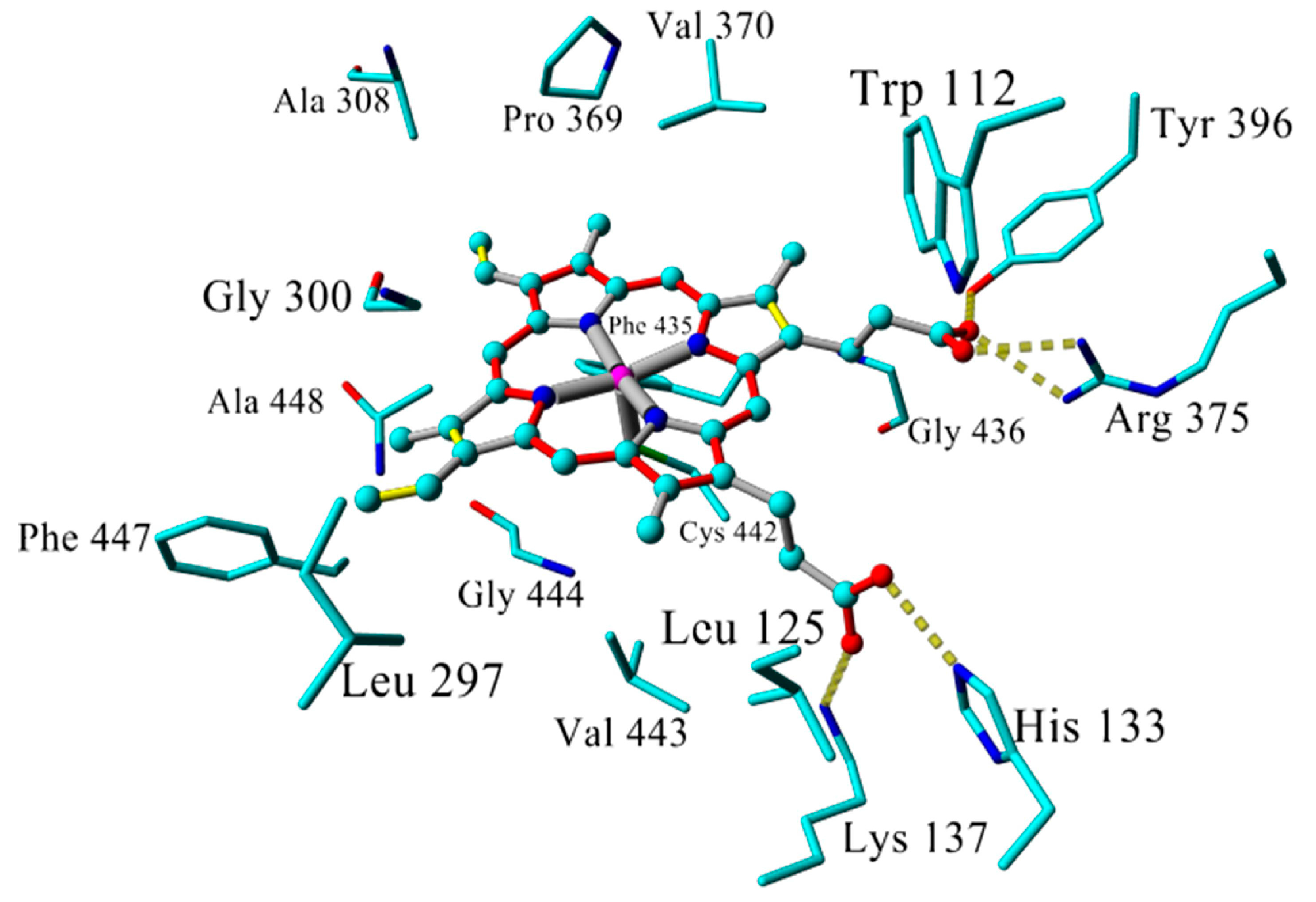
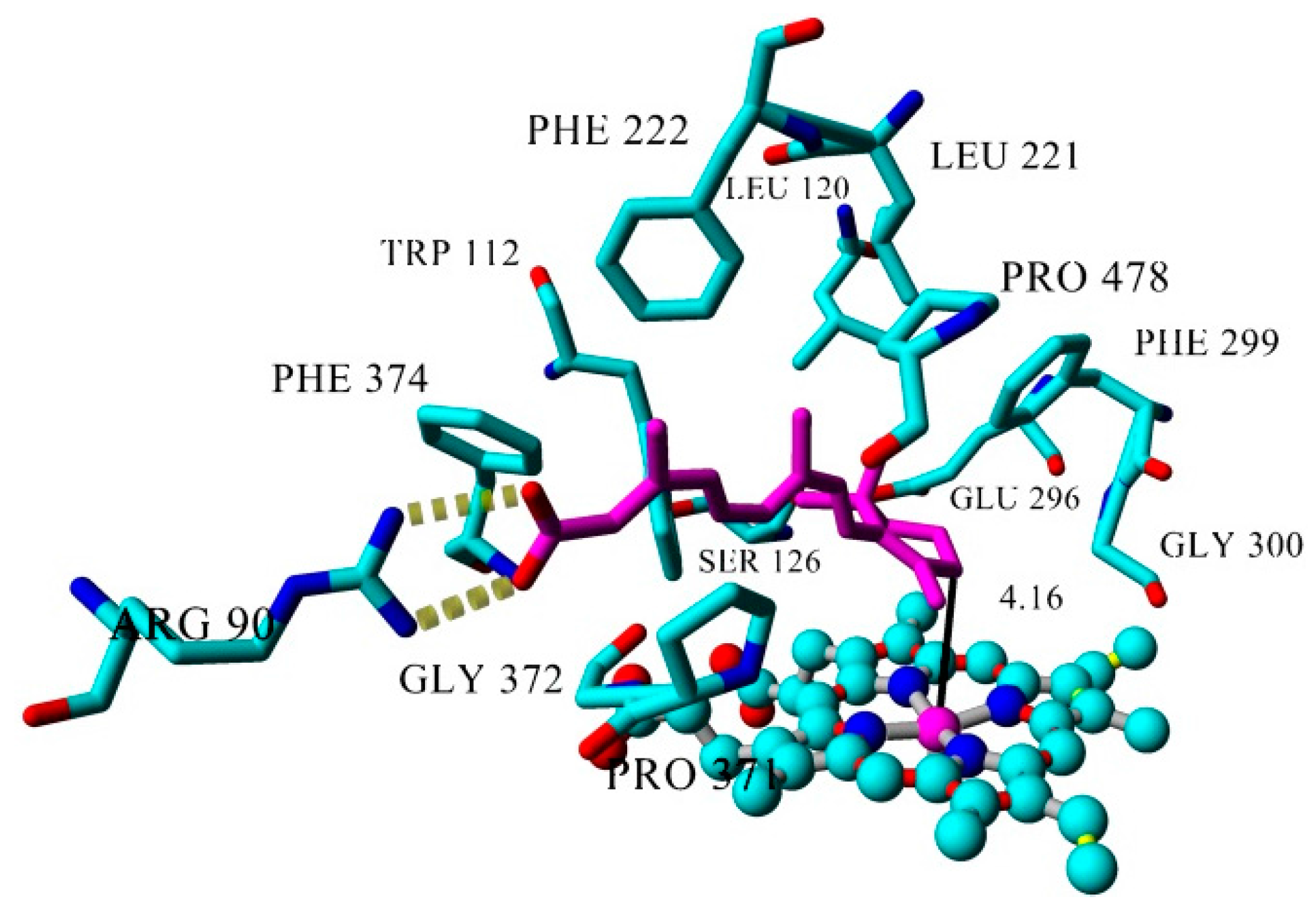
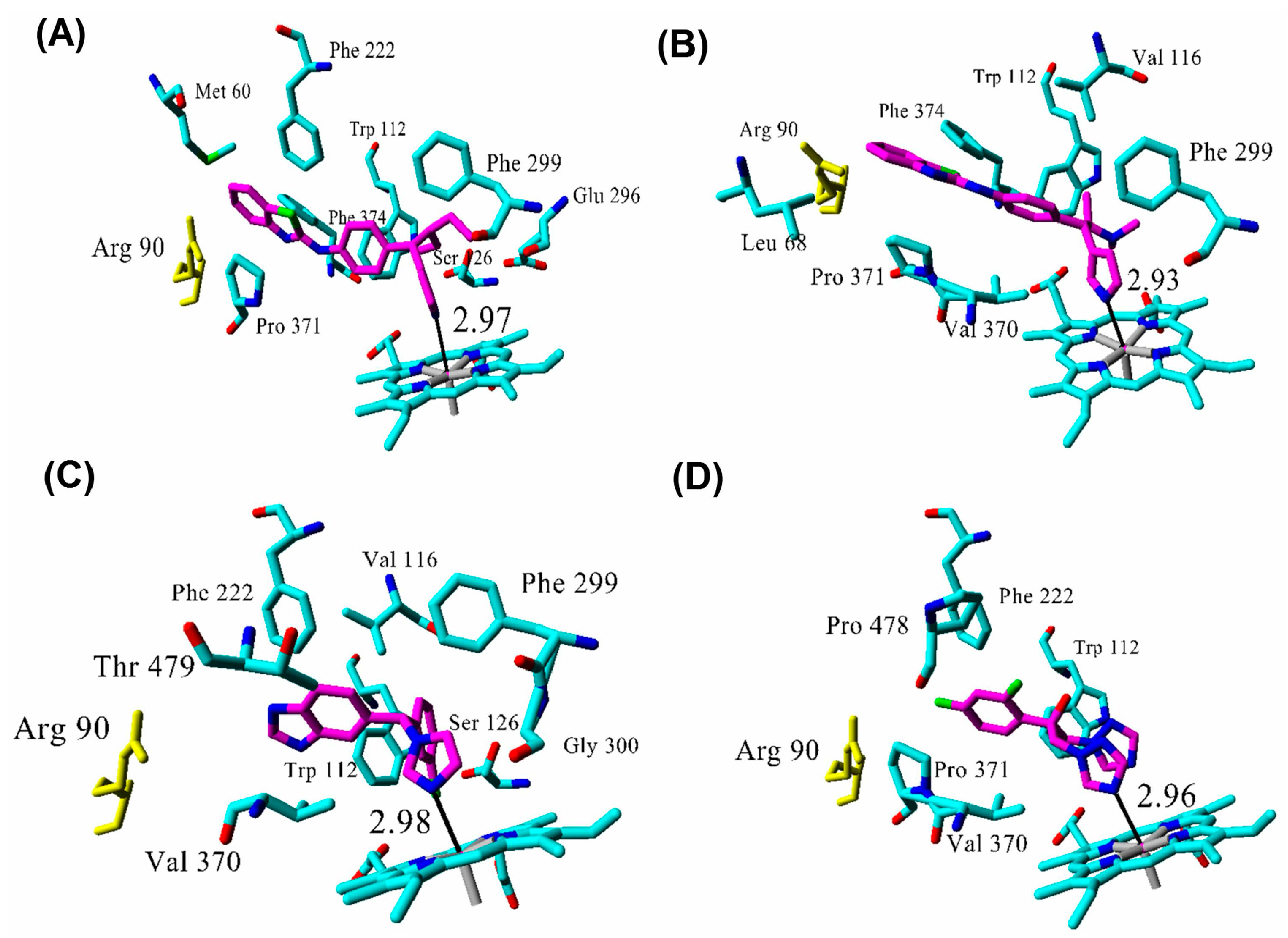
2.3. Docking
2.4. Binding Energy Calculations
3. Materials and Methods
3.1. Computational Approaches
3.2. Model Building
3.3. Model Quality Assessment
3.4. Ligands
3.5. Docking
3.6. Binding Energy Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.A.; Folias, A.E.; Wang, C.; Napoli, J.L. Quantitative Profiling of Endogenous Retinoic Acid in Vivo and in Vitro by Tandem Mass Spectrometry. Anal. Chem. 2008, 80, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Marill, J.; Cresteil, T.; Lanotte, M.; Chabot, G.G. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol. Pharmacol. 2000, 58, 1341–1348. [Google Scholar] [PubMed]
- McSorley, L.C.; Daly, A.K. Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem. Pharmacol. 2000, 60, 517–526. [Google Scholar] [CrossRef]
- White, J.A.; Beckett-Jones, B.; Guo, Y.D.; Dilworth, F.J.; Bonasoro, J.; Jones, G.; Petkovich, M. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J. Biol. Chem. 1997, 272, 18538–18541. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Ramshaw, H.; Taimi, M.; Stangle, W.; Zhang, A.; Everingham, S.; Creighton, S.; Tam, S.-P.; Jones, G.; Petkovich, M. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 6403–6408. [Google Scholar] [CrossRef] [PubMed]
