
Azidation in the Difunctionalization of Olefins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
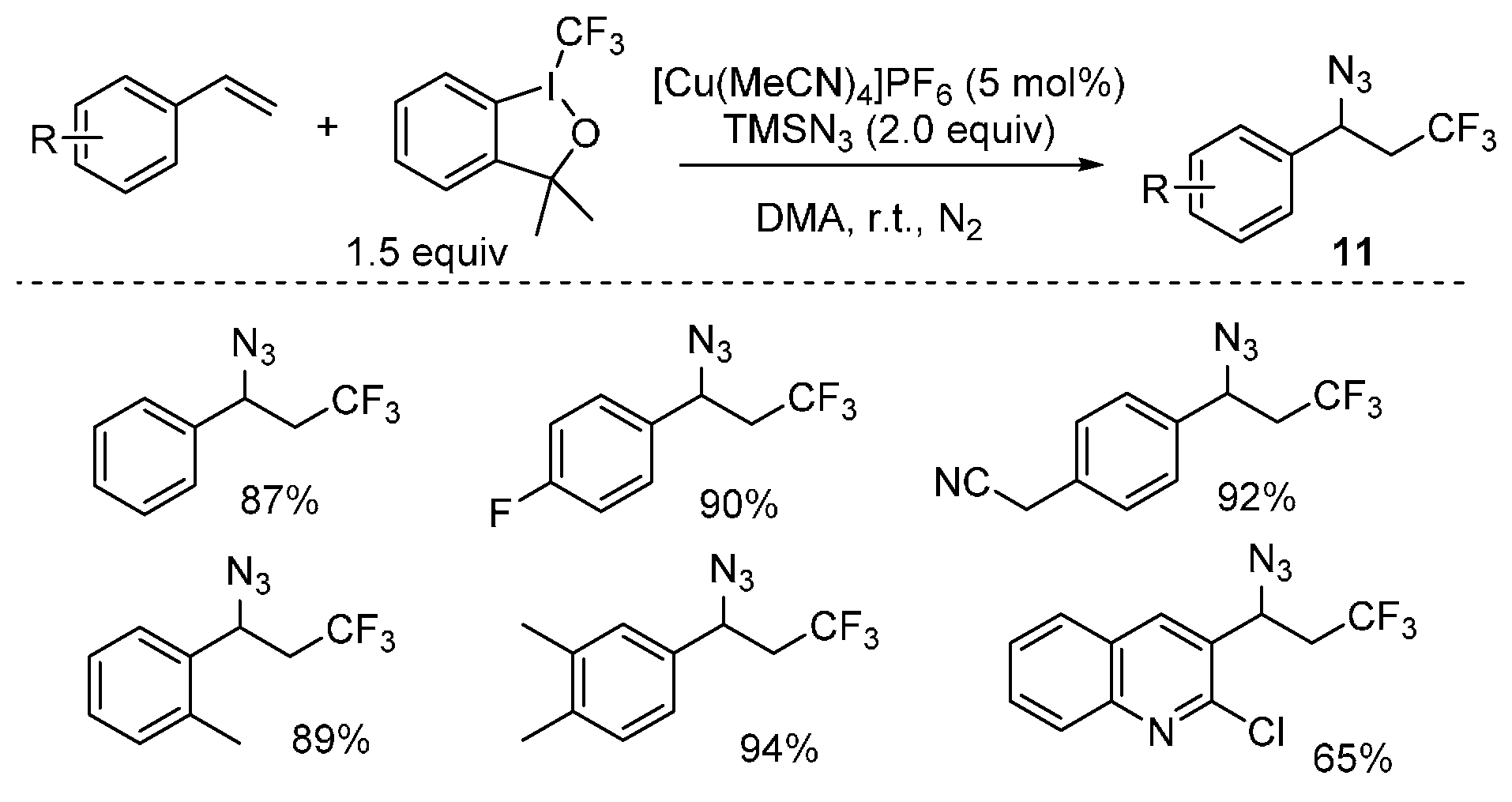{kind=link}
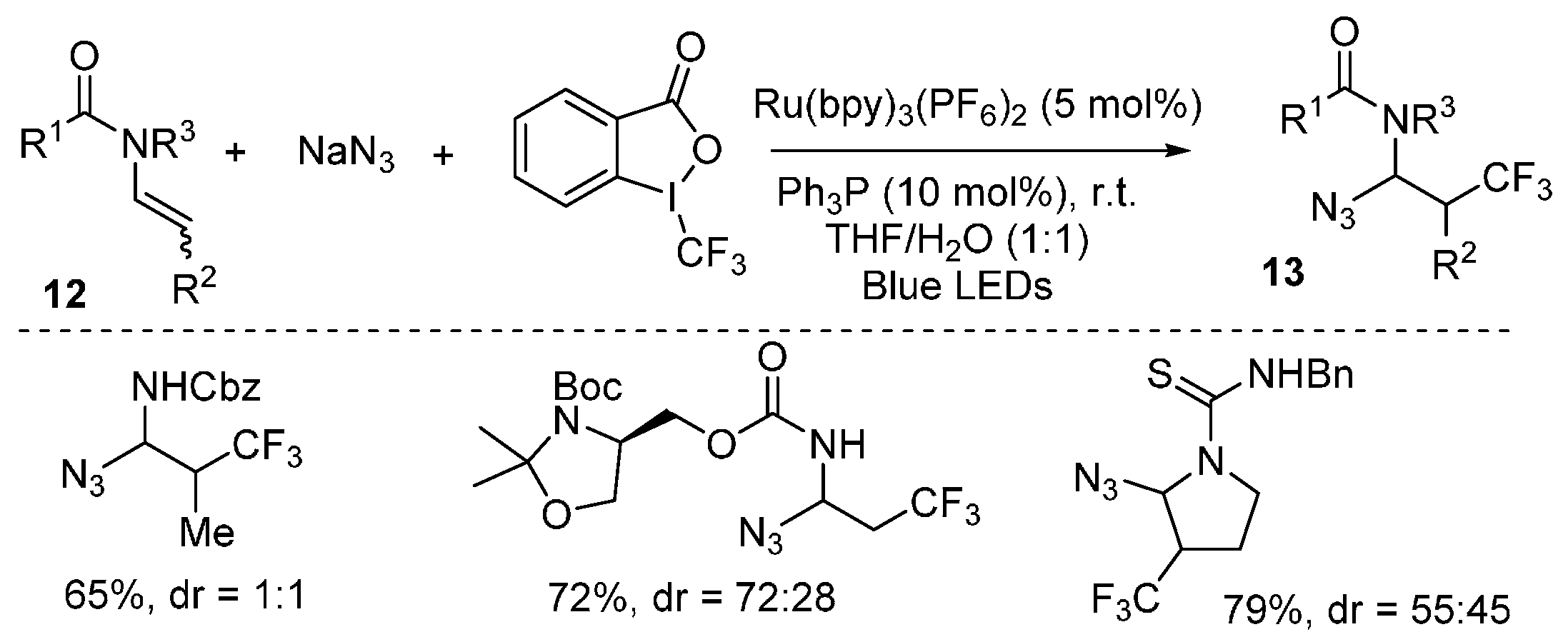{kind=link}
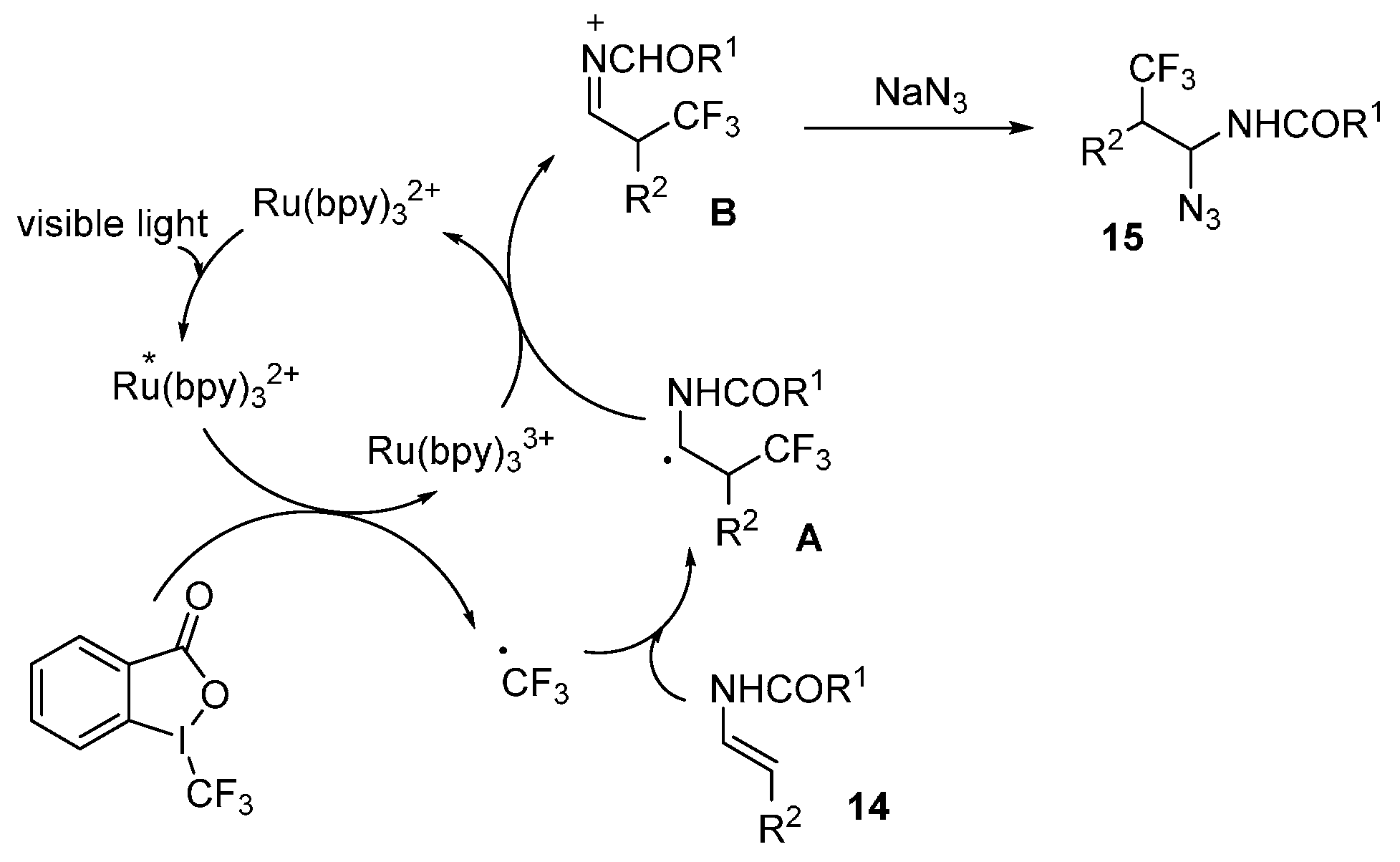{kind=link}
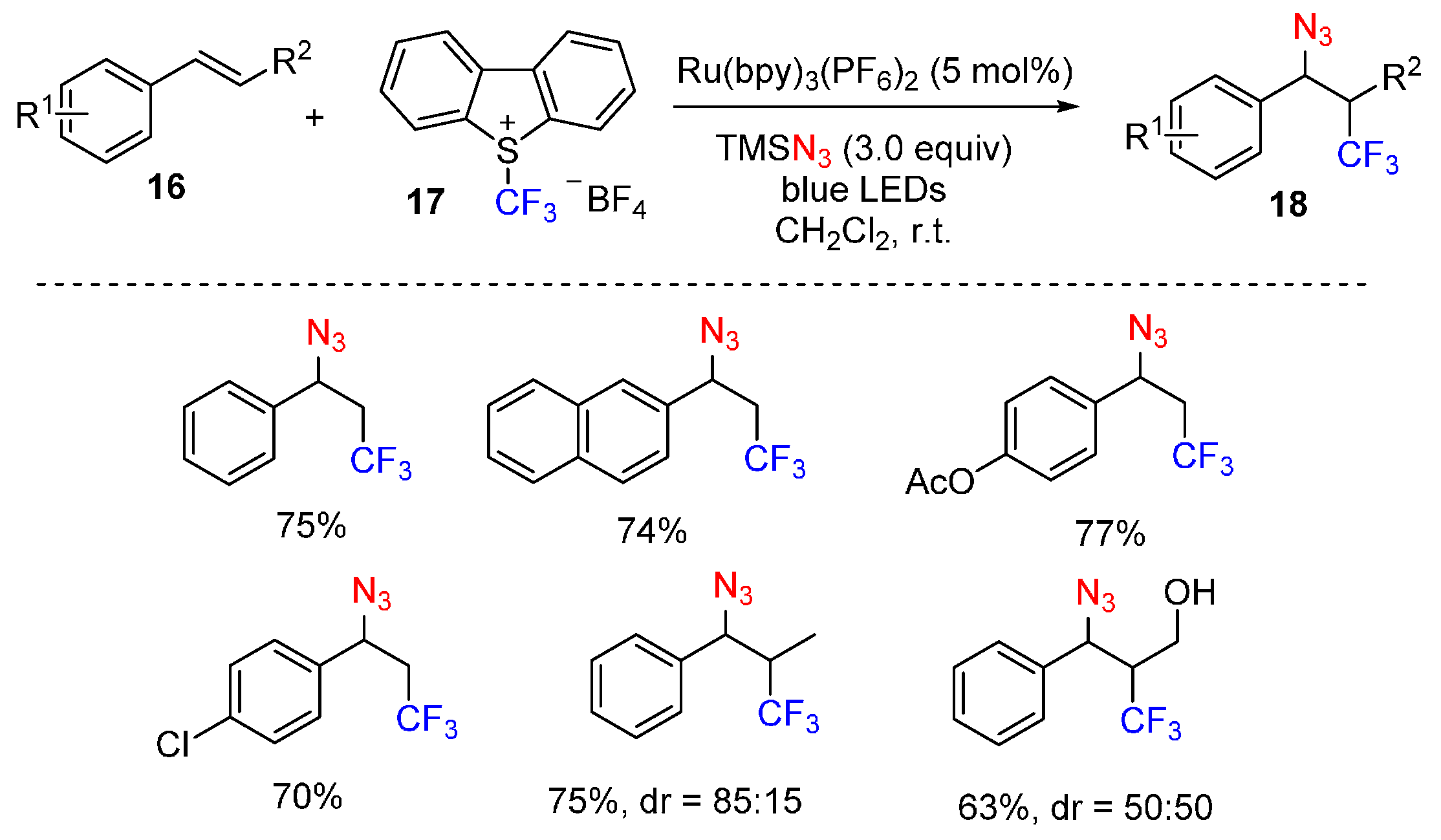{kind=link}
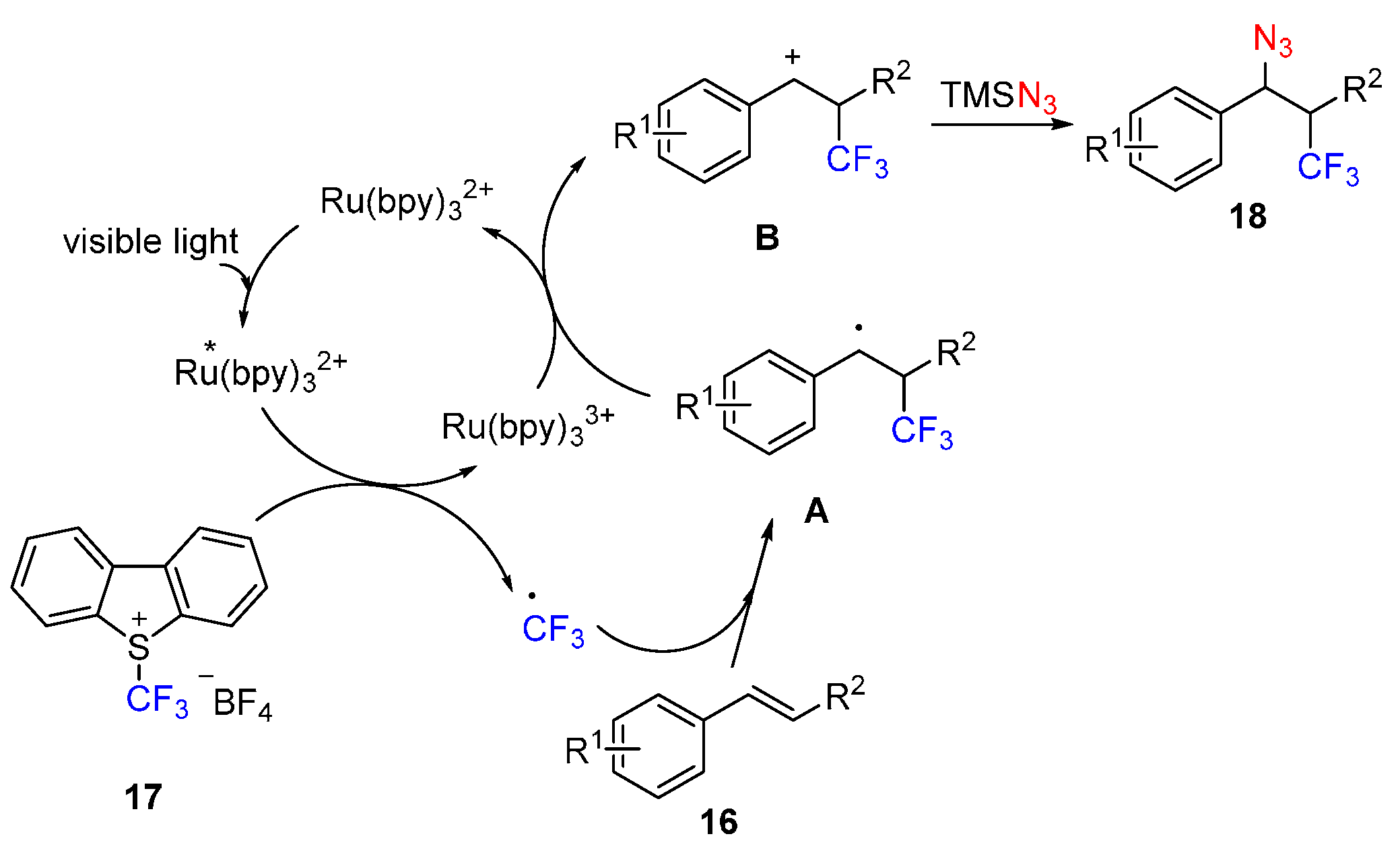{kind=link}
{kind=link}
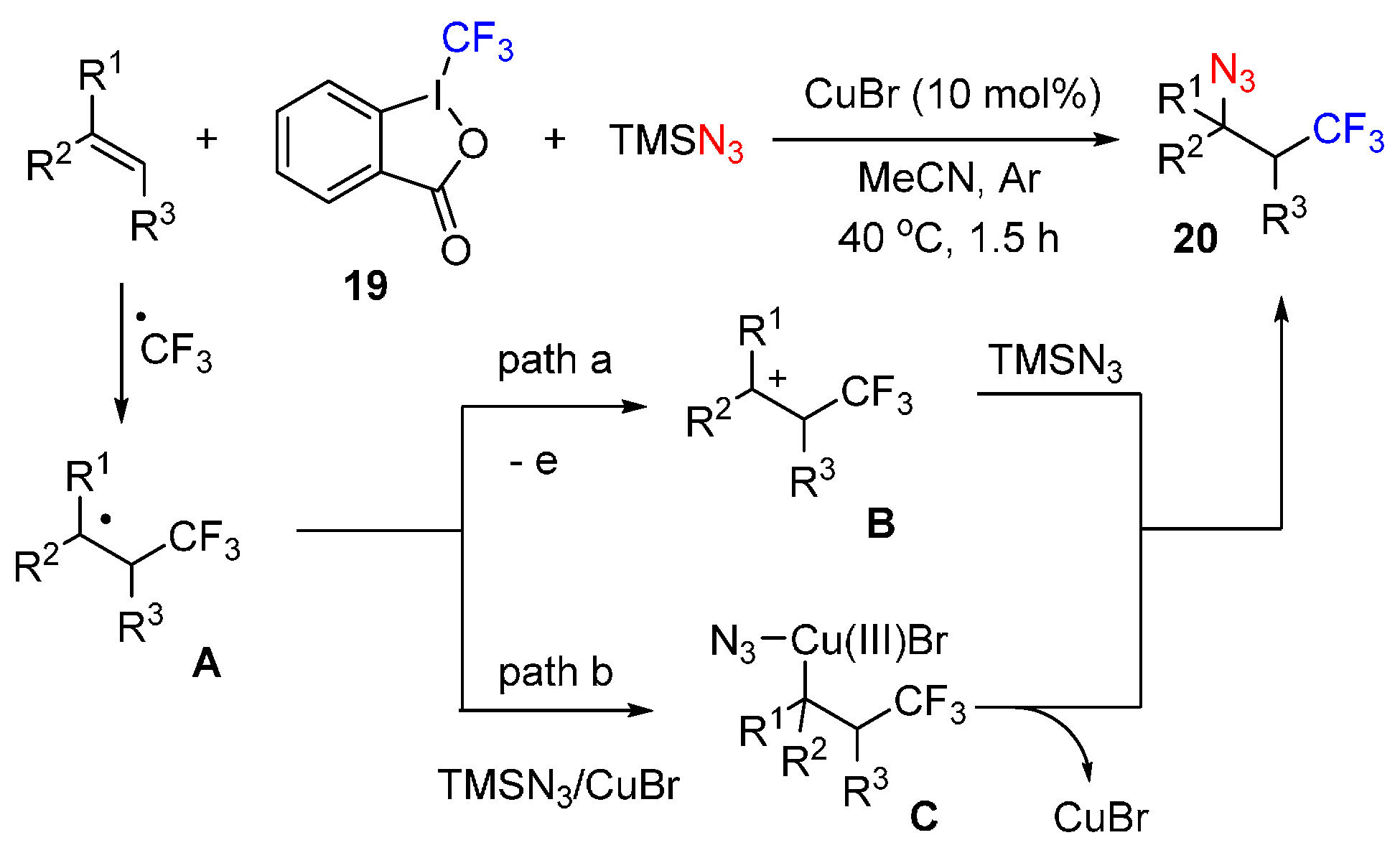
{kind=link}
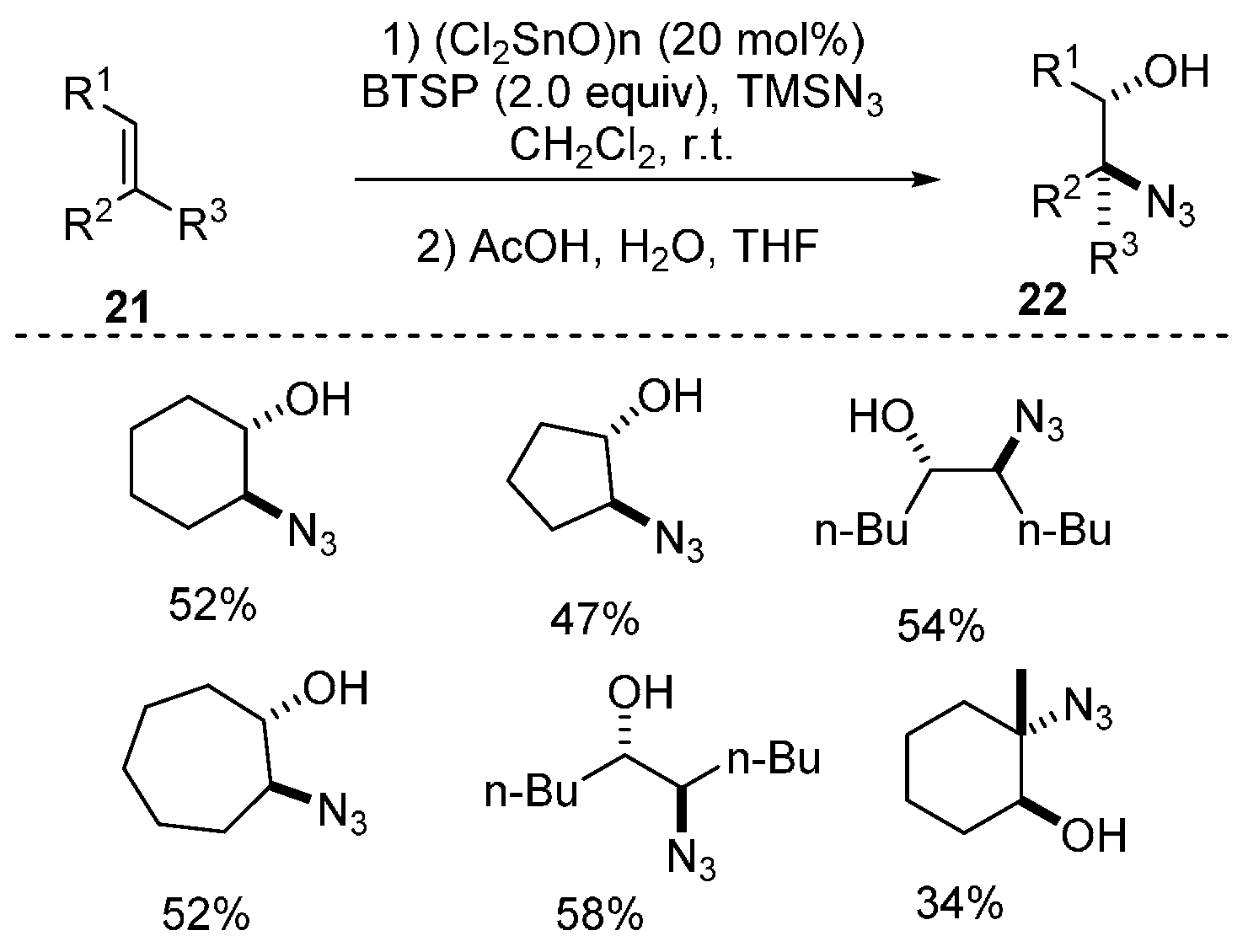
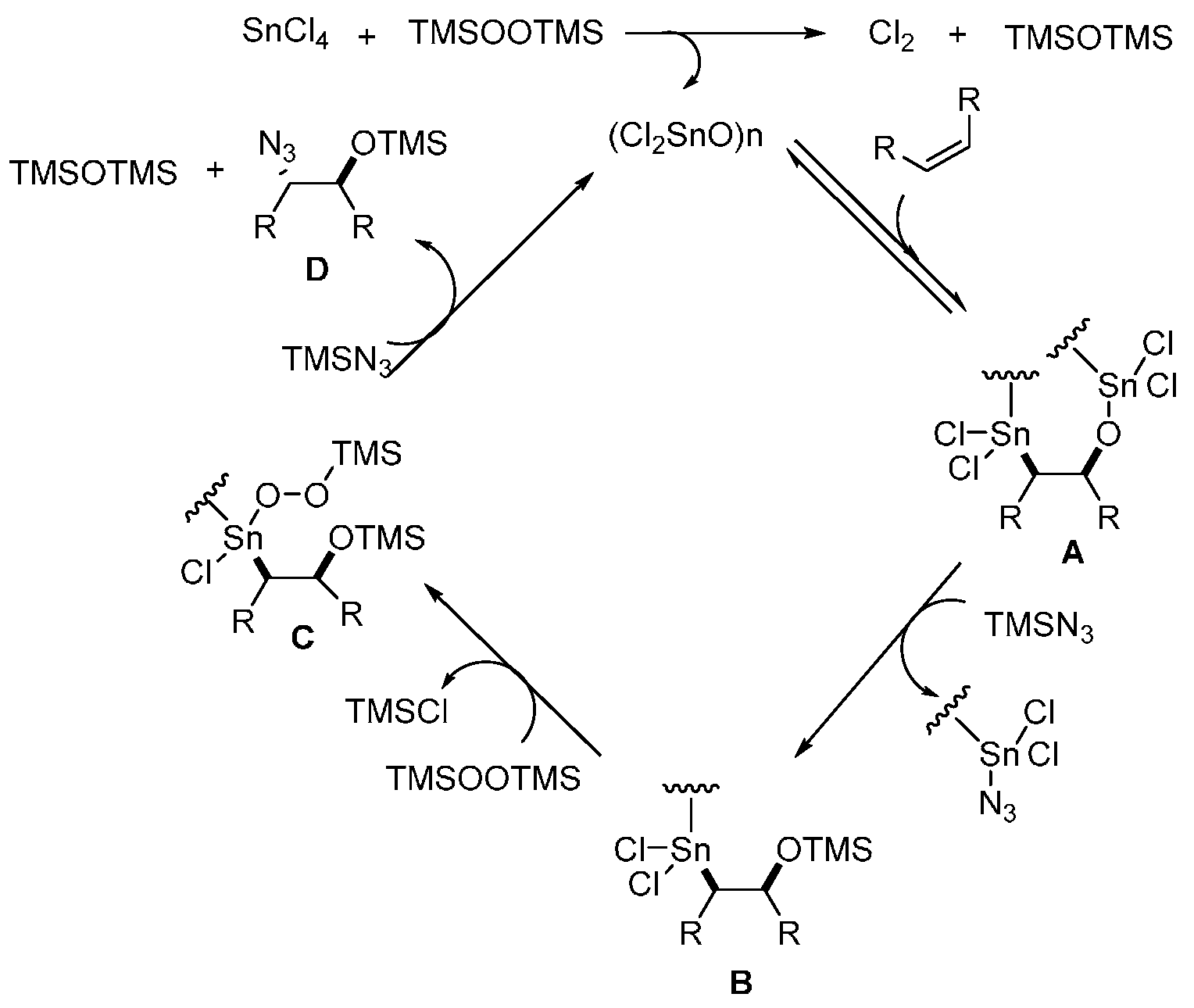{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
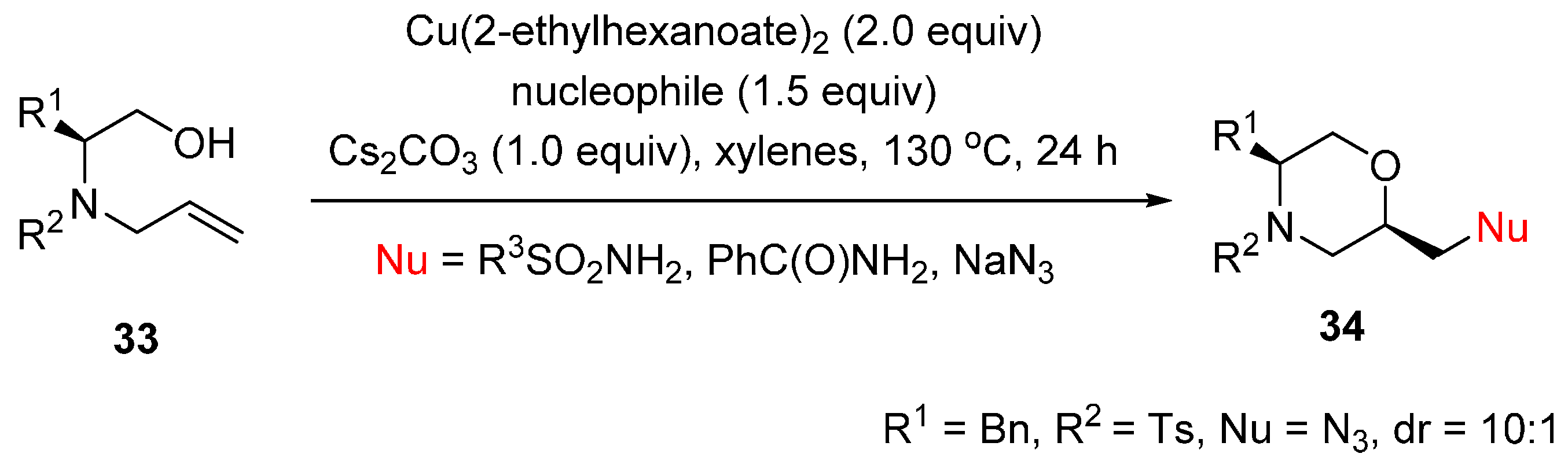{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Application of Organic Azides in Difunctionalization of Olefins
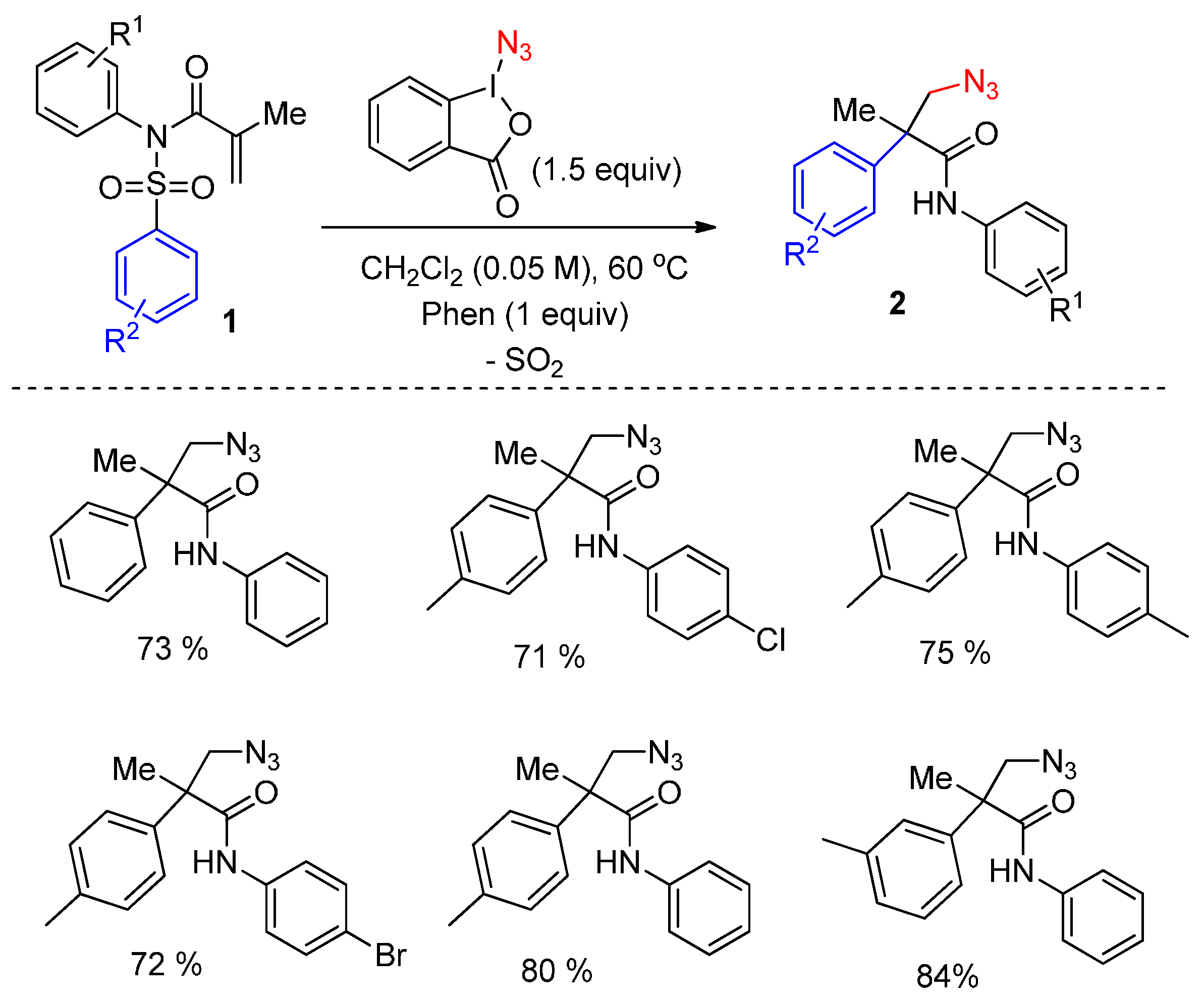
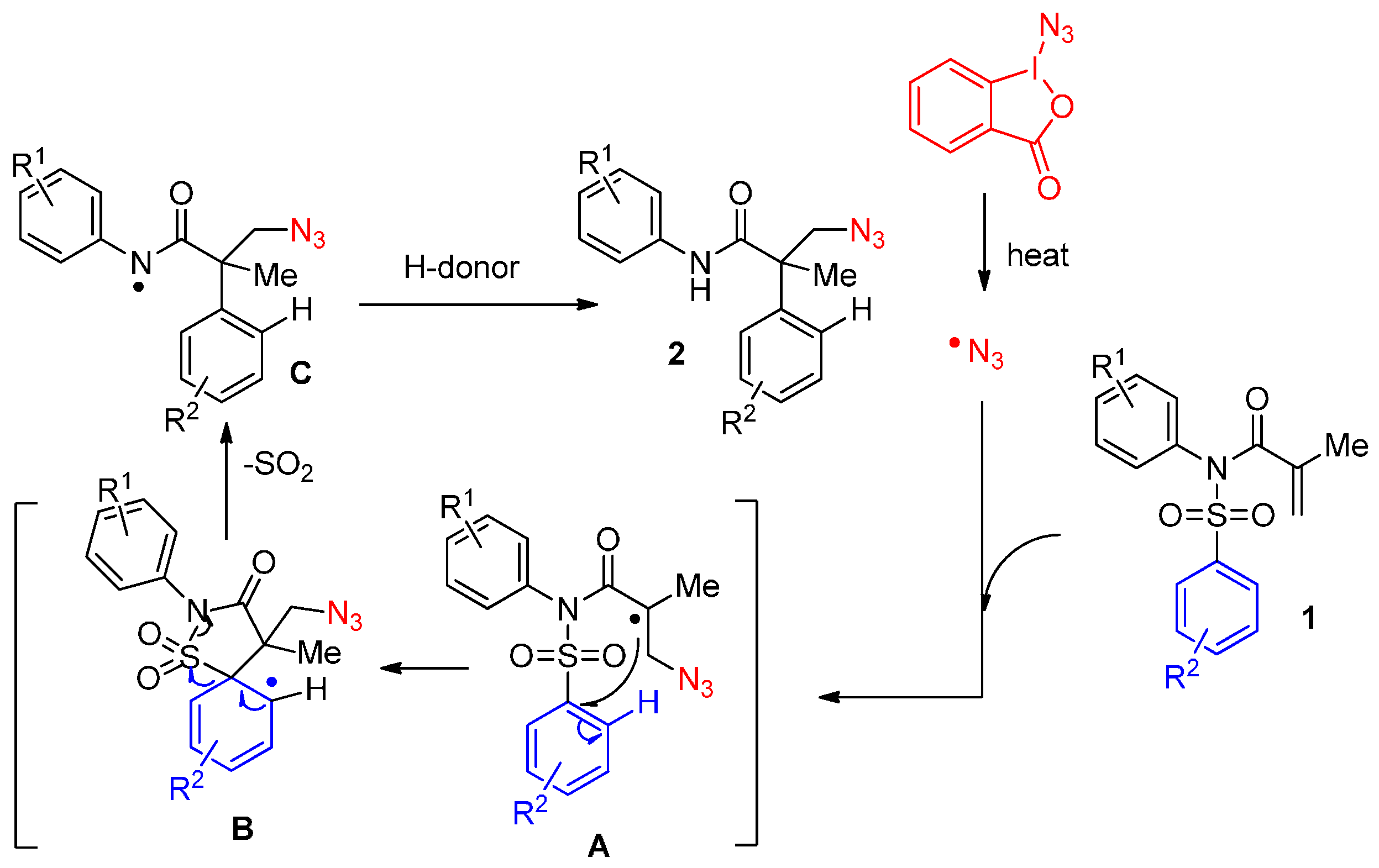
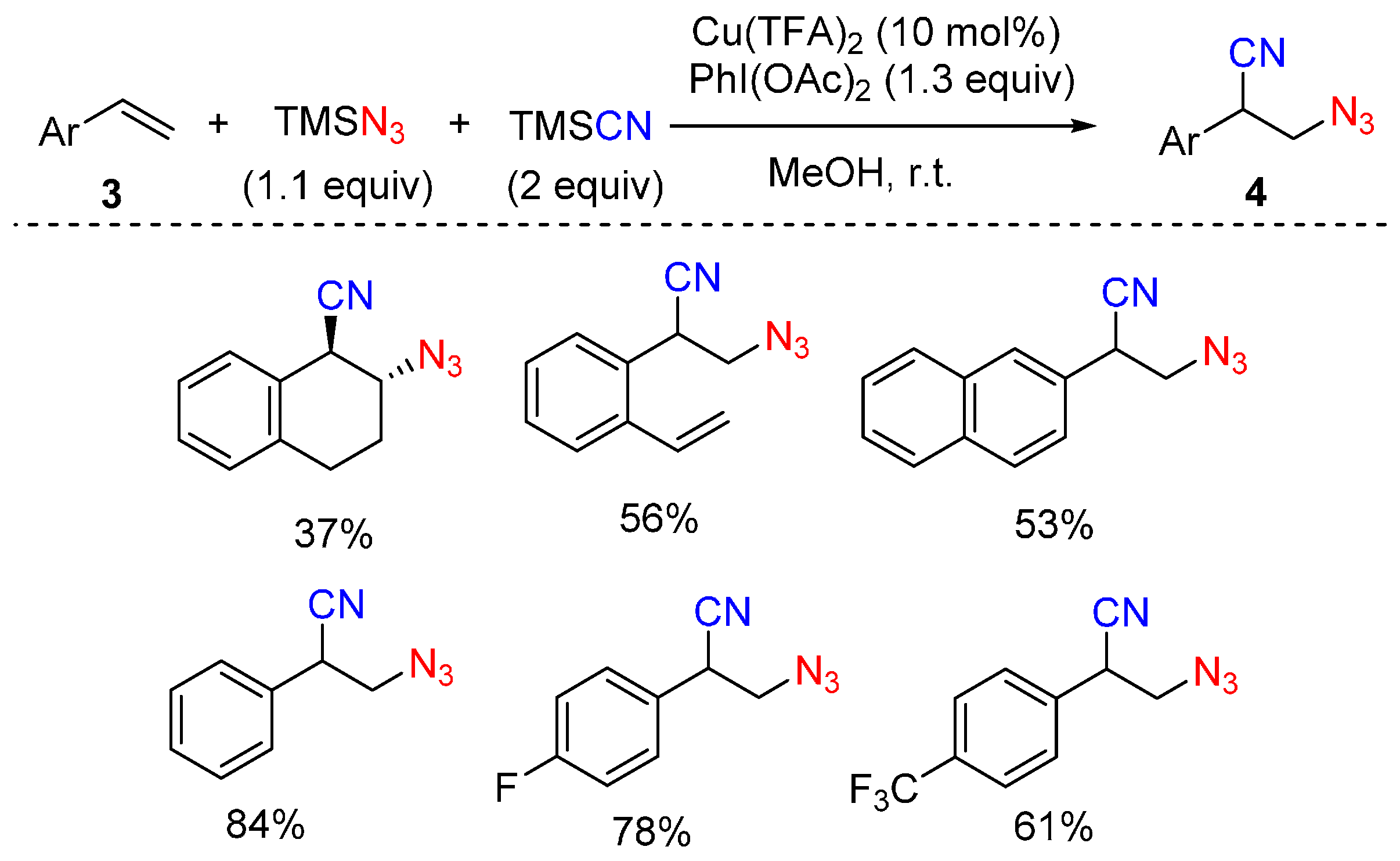
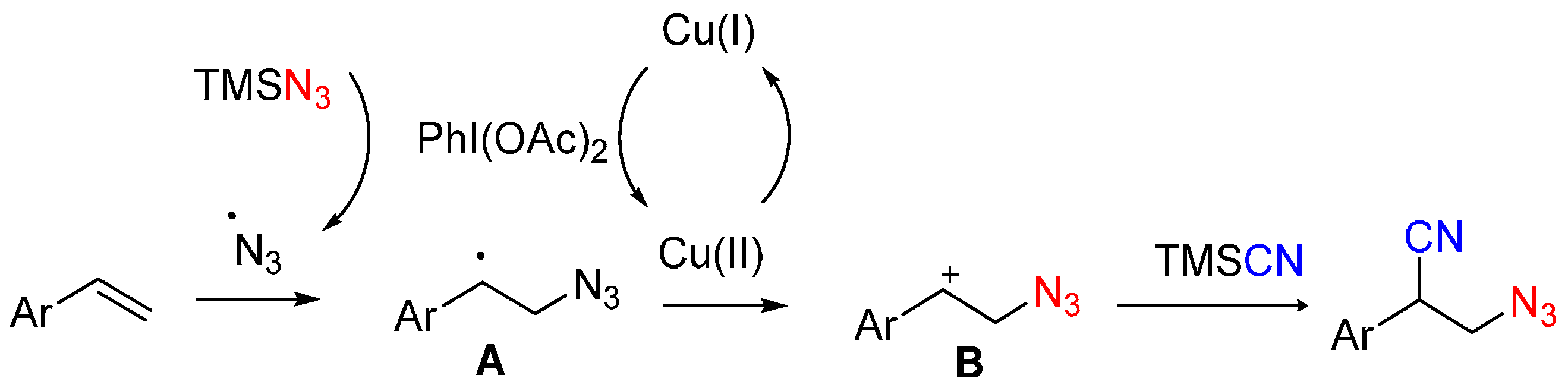
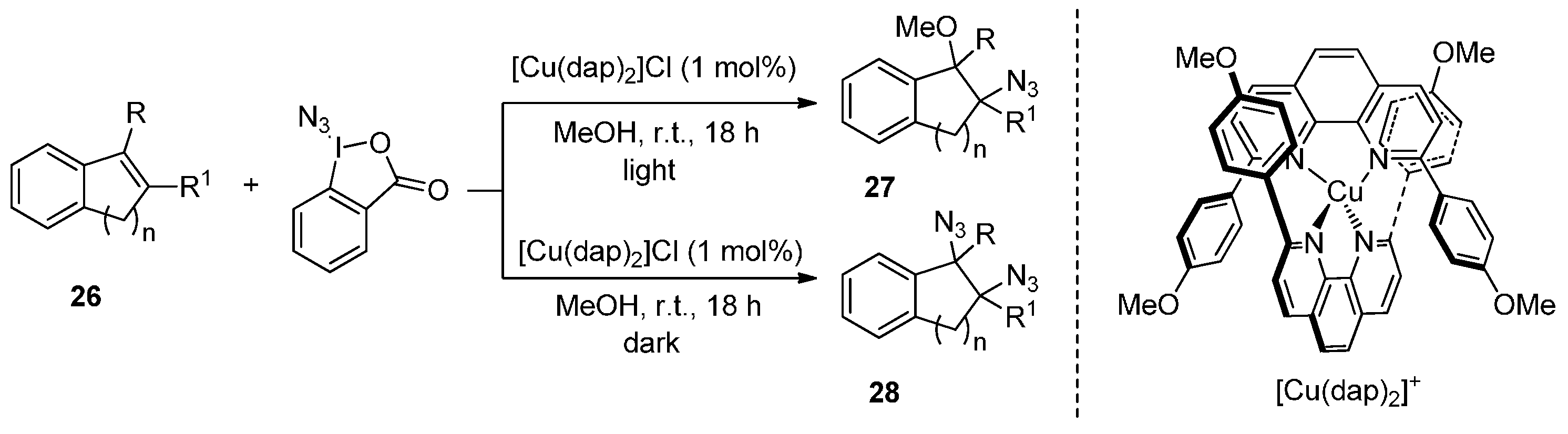
2.1. Construction of C-N3 Bond and C-C Bond
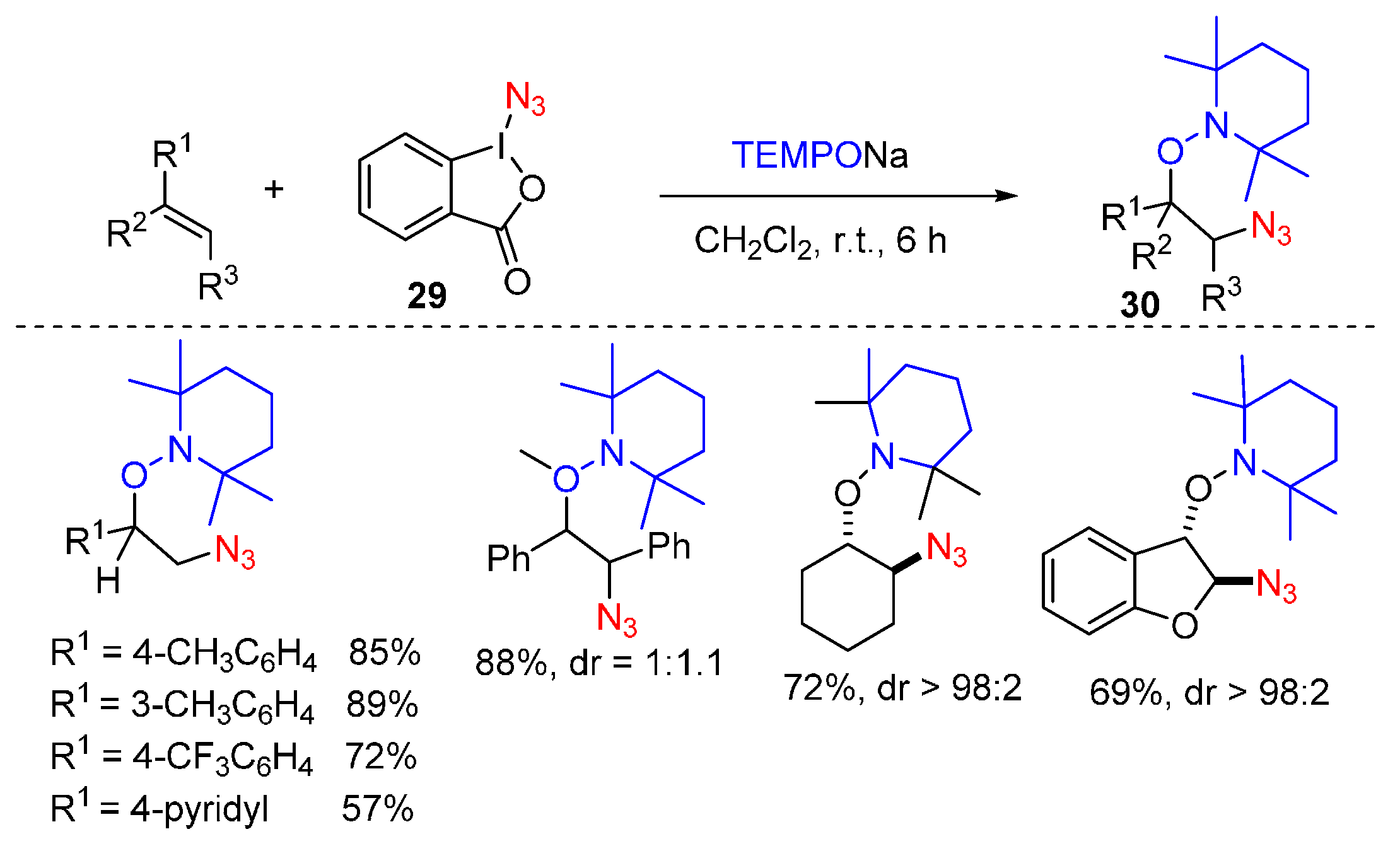
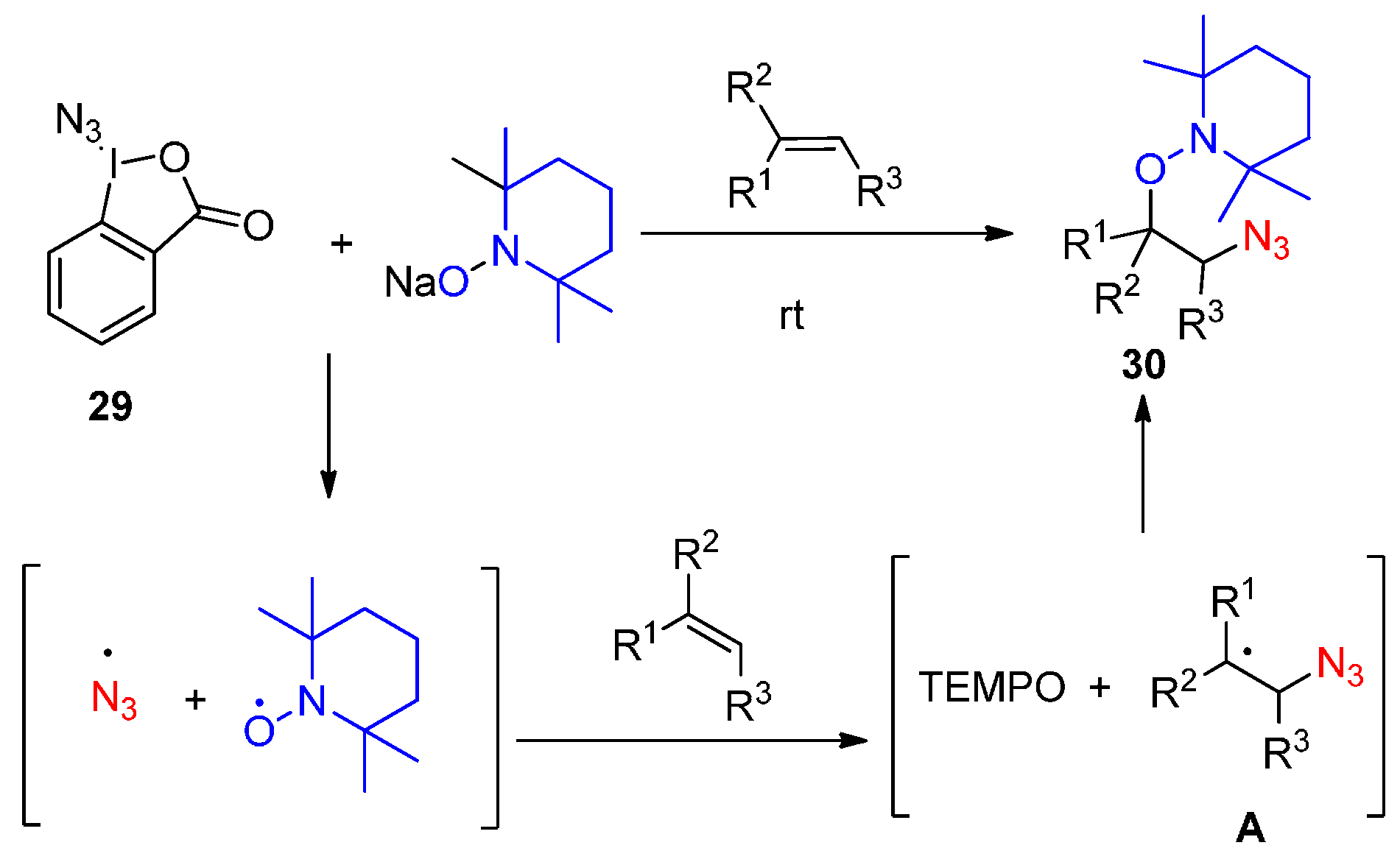
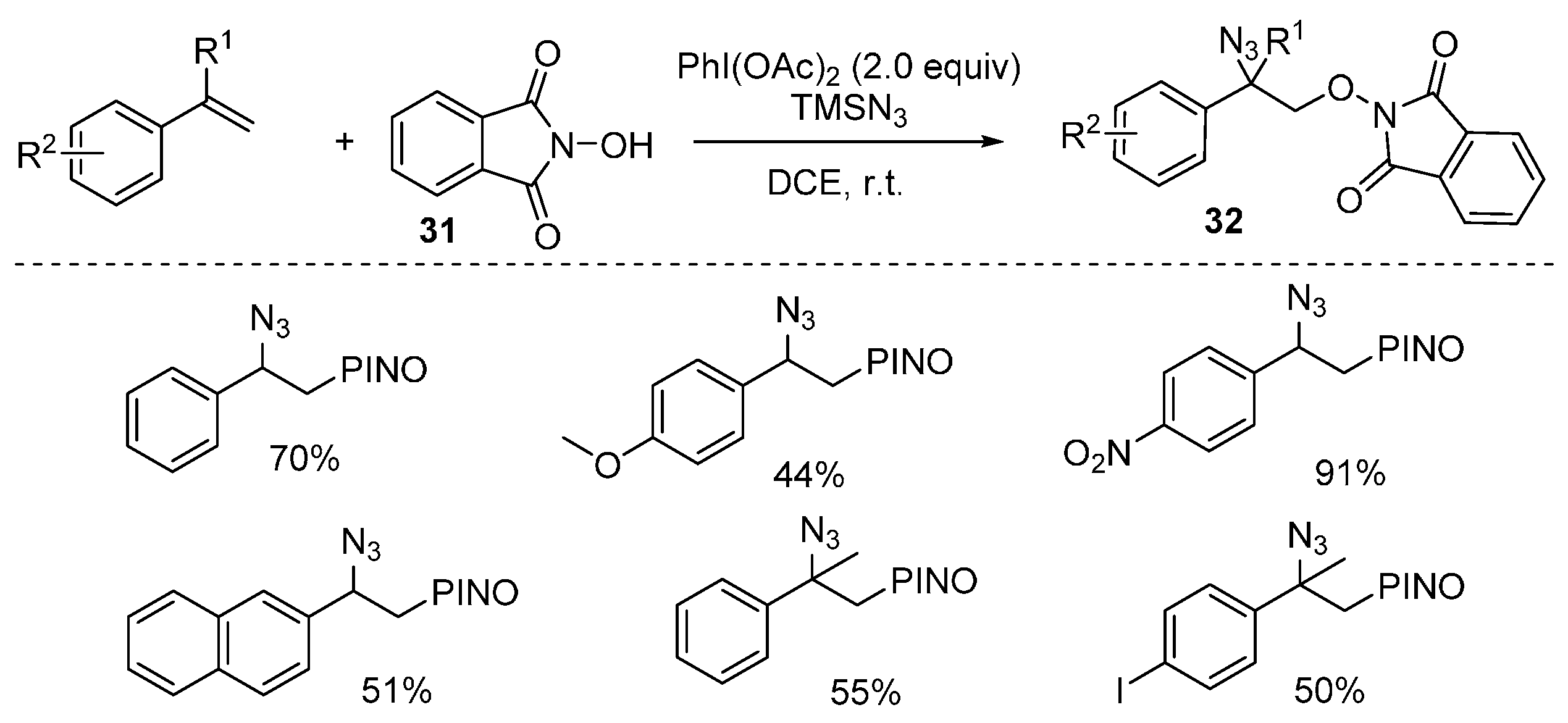
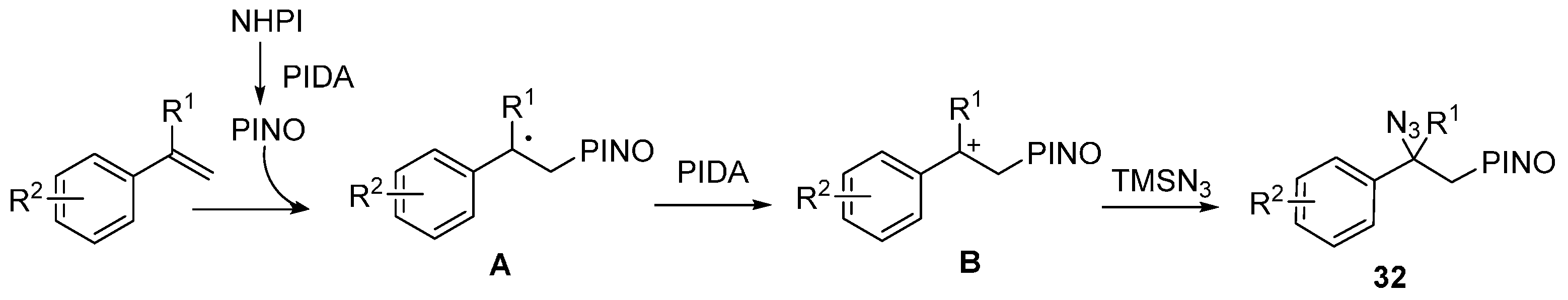
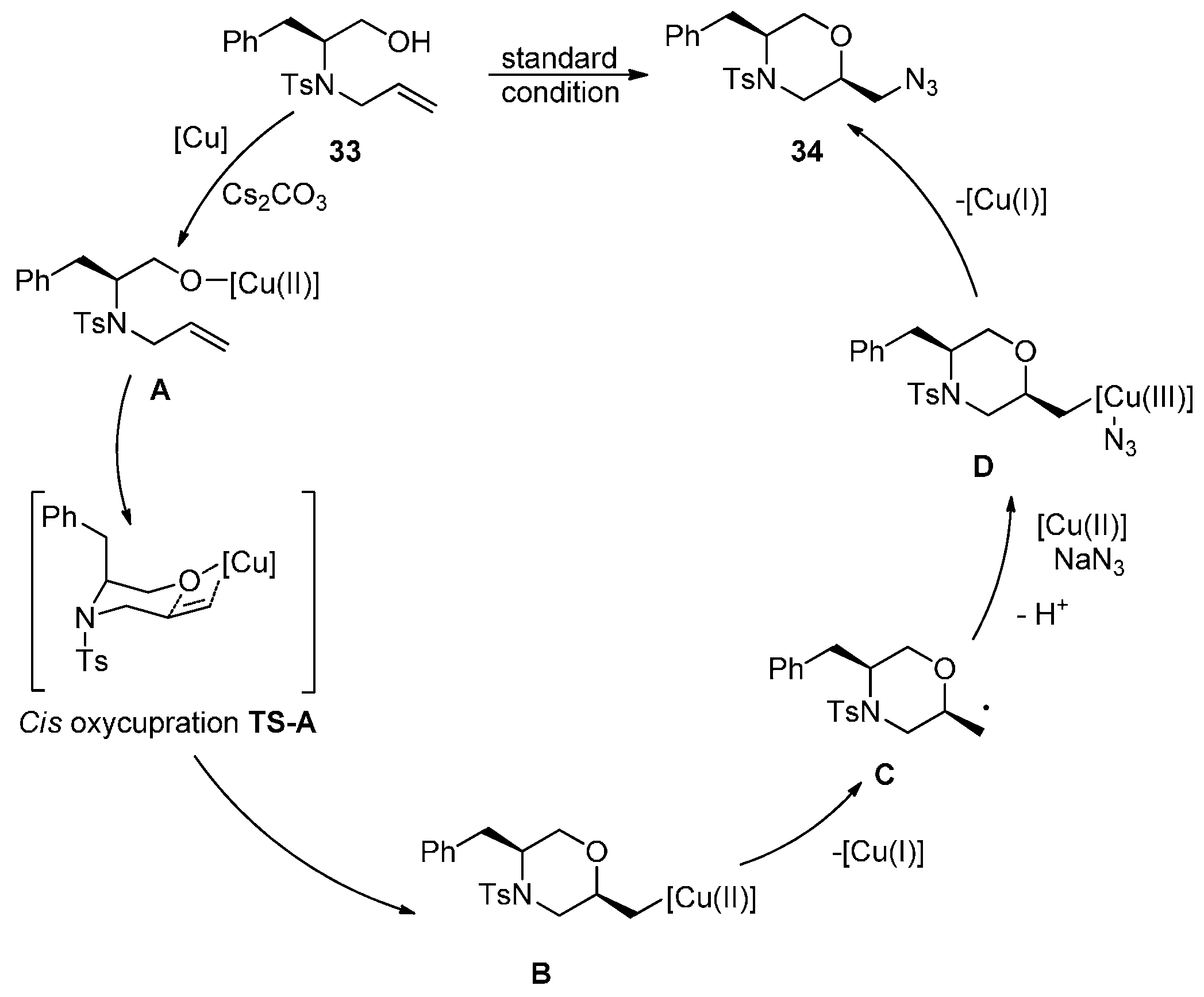
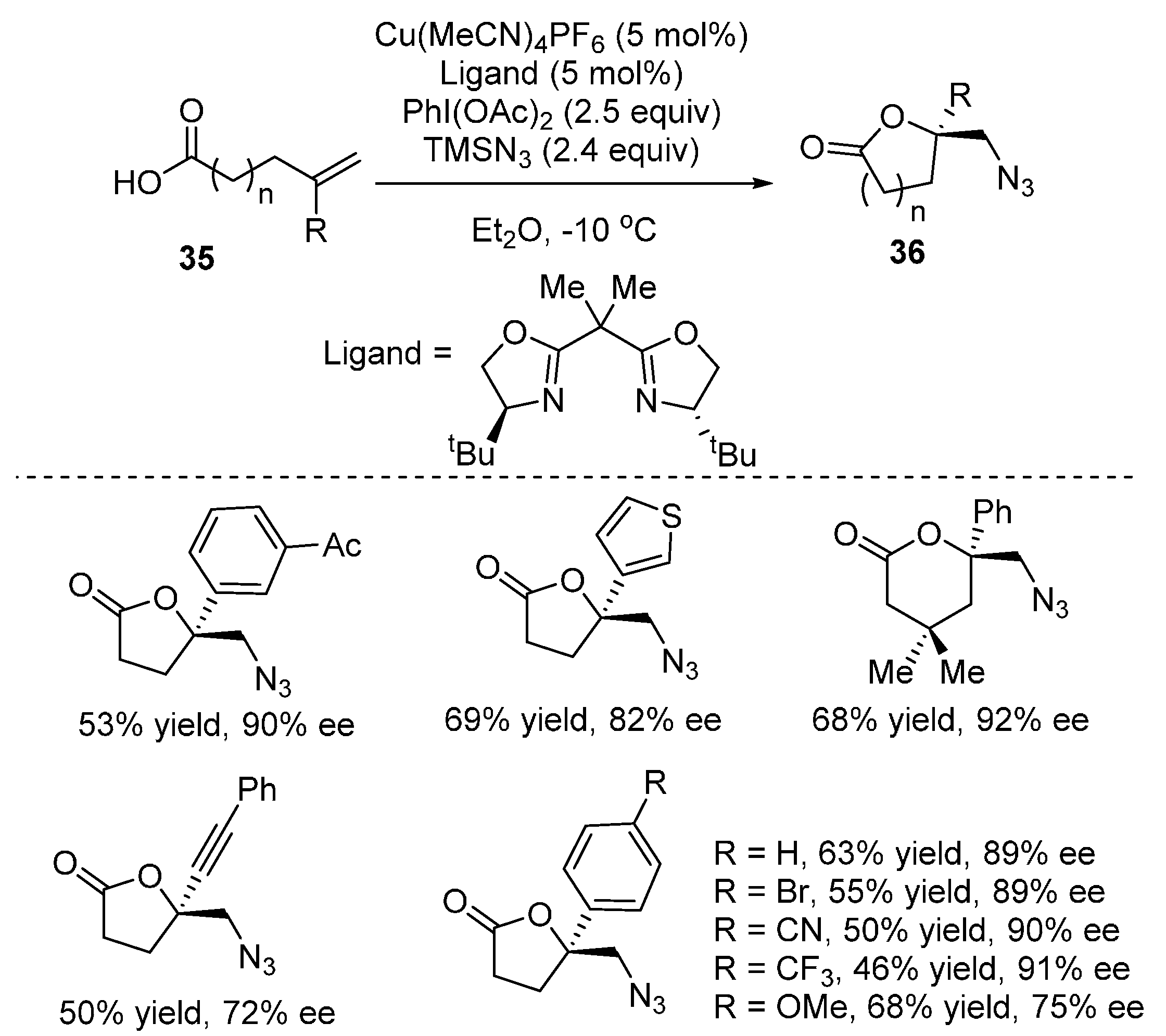
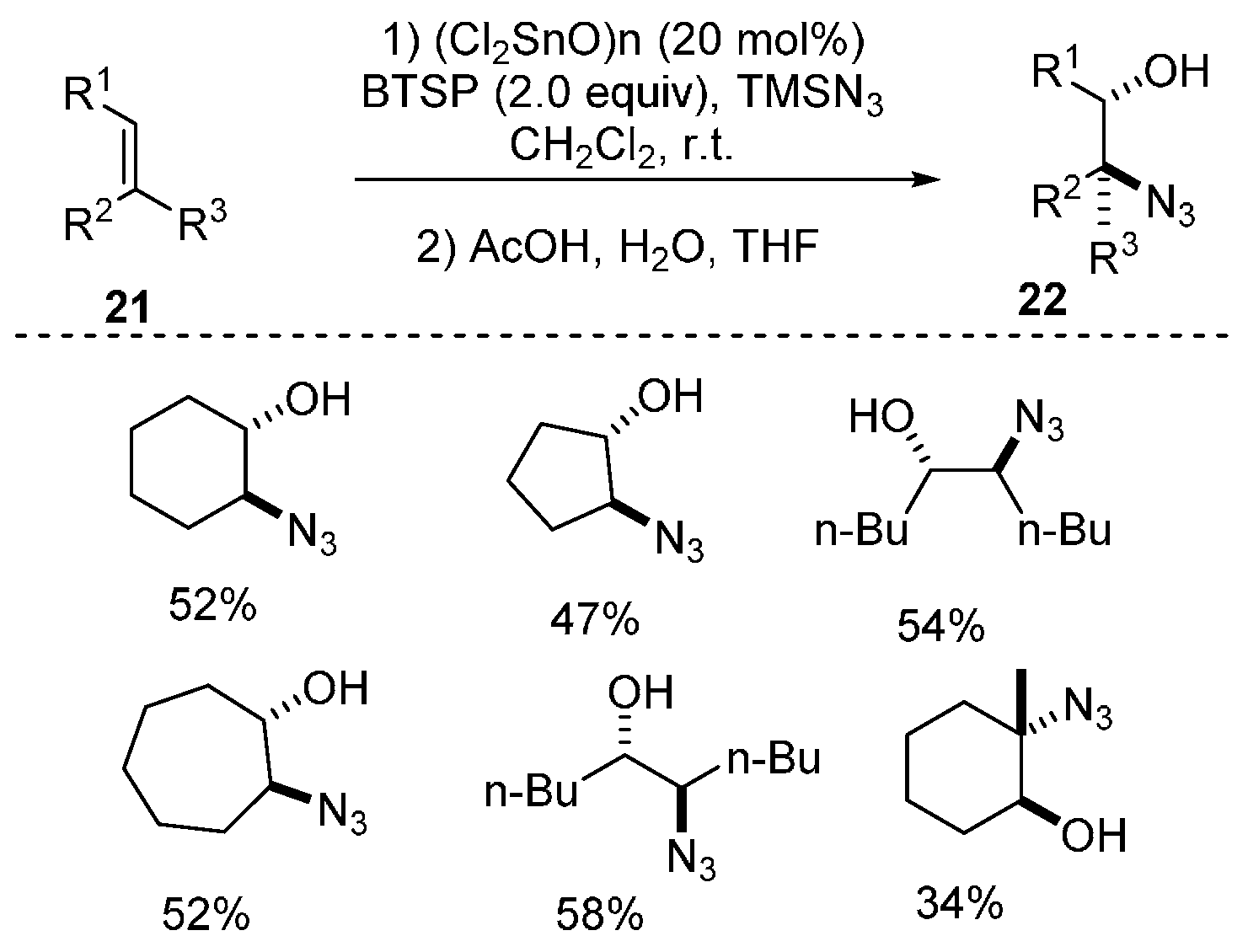
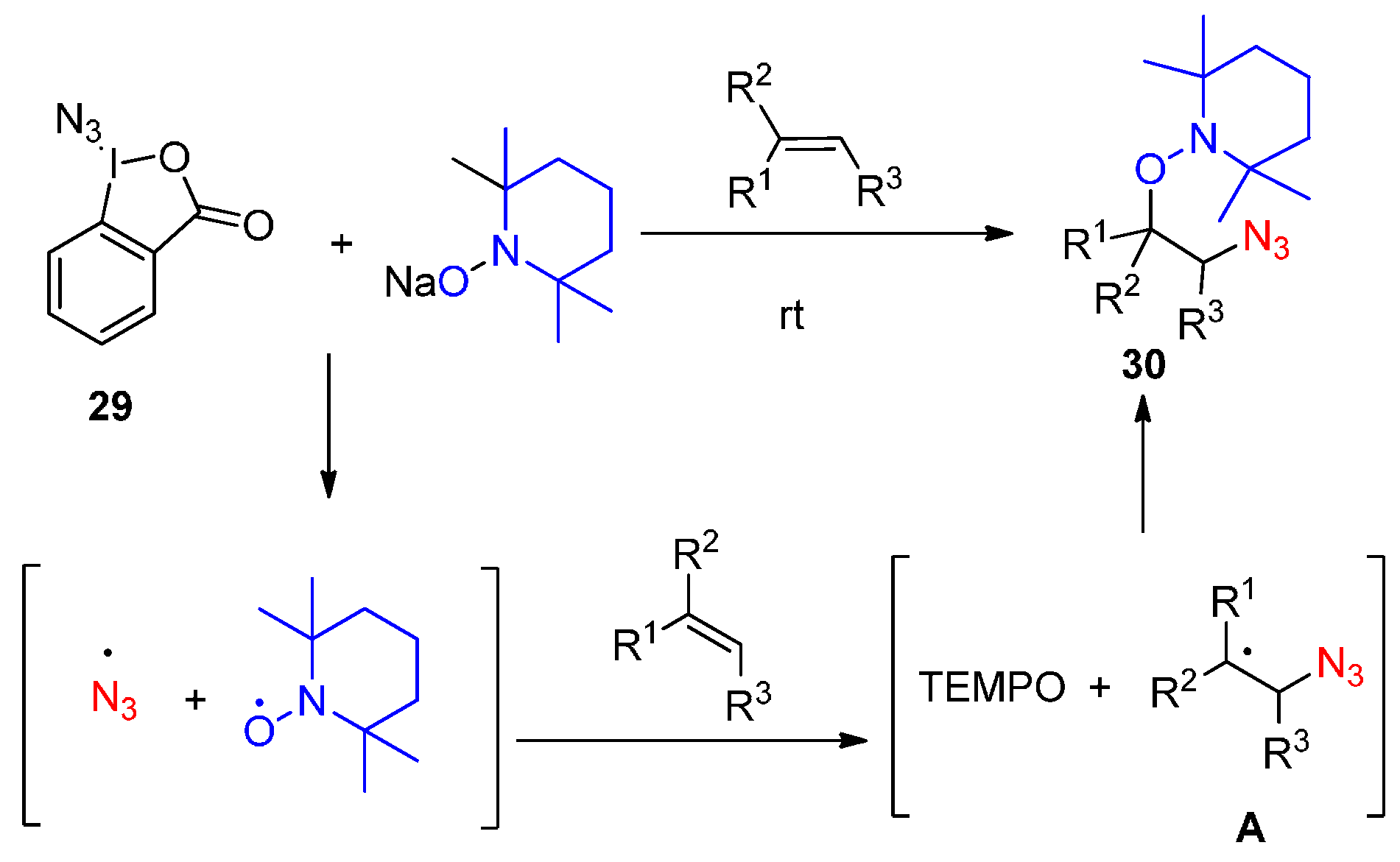
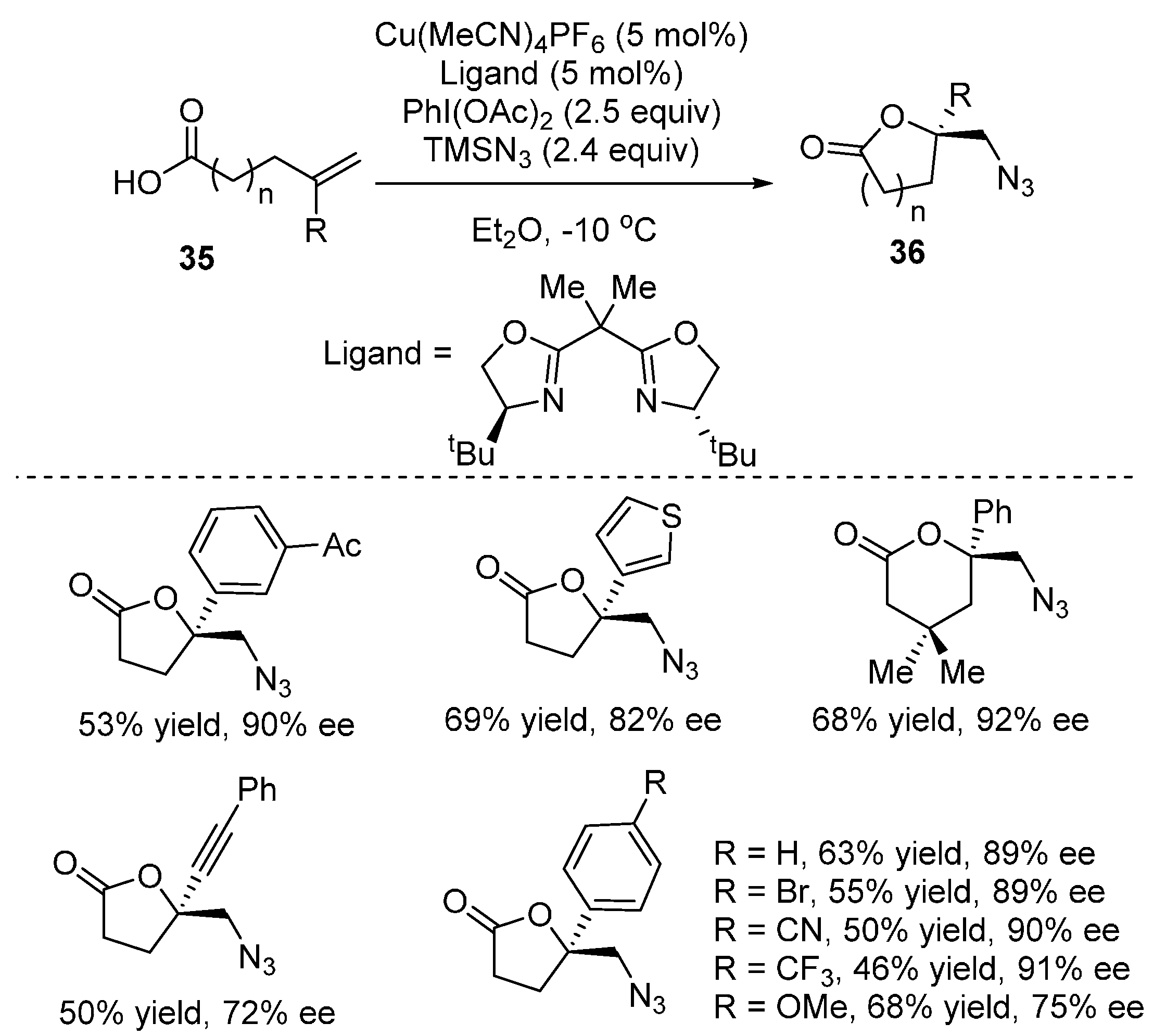
2.2. Construction of C-N3 Bond and C-O Bond
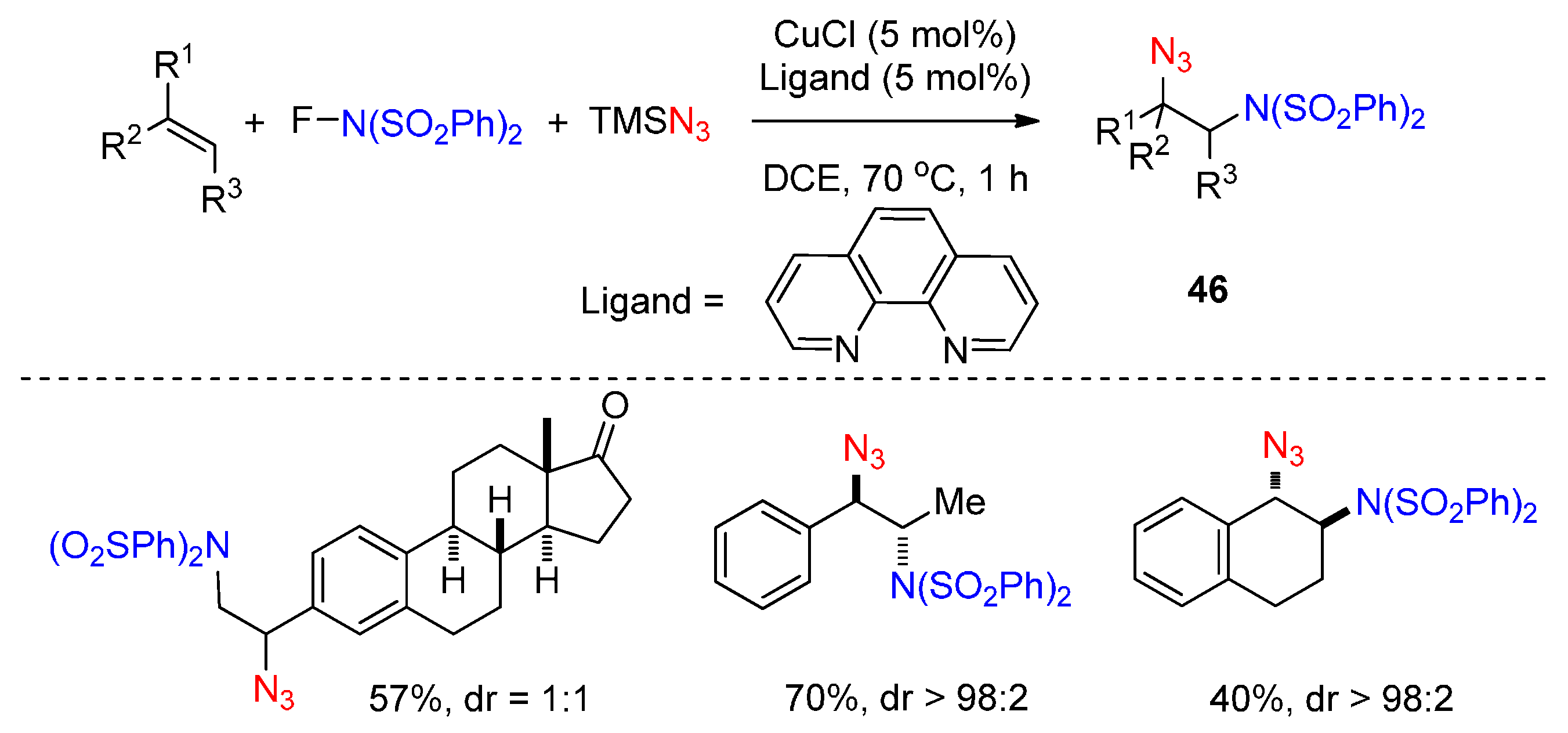
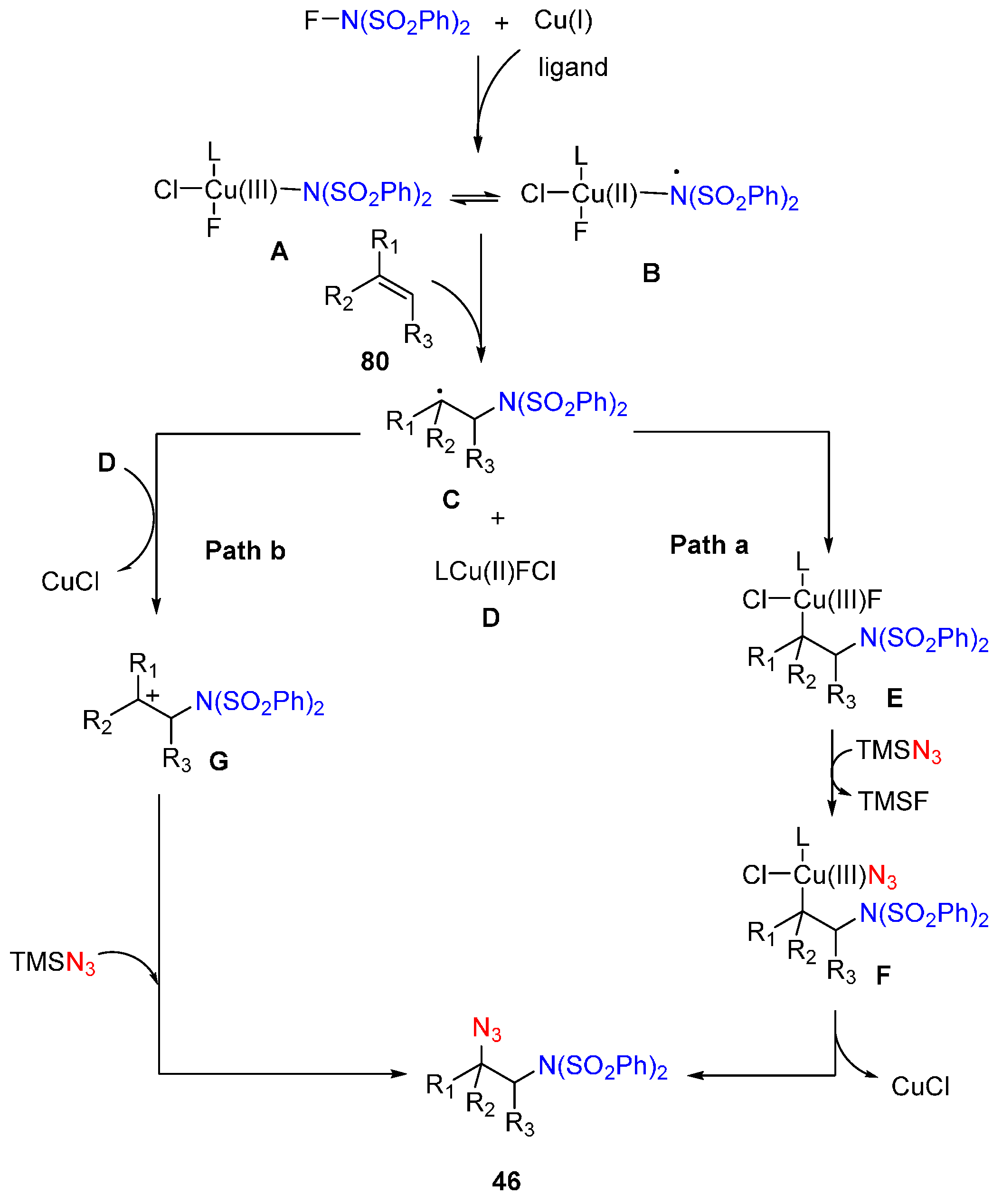
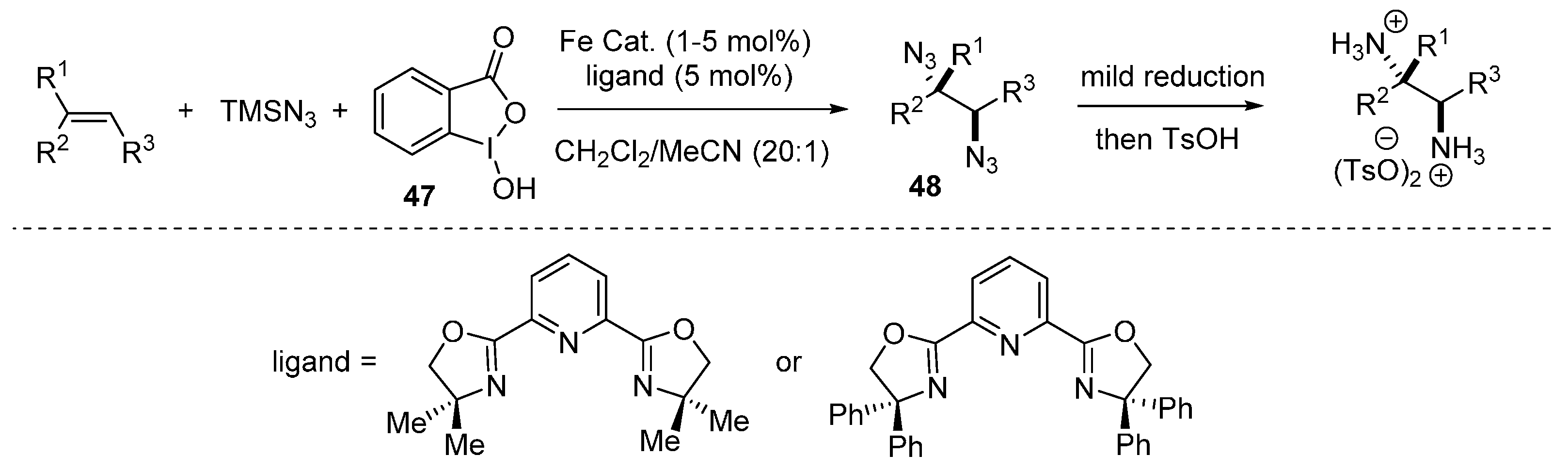
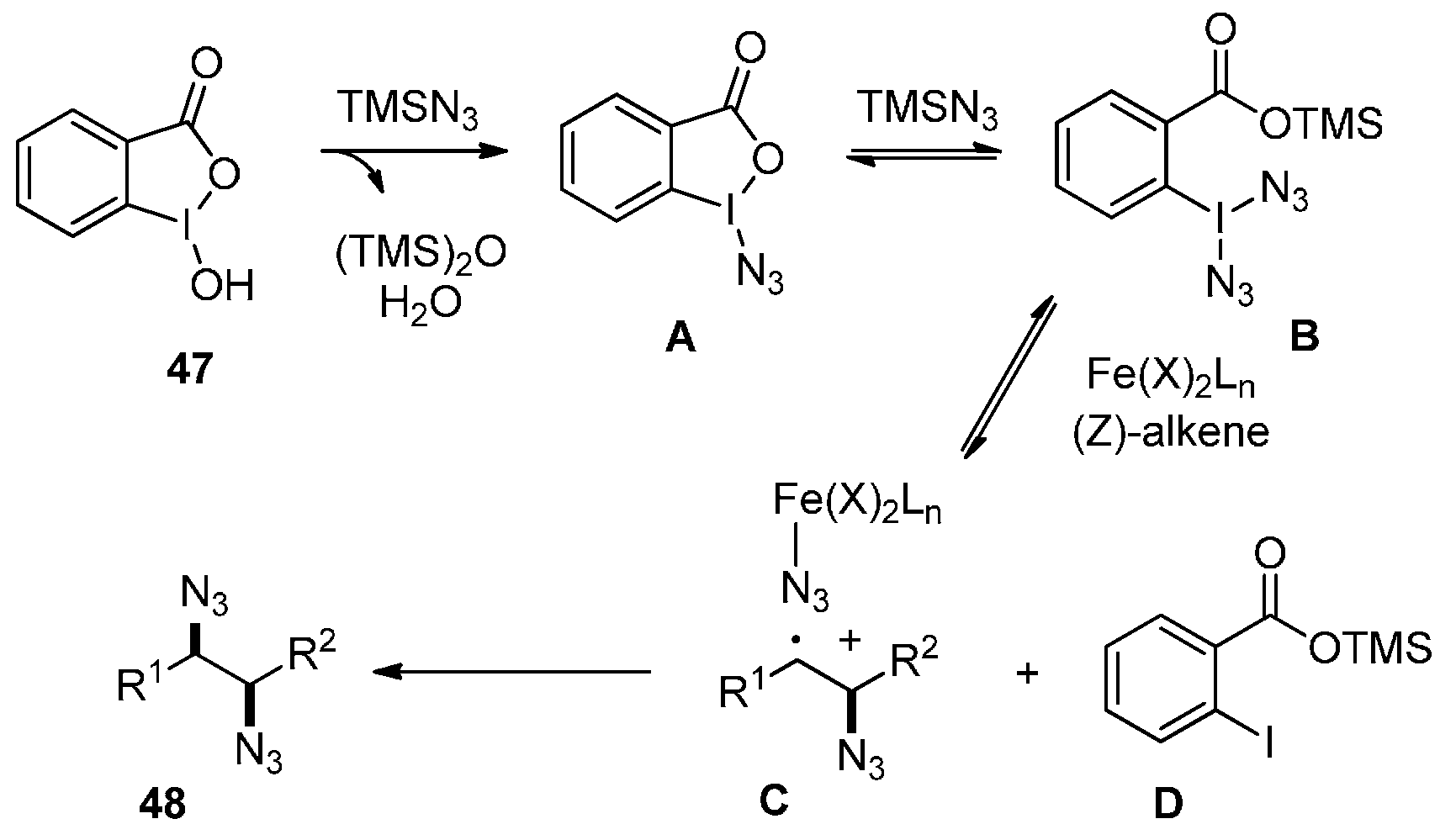
2.3. Construction of C-N3 Bond and C-N Bond
2.4. Construction of C-N3 Bond and C-P Bond
2.5. Construction of C-N3 Bond and C-Se Bond
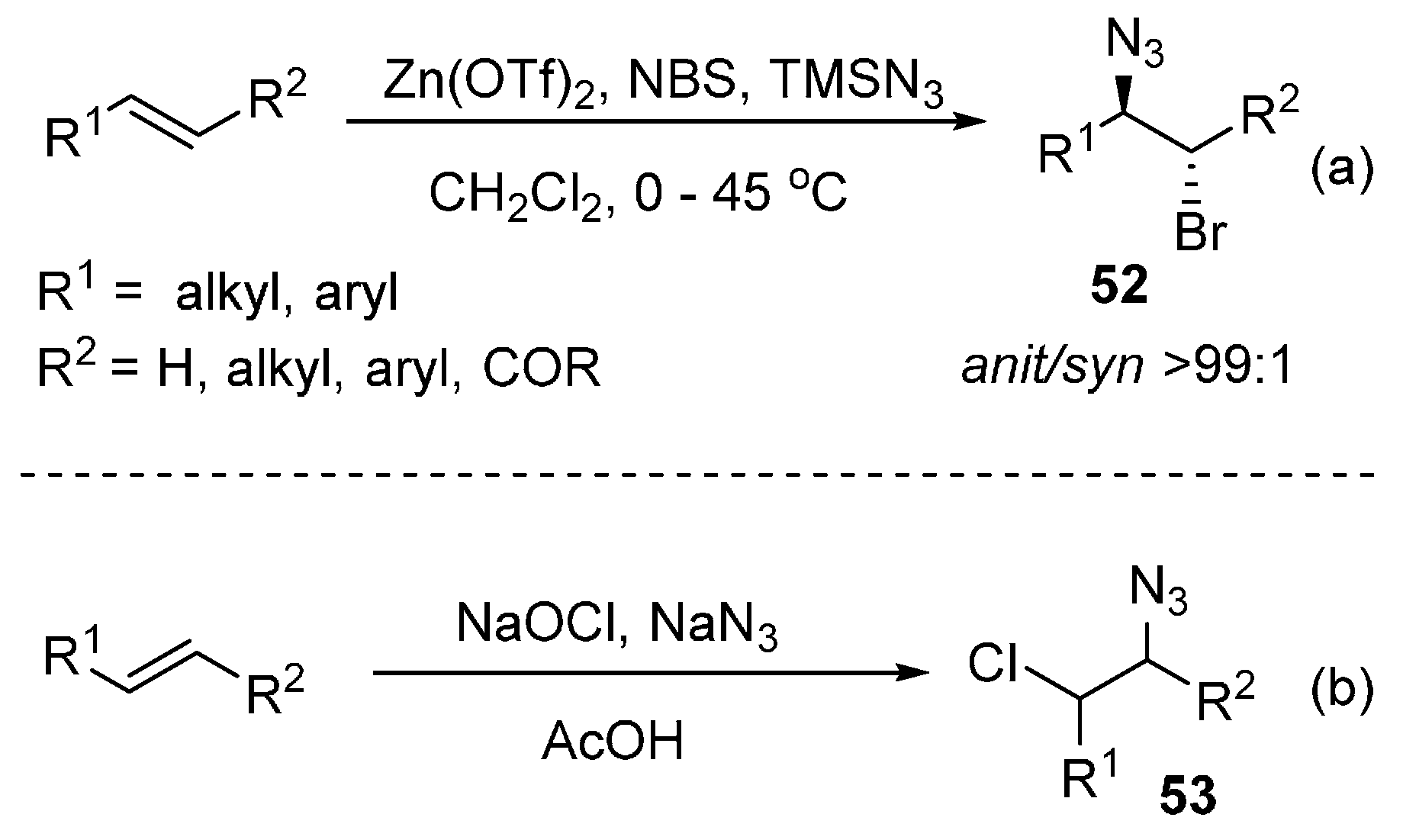
2.6. Construction of C-N3 Bond and C-Halogen Bond
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; Alonso, C.; Aparicio, D.; Rubiales, G.; Santos, J.M.D.L. The aza-wittig reaction: An efficient tool for the construction of carbon-nitrogen double bonds. Tetrahedron 2007, 63, 523–575. [Google Scholar] [CrossRef]
- Curtius, T. Ueber Stickstoffwasserstoffsäure (Azoimid) N3H. Ber. Dtsch. Chem. Ges. 1890, 23, 3023–3033. [Google Scholar] [CrossRef]
- Curtius, T. Hydrazide und Azide organischer Säuren. J. Prakt. Chem. 1894, 50, 275–294. [Google Scholar] [CrossRef]
- Grecian, S.; Aubé, J. Schmidt rearrangement reactions with alkyl azides. In Organic Azides, Syntheses and Applications; Bräse, S., Banert, K., Eds.; John Wiley & Sons: Chichester, UK, 2010; pp. 191–237. [Google Scholar]
- Vital, P.; Tanner, D. Efficient and highly enantioselective formation of the all-carbon quaternary stereocentre of lyngbyatoxin A. Org. Biomol. Chem. 2006, 4, 4292–4298. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar]
- Ritschel, J.; Sasse, F.; Maier, M.E. Synthesis of a benzolactone collection using click chemistry. Eur. J. Org. Chem. 2007, 2007, 78–87. [Google Scholar] [CrossRef]
- Franc, G.; Kakkar, A.K. "Click" methodologies: Efficient, simple and greener routes to design dendrimers. Chem. Soc. Rev. 2010, 39, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Aragão-Leoneti, V.; Campo, V.L.; Gomes, A.S.; Field, R.A.; Carvalho, I. Application of copper(I)-catalysed azide/alkyne cycloaddition (CuAAC) ‘click chemistry’ in carbohydrate drug and neoglycopolymer synthesis. Tetrahedron 2010, 66, 9475–9492. [Google Scholar] [CrossRef]
- Wang, T.; Jiao, N. Direct approaches to nitriles via highly efficient nitrogenation strategy through C-H or C-C bond cleavage. Acc. Chem. Res. 2014, 47, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Application of organic azides for the synthesis of nitrogen-containing molecules. Synlett 2012, 23, 21–44. [Google Scholar] [CrossRef]
- Shin, K.; Kim, H.; Chang, S. Transition-metal-catalyzed C-N bond forming reactions using organic azides as the nitrogen source: A journey for the mild and versatile C-H amination. Acc. Chem. Res. 2015, 48, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Intrieri, D.; Zardi, P.; Caselli, A.; Gallo, E. Organic azides: "Energetic reagents" for the intermolecular amination of C-H bonds. Chem. Commun. 2014, 50, 11440–11453. [Google Scholar] [CrossRef] [PubMed]
- Sebeika, M.M.; Jones, G.B. Safer, greener, and more facile alternatives for synthesis with organic azides. Curr. Org. Synth. 2014, 11, 732–750. [Google Scholar] [CrossRef]
- Tanimoto, H.; Kakiuchi, K. Recent applications and developments of organic azides in total synthesis of natural products. Nat. Prod. Commun. 2013, 8, 1021–1034. [Google Scholar] [PubMed]
- Tuktarov, A.R.; Akhmetov, A.R.; Dzhemilev, U.M. Organic and inorganic azides in fullerene chemistry: Achievements and trends. Mater. Sci. Res. J. 2014, 8, 123–166. [Google Scholar]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Hahn, U.; Elhabiri, M.; Trabolsi, A.; Herschbach, H.; Leize, E.; Dorsselaer, A.V.; Albrecht-Gary, A.M.; Nierengarten, J.F. Supramolecular click chemistry with a bisammonium-C60 substrate and a ditopic crown ether host. Angew. Chem. Int. Ed. 2005, 44, 5338–5341. [Google Scholar] [CrossRef] [PubMed]