- Taimi, M.; Helvig, C.; Wisniewski, J.; Ramshaw, H.; White, J.; Amad, M.; Korczak, B.; Petkovich, M. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J. Biol. Chem. 2004, 279, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Topletz, A.R.; Thatcher, J.E.; Zelter, A.; Lutz, J.D.; Tay, S.; Nelson, W.L.; Isoherranen, N. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem. Pharmacol. 2012, 83, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Yang, Z. Expression of RALDHs (ALDH1As) and CYP26s in human tissues and during the neural differentiation of P19 embryonal carcinoma stem cell. Gene Expr. Patterns 2008, 8, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Helvig, C.; Taimi, M.; Cameron, D.; Jones, G.; Petkovich, M. Functional properties and substrate characterization of human CYP26A1, CYP26B1, and CYP26C1 expressed by recombinant baculovirus in insect cells. J. Pharmacol. Toxicol. Methods 2011, 64, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Grimwade, D.; Tallman, M.S.; Lowenberg, B.; Fenaux, P.; Estey, E.H.; Naoe, T.; Lengfelder, E.; Büchner, T.; Döhner, H.; et al. Management of acute promyelocytic leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009, 113, 1875–1891. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.S. Vitamin a: History, current uses, and controversies. Semin. Cutan. Med. Surg. 2012, 31, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Oehlen, B.; Ma, G.; McCormack, S.; Sung Lim, D.; Tarrant, J.; Zhu, X.; Panicker, B.; Goldberg, I.D. Identification of new CYP26 inhibitors with efficacy in breast cancer xenograft models. Mol. Cancer Ther. 2013, 12, B257. [Google Scholar] [CrossRef]
- Muindi, J.; Frankel, S.R.; Miller, W.H.; Jakubowski, A.; Scheinberg, D.A.; Young, C.W.; Dmitrovsky, E.; Warrell, R.P. Continuous Treatment with All-Trans Retinoic Acid Causes a Progressive Reduction in Plasma Drug Concentrations—Implications for Relapse and Retinoid Resistance in Patients with Acute Promyelocytic Leukemia. Blood 1992, 79, 299–303. [Google Scholar] [PubMed]
- Thatcher, J.E.; Isoherranen, N. The role of CYP26 enzymes in retinoic acid clearance. Expert. Opin. Drug Metab. Toxicol. 2009, 5, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Njar, V.C.O.; Gediya, L.; Purushottamachar, P.; Chopra, P.; Vasaitis, T.S.; Khandelwal, A.; Mehta, J.; Huynh, C.; Belosay, A.; Patel, J. Retinoic acid metabolism blocking agents (RAMBAs) for treatment of cancer and dermatological diseases. Bioorg. Med. Chem. 2006, 14, 4323–4340. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.H.; Buttrick, B.R.; Isoherranen, N. Therapeutic potential of the inhibition of the retinoic acid hydroxylases CYP26A1 and CYP26B1 by xenobiotics. Curr. Top. Med. Chem. 2013, 13, 1402–1428. [Google Scholar] [CrossRef] [PubMed]