- Trabolsi, A.; Elhabiri, M.; Urbani, M.; Delgado de la Cruz, J.L.; Ajamaa, F.; Solladié, N.; Albrecht-Gary, A.M.; Nierengarten, J.F. Supramolecular click chemistry for the self-assembly of a stable Zn(II)–porphyrin–C60 conjugate. Chem. Commun. 2005, 46, 5736–5738. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, R.; Moore, B.C.; Schubert, U.S. Synthesis of star-shaped poly(ε-caprolactone) via “click” chemistry and “supramolecular click” chemistry. Chem. Commun. 2006, 38, 4010–4012. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Costa, J.S.; Aromí, G.; Mutikainen, I.; Turpeinen, U.; Gamez, P.; Reedijk, J. Supramolecular click assembly of a fused double-stranded [MnII3] dihelicate. Eur. J. Inorg. Chem 2007, 26, 4119–4122. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef] [PubMed]
- Kluger, R. Click chemistry for biotechnology and materials science. J. Am. Chem. Soc. 2010, 132, 6611–6612. [Google Scholar] [CrossRef]
- Muñiz, K.; Martínez, C. Development of intramolecular vicinal diamination of alkenes: From palladium to bromine catalysis. J. Org. Chem. 2013, 78, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of heterocycles via palladium pi-olefin and pi-alkyne chemistry. Chem. Rev. 2004, 104, 2285–2309. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K. Imido-osmium(VIII) compounds in organic synthesis: Aminohydroxylation and diamination reactions. Chem. Soc. Rev. 2004, 33, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Minatti, A.; Muñiz, K. Intramolecular aminopalladation of alkenes as a key step to pyrrolidines and related heterocycles. Chem. Soc. Rev. 2007, 36, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.H.; Sigman, M.S. Mechanistic approaches to palladium-catalyzed alkene difunctionalization reactions. Org. Biomol. Chem. 2008, 6, 4083–4088. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.R. Intermolecular olefin functionalisation involving aryl radicals generated from arenediazonium salts. Chem. Eur. J. 2009, 15, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Goti, A. Metal-catalysed 1,2-diamination reactions. Nat. Chem. 2009, 1, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Mcdonald, R.I.; Liu, G.; Stahl, S.S. Palladium(II)-catalyzed alkene functionalization via nucleopalladation: Stereochemical pathways and enantioselective catalytic applications. Chem. Rev. 2011, 111, 2981–3019. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, T.J.; Callens, C.K.A.; Flores, A.; Lacy, A.R.; Rathi, A.H. Recent developments in methodology for the direct oxyamination of olefins. Chem. Eur. J. 2011, 17, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.; Nevado, C. Addition of CF3 across unsaturated moieties: A powerful functionalization tool. Chem. Soc. Rev. 2014, 43, 6598–6608. [Google Scholar] [CrossRef] [PubMed]
- Chemler, S.R.; Bovino, M.T. Catalytic aminohalogenation of alkenes and alkynes. ACS Catal. 2013, 3, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Sodeoka, M. Trifluoromethylation of alkenes with concomitant introduction of additional functional groups. Angew. Chem. Int. Ed. 2014, 53, 8294–8308. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.M.; Wöste, T.H.; Muñiz, K. Vicinal difunctionalization of alkenes with iodine (III) reagents and catalysts. Chem. Asian J. 2014, 9, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Kanai, M. Recent progress in copper-catalyzed difunctionalization of unactivated carbon-carbon multiple bonds. Tetrahedron Lett. 2014, 55, 3727–3737. [Google Scholar] [CrossRef]
- Wolfe, J.P. Intramolecular alkoxycyanation and alkoxyacylation reactions: New types of alkene difunctionalizations for the construction of oxygen heterocycles. Angew. Chem. Int. Ed. 2012, 51, 10224–10225. [Google Scholar] [CrossRef] [PubMed]
- Waser, J.; Nambu, H.; Carreira, E.M. Cobalt-catalyzed hydroazidation of olefins. J. Am. Chem. Soc. 2005, 127, 8294–8295. [Google Scholar] [CrossRef] [PubMed]
- Waser, J.; Gaspar, B.; Nambu, H.; Carreira, E.M. Hydrazines and azides via the metal-catalyzed hydrohydrazination and hydroazidation of olefins. J. Am. Chem. Soc. 2006, 128, 11693–11712. [Google Scholar] [CrossRef] [PubMed]
- Kapat, A.; König, A.; Montermini, F.; Renaud, P. A radical procedure for the anti-Markovnikov hydroazidation of alkenes. J. Am. Chem. Soc. 2011, 133, 13890–13893. [Google Scholar] [CrossRef] [PubMed]
- Leggans, E.K.; Barker, T.J.; Duncan, K.K.; Boger, D.L. Iron(III)/NaBH4-mediated additions to unactivated alkenes: Synthesis of novel 200-vinblastine analogues. Org. Lett. 2012, 14, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Glavee, G.N.; Klabunde, K.J.; Sorensen, C.M.; Hadjipanayis, G.C. Chemistry of borohydride reduction of iron(II) and iron(III) ions in aqueous and nonaqueous media. Formation of nanoscale Fe, FeB, and Fe2B powders. Inorg. Chem. 1995, 34, 28–35. [Google Scholar] [CrossRef]
- Bellavista, T.; Meninno, S.; Lattanzi, A.; Sala, G.D. Asymmetric hydroazidation of nitroalkenes promoted by a secondary amine-thiourea catalyst. Adv. Synth. Catal. 2015, 357, 3365–3373. [Google Scholar] [CrossRef]
- Kong, W.; Merino, E.; Nevado, C. Arylphosphonylation and arylazidation of activated alkenes. Angew. Chem. Int. Ed. 2014, 53, 5078–5082. [Google Scholar]
- Xu, L.; Mou, X.Q.; Chen, Z.M.; Wang, S.H. Copper-catalyzed intermolecular azidocyanation of aryl alkenes. Chem. Commun. 2014, 50, 10676–10679. [Google Scholar] [CrossRef] [PubMed]
- Mantrand, N.; Renaud, P. Azidosulfonylation of alkenes, dienes, and enynes. Tetrahedron 2008, 64, 11860–11864. [Google Scholar] [CrossRef]
- Klein, J.E.M.N.; Taylor, R.J.K. Transition-metal-mediated routes to 3,3-disubstituted oxindoles through anilide cyclisation. Eur. J. Org. Chem. 2011, 2011, 6821–6841. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.L.; Zhou, J. Catalytic asymmetric synthesis of oxindoles bearing a tetrasubstituted stereocenter at the C-3 position. Adv. Synth. Catal. 2010, 352, 1381–1407. [Google Scholar] [CrossRef]
- Matcha, K.; Narayan, R.; Antonchick, A.P. Metal-free radical azidoarylation of alkenes: Rapid access to oxindoles by cascade C-N and C-C bond-forming reactions. Angew. Chem. Int. Ed. 2013, 52, 7985–7989. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, R. Transition-metal-free oxidative carboazidation of acrylamides via cascade C-N and C-C bond forming reactions. Org. Biomol. Chem. 2014, 12, 4329–4334. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.H.; Li, Y.M.; Zhou, A.X.; Yang, T.T.; Yang, S.D. Silver-catalyzed carboazidation of arylacrylamides. Org. Lett. 2013, 15, 4158–4161. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shen, T.; Wang, K.; Jiao, N. Ag-promoted azido-carbocyclization of activated alkenes via C-H bond cleavage. Chem. Asian J. 2013, 8, 2932–2935. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Taguchi, T.; Ojima, I. Unique properties of fluorine and their relevance to medicinal chemistry and chemical biology. In Fluorine in Medical Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009; pp. 1–46. [Google Scholar]