- Soppie, P.; Borgers, M.; Borghgraef, P.; Dillen, L.; Goossens, J.; Sanz, J.; Szel, H.; van Hove, C.; van Nyen, G.; Nobels, G.; et al. R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodents. J. Pharmacol. Exp. Ther. 2000, 293, 304–312. [Google Scholar]
- Van Heusden, J.; Van Ginckel, R.; Bruwiere, H.; Moelans, P.; Janssen, B.; Floren, W.; van der Leede, B.J.; van Dun, J.; Sanz, G.; Venet, M.; et al. Inhibition of all-TRANS-retinoic acid metabolism by R116010 induces antitumour activity. Br. J. Cancer 2002, 86, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Van Wauwe, J.; Van Nyen, G.; Coene, M.C.; Stoppie, P.; Cools, W.; Goossens, J.; Borghgraef, P.; Janssen, P.A. Liarozole, an inhibitor of retinoic acid metabolism, exerts retinoid-mimetic effects in vivo. J. Pharmacol. Exp. Ther. 1992, 261, 773–779. [Google Scholar] [PubMed]
- Barrier Therapeutics Granted European Orphan Drug Status for Liarozole. Available online: http://www.prnewswire.com/news-releases/71303547.html (accessed on 10 August 2015).
- Barrier Therapeutics’ Liarozole Receives FDA Orphan Drug Status. Available online: http://www.prnewswire.com/news-releases/barrier-therapeutics-liarozole-receives-fda-orphan-drug-status-75105912.html (accessed on 10 August 2015).