- Wang, F.; Qi, X.; Liang, Z.; Chen, P.; Liu, G. Copper-catalyzed intermolecular trifluoromethylazidation of alkenes: Convenient access to CF3-containing alkyl azides. Angew. Chem. Int. Ed. 2014, 126, 1912–1917. [Google Scholar] [CrossRef]
- Carboni, A.; Dagousset, G.; Magnier, E.; Masson, G. Photoredox-induced three-component oxy-, amino-, and carbotrifluoromethylation of enecarbamates. Org. Lett. 2014, 16, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Dagousset, G.; Carboni, A.; Magnier, E.; Masson, G. Photoredox-induced three-component azido- and aminotrifluoromethylation of alkenes. Org. Lett. 2014, 16, 4340–4343. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, W.; Liu, Y.; Feng, L.; Ju, X.; Yang, M. Copper-catalyzed three-component azidotrifluoromethylation/difunctionalization of alkenes. Chin. J. Chem. 2014, 32, 833–837. [Google Scholar] [CrossRef]
- Sakurada, I.; Yamasaki, S.; Kanai, M.; Shibasaki, M. Dichlorotin oxide-catalyzed new direct functionalization of olefins: Synthesis of trans β-azidohydrins and 1,2-diols. Tetrahedron Lett. 2000, 41, 2415–2418. [Google Scholar] [CrossRef]
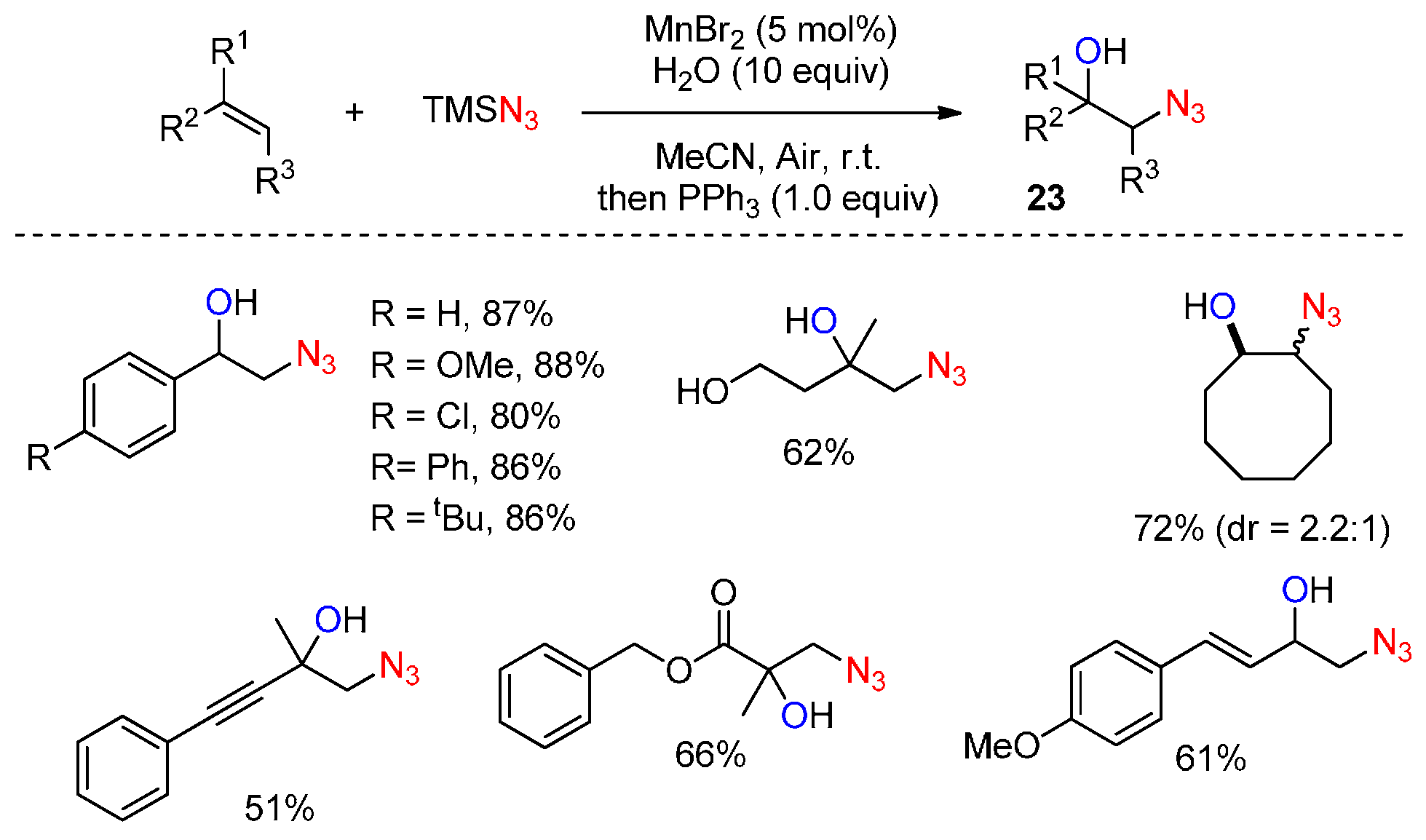
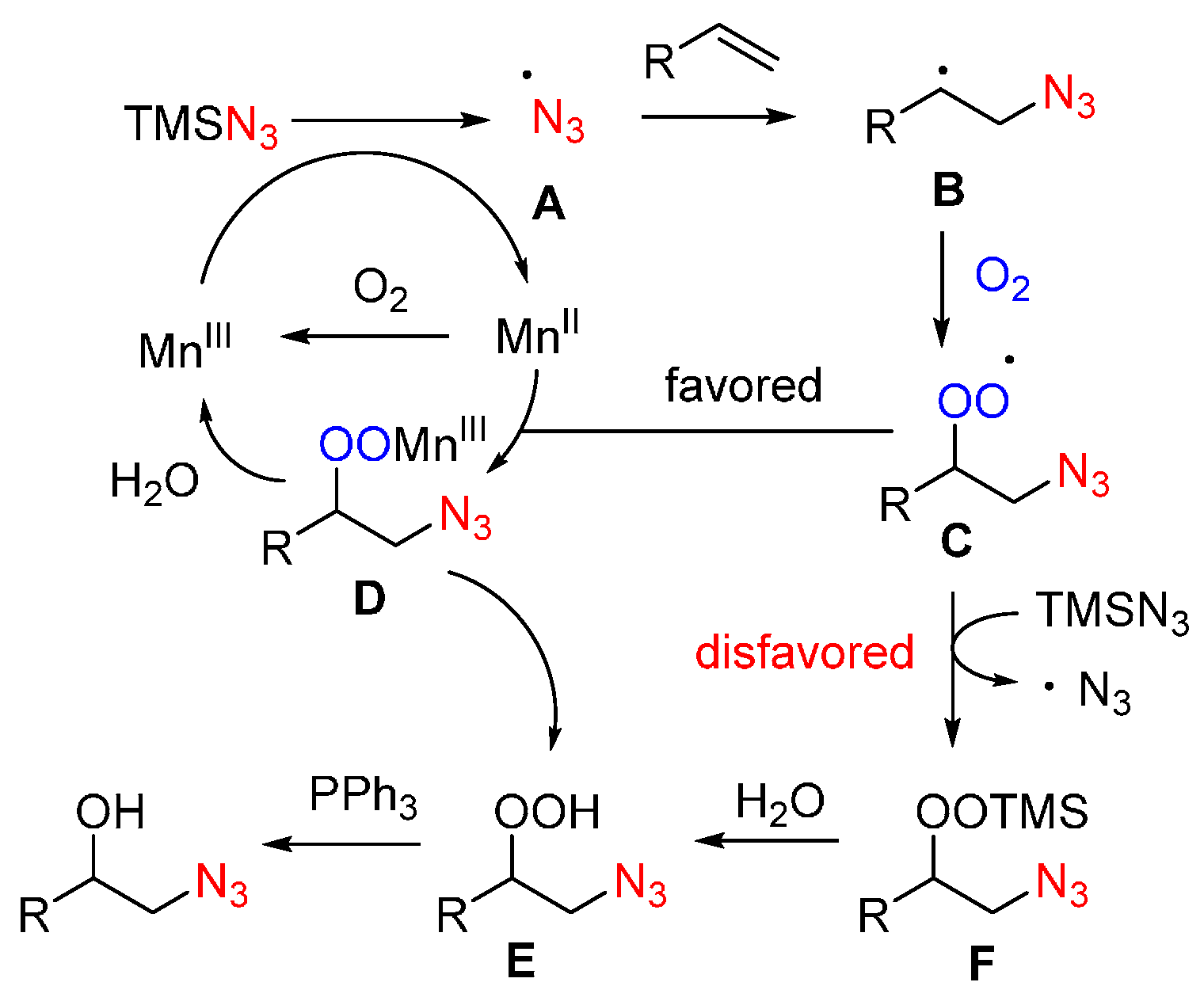
- Sun, X.; Li, X.; Song, S.; Zhu, Y.; Liang, Y.F.; Jiao, N. Mn-catalyzed highly efficient aerobic oxidative hydroxyazidation of olefins: A direct approach to β-azido alcohols. J. Am. Chem. Soc. 2015, 137, 6059–6066. [Google Scholar] [CrossRef] [PubMed]
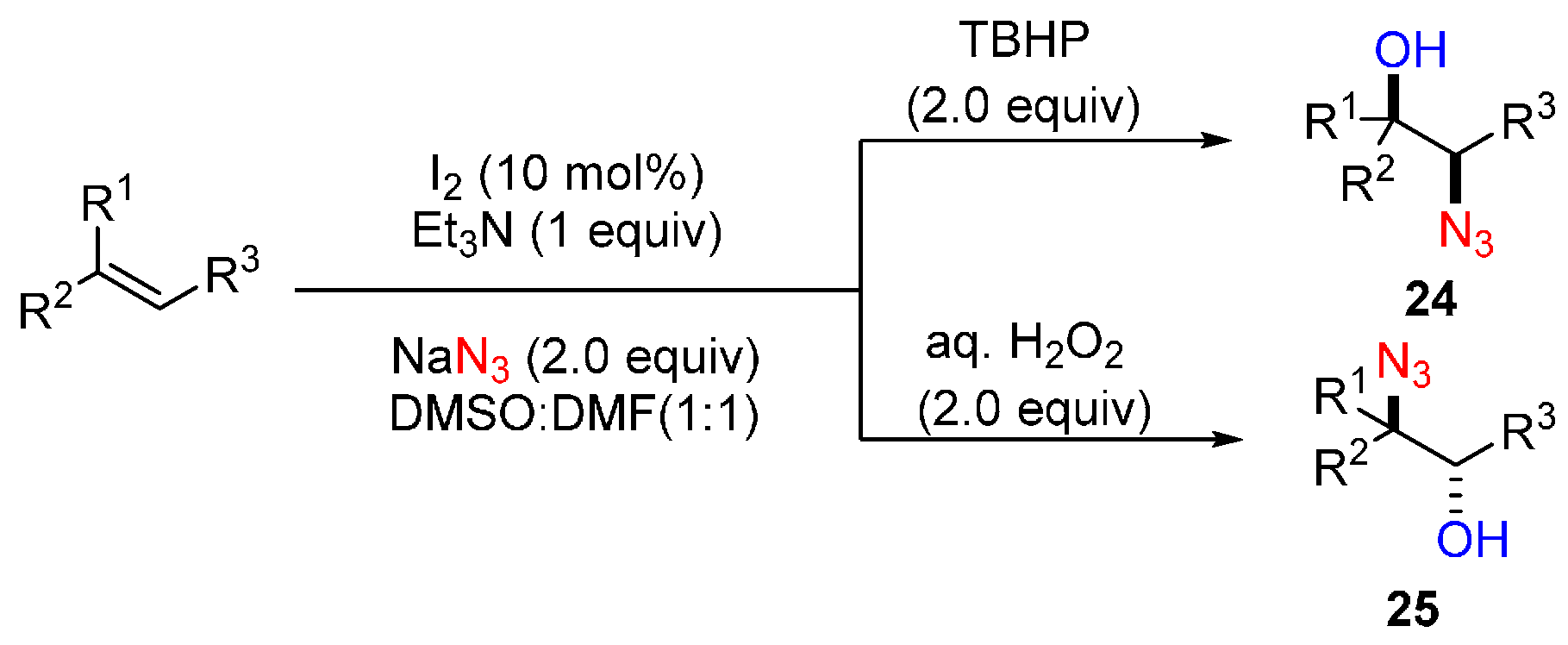
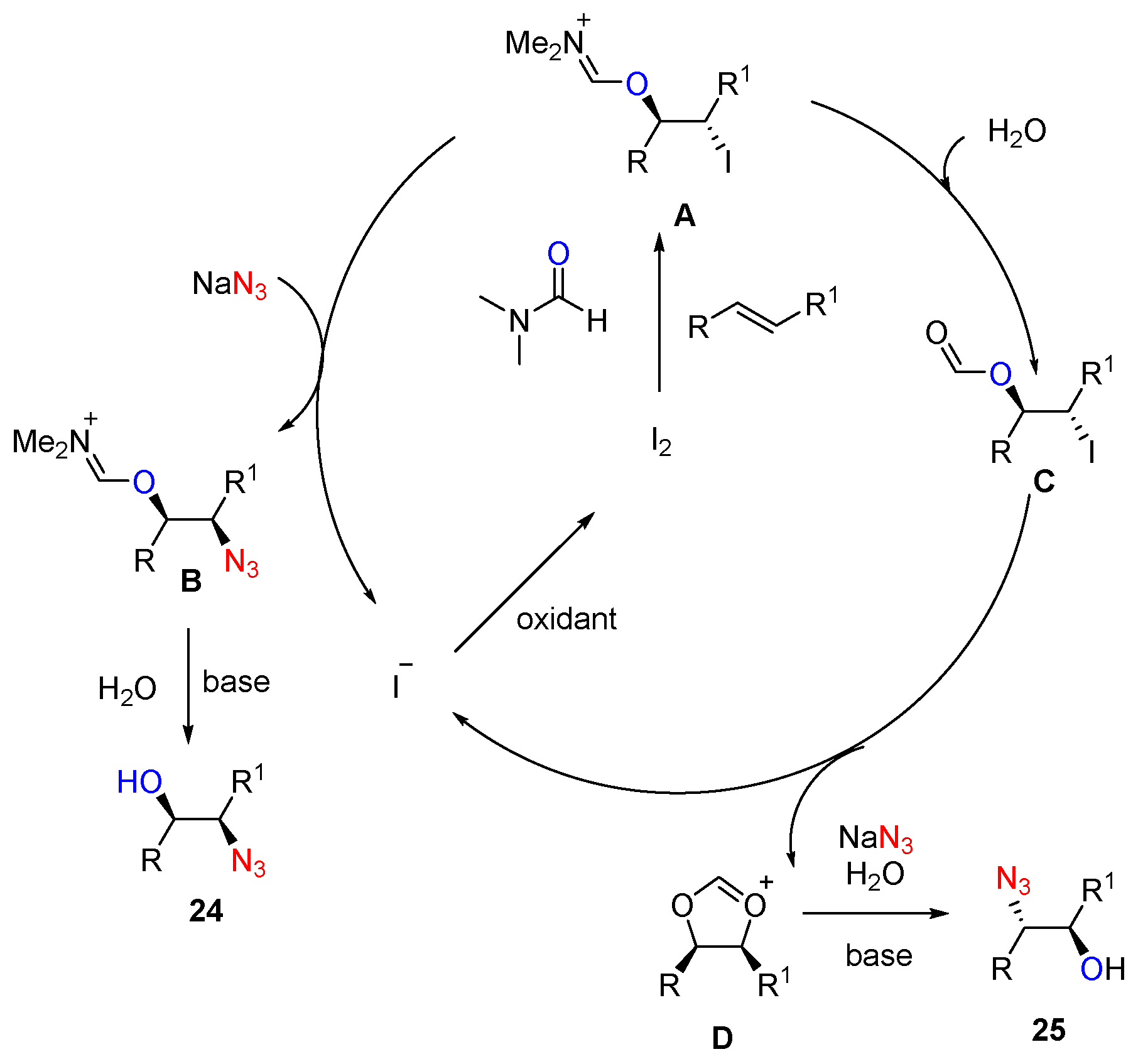
- Prasad, P.K.; Reddi, R.N.; Sudalai, A. Oxidant controlled regio- and stereodivergent azidohydroxylation of alkenes via I2 catalysis. Chem. Commun. 2015, 51, 10276–10279. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, G.; Rabet, P.T.G.; Boyd, S.; Greaney, M.F. Three-component azidation of styrene-type double bonds: Light-switchable behavior of a copper photoredox catalyst. Angew. Chem. Int. Ed. 2015, 54, 11481–11484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Studer, A. Stereoselective radical oxyazidation of alkenes. Org. Lett. 2013, 15, 4548–4551. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Studer, A. Transition-metal-free trifluoromethylaminoxylation of alkenes. Angew. Chem. Int. Ed. 2012, 51, 8221–8224. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.F.; Gu, Z.; Liu, W.; Wang, H.; Xia, Y.; Gao, H.; Liu, X.; Liang, Y.M. Metal-free three-component oxyazidation of alkenes with trimethylsilyl azide and N-hydroxyphthalimide. J. Org. Chem. 2015, 80, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, F.C.; Chemler, S.R. Stereoselective synthesis of morpholines via copper-promoted oxyamination of alkenes. Org. Lett. 2012, 14, 4482–4485. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Buchwald, S.L. Versatile enantioselective synthesis of functionalized lactones via copper-catalyzed radical oxyfunctionalization of alkenes. J. Am. Chem. Soc. 2015, 137, 8069–8077. [Google Scholar] [CrossRef] [PubMed]
- Carre, G.; Ottoli, G. Stereoselective synthesis of drugs and drug precursors by hydrolases and oxidoreductases. Biocatal. Biotransfor. 2000, 18, 119–132. [Google Scholar] [CrossRef]
- Zhu, L.; Yu, H.; Xu, Z.; Jiang, X.; Lin, L.; Wang, R. Copper-catalyzed oxyazidation of unactivated alkenes. Org. Lett. 2014, 16, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, F.C.; Turnpenny, B.W.; Chemler, S.R. Copper-promoted and copper-catalyzed intermolecular alkene diamination. Angew. Chem. Int. Ed. 2010, 49, 6365–6368. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Li, W.; Xuan, Z.; Yu, W. Copper-catalyzed cyclization and azidation of γ, δ-unsaturated ketone o-benzoyl oximes. Adv. Synth. Catal. 2015, 357, 64–70. [Google Scholar] [CrossRef]
- Zhang, B.; Studer, A. Copper-catalyzed intermolecular aminoazidation of alkenes. Org. Lett. 2014, 16, 1790–1793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pu, W.; Xiong, T.; Li, Y.; Zhou, X.; Sun, K.; Liu, Q.; Zhang, Q. Copper-catalyzed intermolecular aminocyanation and diamination of alkenes. Angew. Chem. Int. Ed. 2013, 52, 2529–2533. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Yoshino, T.; Matsunaga, S.; Kanai, M. Sultam synthesis via Cu-catalyzed intermolecular carboamination of alkenes with N-fluorobenzenesulfonimide. Org. Lett. 2013, 15, 2502–2505. [Google Scholar] [CrossRef] [PubMed]
- Snider, B.B.; Lin, H. An improved procedure for the conversion of alkenes and glycals to 1,2-diazides using Mn(OAc)3·2H2O in acetonitrile containing trifluoroacetic acid. Synth. Commun. 1998, 28, 1913–1922. [Google Scholar] [CrossRef]
- Yuan, Y.A.; Lu, D.F.; Chen, Y.R.; Xu, H. Iron catalyzed direct diazidation of a broad range of olefins. Angew. Chem. Int. Ed. 2016, 55, 534–538. [Google Scholar] [CrossRef] [PubMed]