- Yee, S.W.; Jarno, L.; Gomaa, M.S.; Elford, C.; Ooi, L.L.; Coogan, M.P.; McClelland, R.; Nicholson, R.I.; Evans, B.A.J.; Brancale, A.; et al. Novel tetralone-derived retinoic acid metabolism blocking agents: Synthesis and in vitro evaluation with liver microsomal and MCF-7 CYP26A1 cell assays. J. Med. Chem. 2005, 48, 7123–7131. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, M.S.; Bridgens, C.E.; Aboraia, A.S.; Veal, G.J.; Redern, C.P.F.; Brancale, A.; Armstrong, J.L.; Simons, C. Small Molecule Inhibitors of Retinoic Acid 4-Hydroxylase (CYP26): Synthesis and Biological Evaluation of Imidazole Methyl 3-(4-(aryl-2-ylamino)phenyl)propanoates. J. Med. Chem. 2011, 54, 2778–2791. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, M.S.; Bridgens, C.E.; Illingworth, N.A.; Veal, G.J.; Redfern, C.P.; Brancale, A.; Armstrong, J.L.; Simons, C. Novel retinoic acid 4-hydroxylase (CYP26) inhibitors based on a 3-(1H-imidazol- and triazol-1-yl)-2,2-dimethyl-3-(4-(phenylamino)phenyl)propyl scaffold. Bioorg. Med. Chem. 2012, 20, 4201–4207. [Google Scholar] [CrossRef] [PubMed]
- Kühnel, K.; Ke, N.; Cryle, M.J.; Sligar, S.G.; Schuler, M.A.; Schlichting, I. Crystal Structures of Substrate-Free and Retinoic Acid-Bound Cyanobacterial Cytochrome P450 CYP120A1. Biochemistry 2008, 47, 6552–6559. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, J.E.; Buttrick, B.R.; Shaffer, S.A.; Shimishoni, J.A.; Goodlett, D.R.; Nelson, W.L.; Isoherranen, N. Substrate specificity and ligand interactions of CYP26A1, the human liver retinoic acid hydroxylase. Mol. Pharmacol. 2011, 80, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Strid, Å.; Sirsjö, A.; Eriksson, L.A. Homology Models and Molecular Modeling of Human Retinoic Acid Metabolizing Enzymes Cytochrome P450 26A1 (CYP26A1) and P450 26B1 (CYP26B1). J. Chem. Theory Comput. 2008, 4, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, M.S.; Yee, S.W.; Milbourne, C.E.; Barbera, M.C.; Simons, C.; Brancale, A. Homology model of human retinoic acid metabolising enzyme cytochrome P450 26A1 (CYP26A1): Active site architecture and ligand binding. J. Enzyme Inhib. Med. Chem. 2006, 21, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.H.; Xiong, X.Q.; Sha, Y.; Yan, M.C.; Lin, B.; Wang, J.; Jing, D.M.; Cheng, M.S. Structure prediction and R115866 binding study of human CYP26A1: Homology modelling, fold recognition, molecular docking and MD simulations. Mol. Sim. 2008, 34, 337–346. [Google Scholar] [CrossRef]
- Sun, B.; Song, S.; Hao, C.Z.; Huang, W.X.; Liu, C.C.; Xie, H.L.; Lin, B.; Cheng, M.S.; Zhao, D.M. Molecular recognition of CYP26A1 binding pockets and structure-activity relationship studies for design of potent and selective retinoic acid metabolism blocking agents. J. Mol. Graph. Model. 2015, 56, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Shimishoni, J.A.; Roberts, A.G.; Scian, M.; Topletz, A.R.; Blankert, S.A.; Halpert, J.R.; Nelson, W.L.; Isoherranen, N. Stereoselective formation and metabolism of 4-hydroxy-retinoic Acid enantiomers by cytochrome p450 enzymes. J. Biol. Chem. 2012, 287, 42223–42232. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.S.; Vijayasarathy, K.; Srinivas, E.; Garikapati, M.S.; Sastry, G.N. Homology modeling of membrane proteins: A critical assessment. Comput. Biol. Chem. 2006, 302, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Saenz Méndez, P.; Elmabsout, A.A.; Sävenstrand, H.; Awadalla, M.K.A.; Strid, Å.; Sirsjö, A.; Eriksson, L.A. Ciclohexanona monooxigenasa de Acinetobacter calcoaceticus: Modelo por homología basado en moldes múltiples. J. Chem. Inf. Model. 2012, 52, 2631–2637. [Google Scholar] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z. Advances in Homology Protein Structure Modeling. Curr. Protein Pept. Sci. 2006, 7, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ekross, M.; Sjögren, M. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.C.; Brueggemeier, R.W.; Hadad, C.M. The final catalytic step of cytochrome p450 aromatase: A density functional theory study. J. Am. Chem. Soc. 2005, 127, 5224–5237. [Google Scholar] [CrossRef] [PubMed]
- Balding, P.R.; Porro, C.S.; McLean, K.J.; Sutcliffe, M.J.; Maréchal, J.-D.; Munro, A.W.; de Visser, S.P. How Do Azoles Inhibit Cytochrome P450 Enzymes? A Density Functional Study. J. Chem. Phys. A 2008, 112, 12911–12918. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E. Yet Another Scientific Artificial Reality Application; YASARA: Vienna, Austria, 2004. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A Point-Charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-Phase Quantum Mechanical Calculations. J. Comp. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Dunbrack, R.L.J.; Hooft, R.W.; Kieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- RAMPAGE Server. Available online: http://mordred.bioc.cam.ac.uk/~rapper/rampage.php (accessed on 15 March 2015).