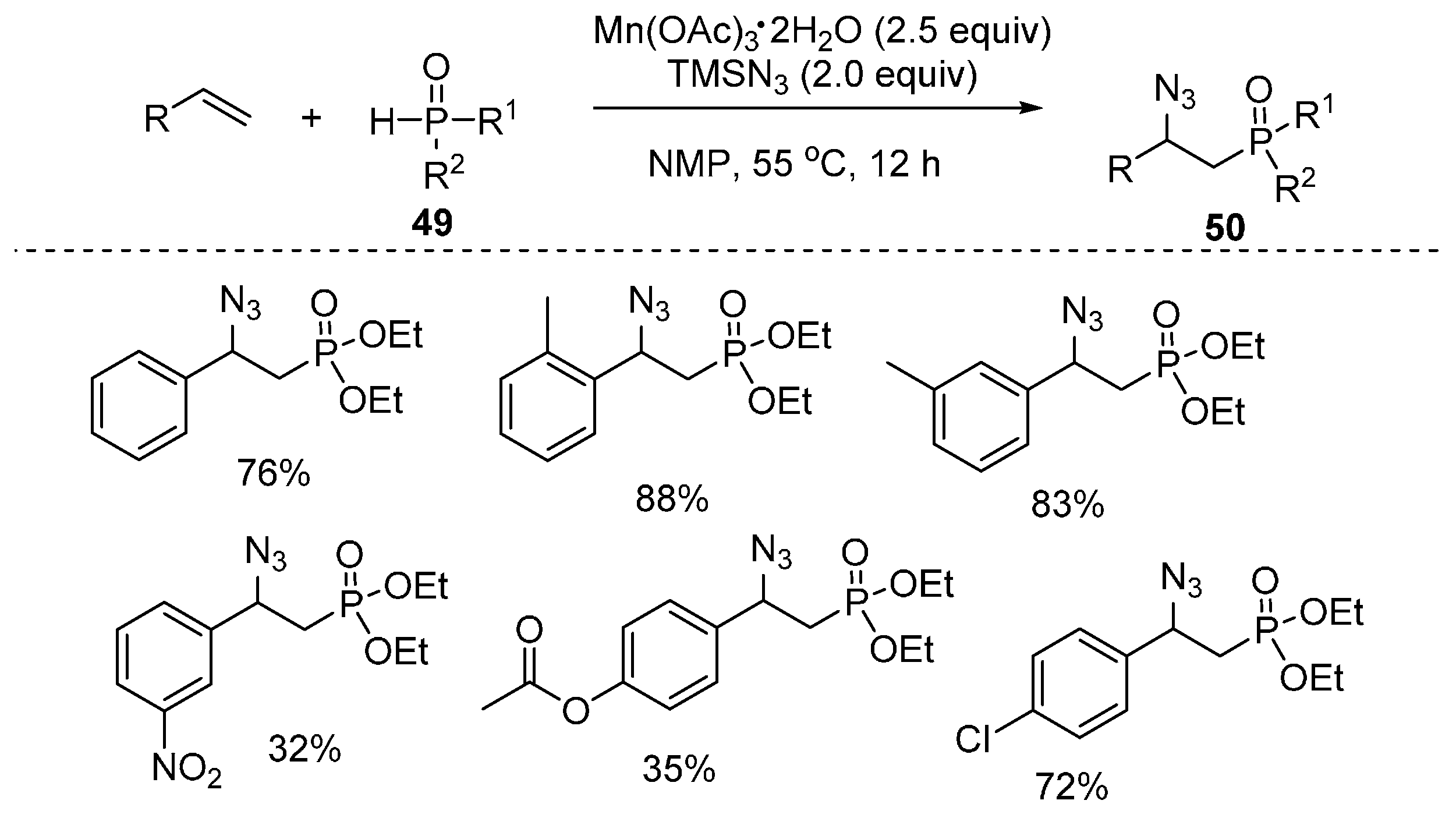
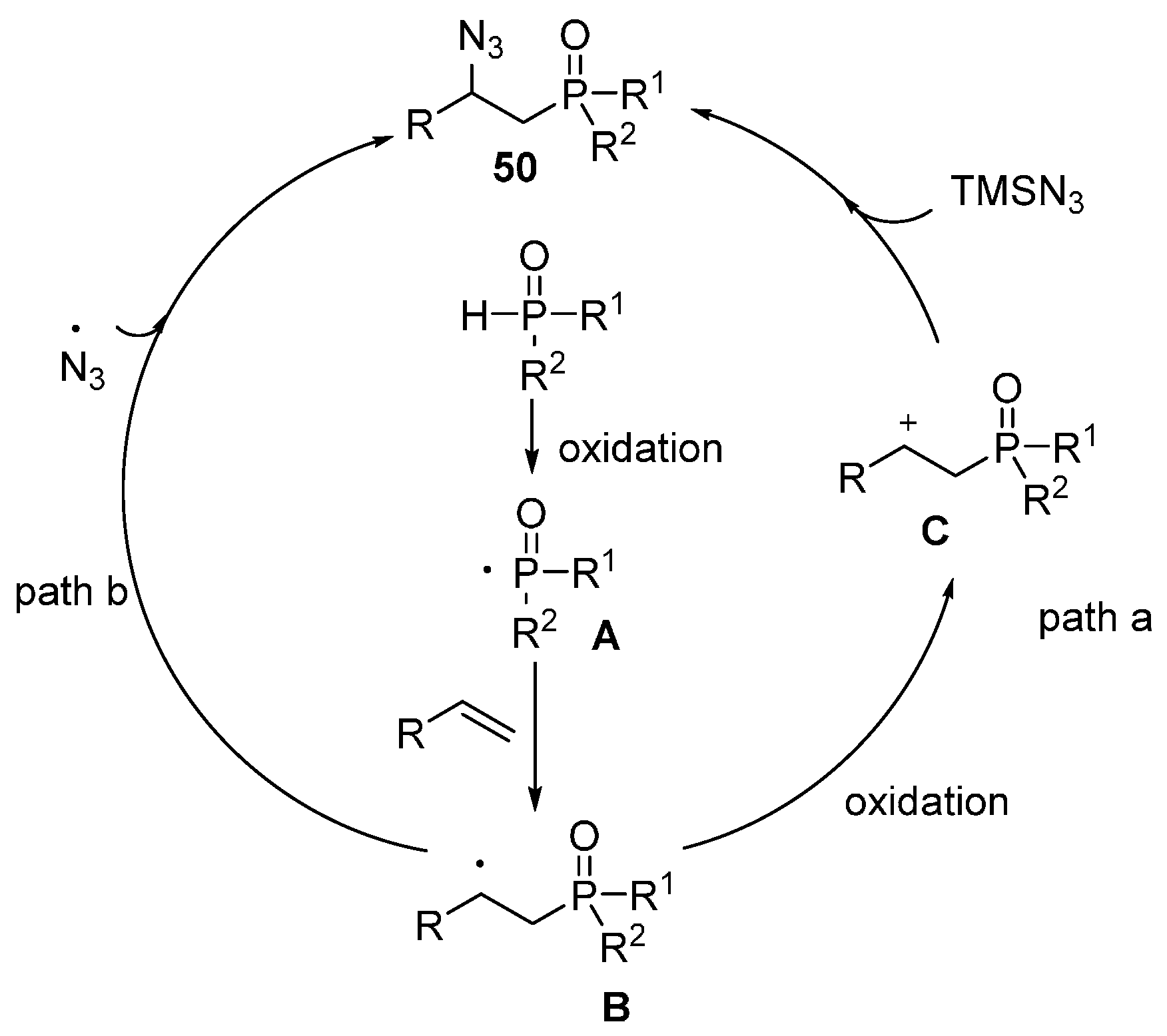
- Xu, J.; Li, X.; Gao, Y.; Zhang, L.; Chen, W.; Fang, H.; Tang, G.; Zhao, Y. Mn(III)-mediated phosphonation–azidation of alkenes: A facile synthesis of β-azidophosphonates. Chem. Commun. 2015, 51, 11240–11243. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, P.; Li, X.; Gao, Y.; Wu, J.; Tang, G.; Zhao, Y. Tetrabutylammonium iodide-catalyzed phosphorylation of benzyl C-H bonds via a cross-dehydrogenative coupling reaction. Adv. Synth. Catal. 2014, 356, 3331–3335. [Google Scholar] [CrossRef]
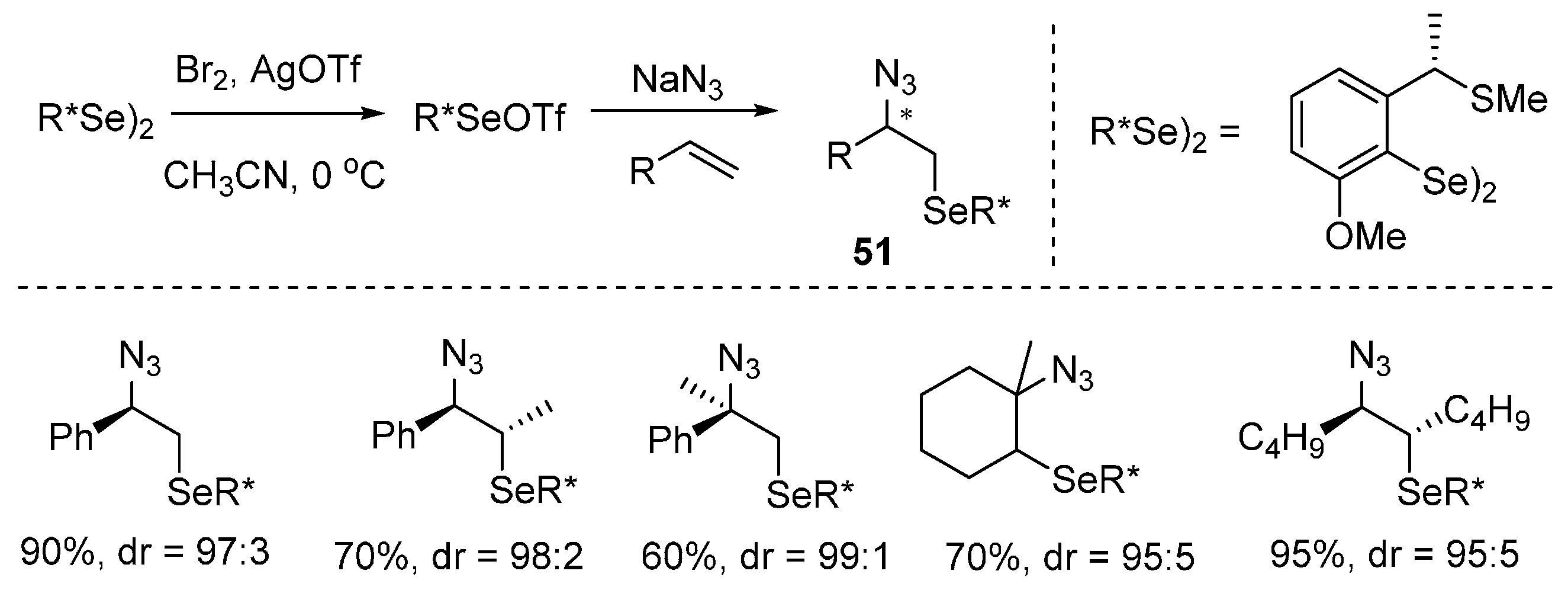
- Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. Asymmetric azidoselenenylation of alkenes: A key step for the synthesis of enantiomerically enriched nitrogen-containing compounds. Angew. Chem. Int. Ed. 2003, 42, 3131–3133. [Google Scholar] [CrossRef] [PubMed]
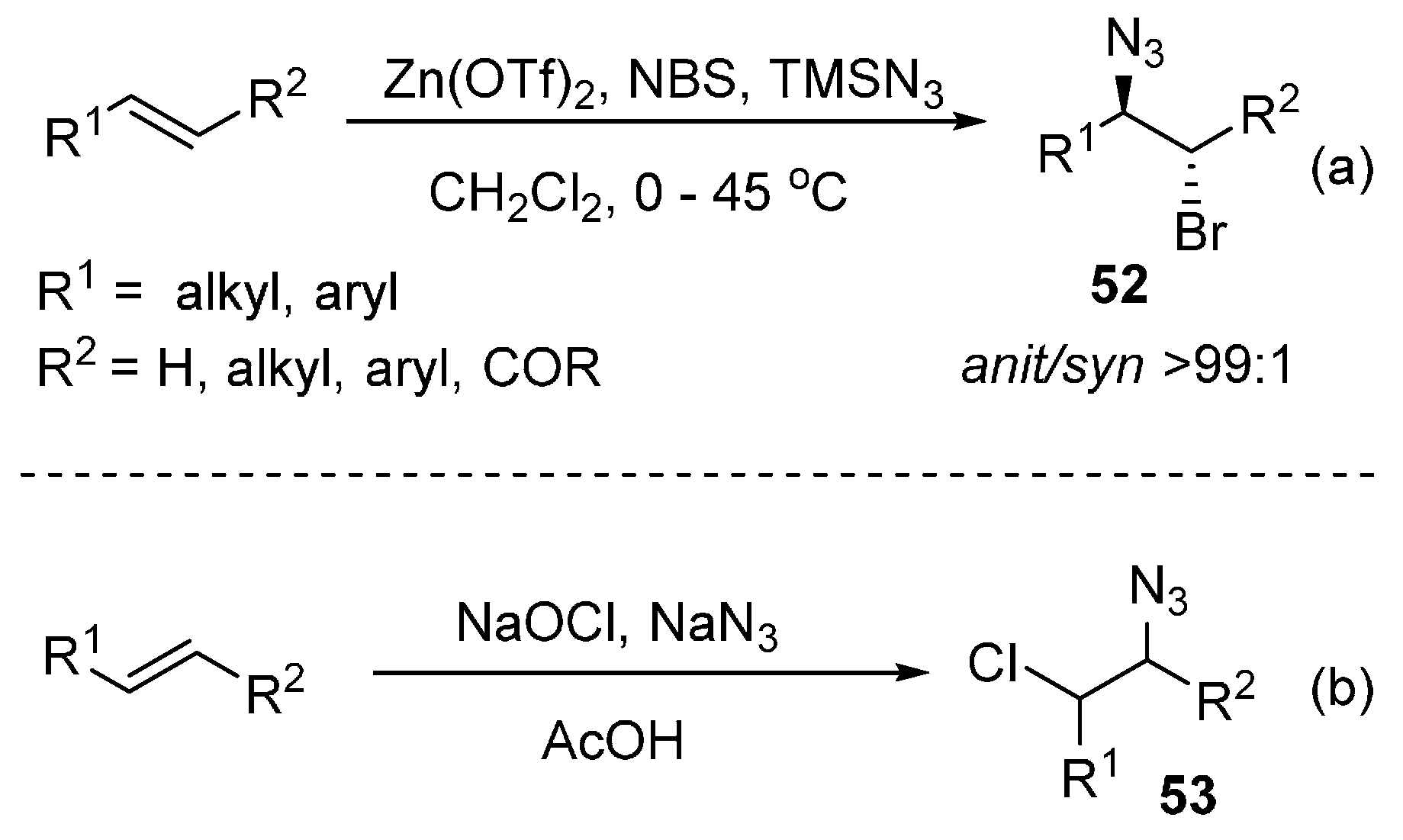
- Hajra, S.; Sinha, D.; Bhowmick, M. Metal triflate catalyzed highly regio- and stereoselective 1,2-bromoazidation of alkenes using NBS and TMSN3 as the bromine and azide sources. Tetrahedron Lett. 2006, 47, 7017–7019. [Google Scholar] [CrossRef]
- Saikia, I.; Phukan, P. Catalyst-free vicinal bromoazidation of olefins using TMSN3 and NBS. C. R. Chim. 2012, 15, 688–692. [Google Scholar] [CrossRef]
- Saikia, I.; Phukan, P. Facile generation of vicinal bromoazides from olefins using TMSN3 and TsNBr2 without any catalyst. Tetrahedron Lett. 2009, 50, 5083–5087. [Google Scholar] [CrossRef]
- Valiulin, R.A.; Mamidyala, S.; Finn, M.G. Taming ghlorine azide: access to 1,2-azidochlorides from alkenes. J. Org. Chem. 2015, 80, 2740–2755. [Google Scholar] [CrossRef] [PubMed]




































© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.; Liang, Y.; Jiao, N. Azidation in the Difunctionalization of Olefins. Molecules 2016, 21, 352. https://doi.org/10.3390/molecules21030352
Wu K, Liang Y, Jiao N. Azidation in the Difunctionalization of Olefins. Molecules. 2016; 21(3):352. https://doi.org/10.3390/molecules21030352
Chicago/Turabian StyleWu, Kai, Yujie Liang, and Ning Jiao. 2016. "Azidation in the Difunctionalization of Olefins" Molecules 21, no. 3: 352. https://doi.org/10.3390/molecules21030352
APA StyleWu, K., Liang, Y., & Jiao, N. (2016). Azidation in the Difunctionalization of Olefins. Molecules, 21(3), 352. https://doi.org/10.3390/molecules21030352