- Verify 3D Structure Evaluation Server. Available online: http://services.mbi.ucla.edu/Verify_3D/ (accessed on 15 March 2015).
- QMEAN Server. Available online: http://swissmodel.expasy.org/qmean/cgi/index.cgi (accessed on 15 March 2015).
- Pence, H.E. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2008.
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Albada, H.B.; Golub, E.; Willner, I. Computational docking simulations of a DNA-aptamer for argininamide and related ligands. J. Comput. Aided Mol. Des. 2015, 29, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, C.E.P.; Silva, P.J. Computational development of rubromycin-based lead compounds for HIV-1 reverse transcriptase inhibition. Peer J. 2014, 2, e470. [Google Scholar] [CrossRef] [PubMed]
- Rua, F.; di Nardo, F.; Sadeghi, S.J.; Gilardi, G. Toward reduction in animal sacrifice for drugs: Molecular modeling of Macaca fascicularis P450 2C20 for virtual screening of Homo sapiens P450 2C8 substrates. Biotechnol. Appl. Biochem. 2012, 59, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (µM) | Binding Energy (kcal·mol−1) | Distance to Heme Iron (Å) |
|---|---|---|---|
| atRA (substrate) | Inducer | 362 | 4.16 |
| R115866 | 0.0043 1 | 102 | 2.97 |
| R116010 | 0.0051 1 | 95 | 2.93 |
| liarozole | 2.1 1 | 63 | 2.98 |
| fluconazole | ≥10 1 | 60 | 2.96 |
| tetralone 1 | 9 2 | 50 | 3.51 |
| tetralone 2 | 30 2 | 48 | 5.82 |
| tetralone 3 | 7 2 | 51 | 3.66 |
| tetralone 4 | 5 2 | 56 | 3.40 |
| tetralone 5 | 9 2 | 62 | 3.62 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awadalla, M.K.A.; Alshammari, T.M.; Eriksson, L.A.; Saenz-Méndez, P. Improved Homology Model of the Human all-trans Retinoic Acid Metabolizing Enzyme CYP26A1. Molecules 2016, 21, 351. https://doi.org/10.3390/molecules21030351
Awadalla MKA, Alshammari TM, Eriksson LA, Saenz-Méndez P. Improved Homology Model of the Human all-trans Retinoic Acid Metabolizing Enzyme CYP26A1. Molecules. 2016; 21(3):351. https://doi.org/10.3390/molecules21030351
Chicago/Turabian StyleAwadalla, Mohamed K. A., Thamir M. Alshammari, Leif A. Eriksson, and Patricia Saenz-Méndez. 2016. "Improved Homology Model of the Human all-trans Retinoic Acid Metabolizing Enzyme CYP26A1" Molecules 21, no. 3: 351. https://doi.org/10.3390/molecules21030351
APA StyleAwadalla, M. K. A., Alshammari, T. M., Eriksson, L. A., & Saenz-Méndez, P. (2016). Improved Homology Model of the Human all-trans Retinoic Acid Metabolizing Enzyme CYP26A1. Molecules, 21(3), 351. https://doi.org/10.3390/molecules21030351