
Iron(III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions



Abstract
:
1. Introduction
2. Results and Discussion
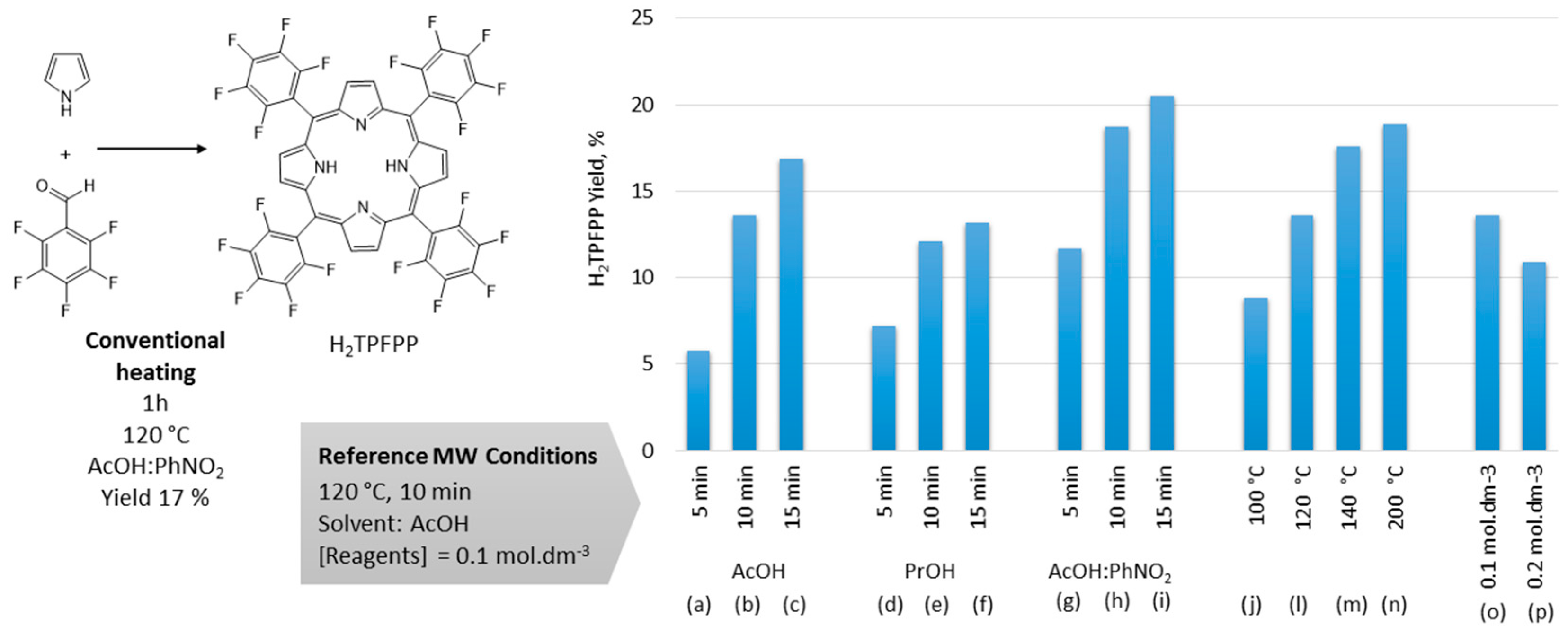
2.1. Optimization of the Microwave Synthesis of H2TPFPP
2.2. Optimization of the Iron Insertion Reaction Using Microwave Heating Conditions
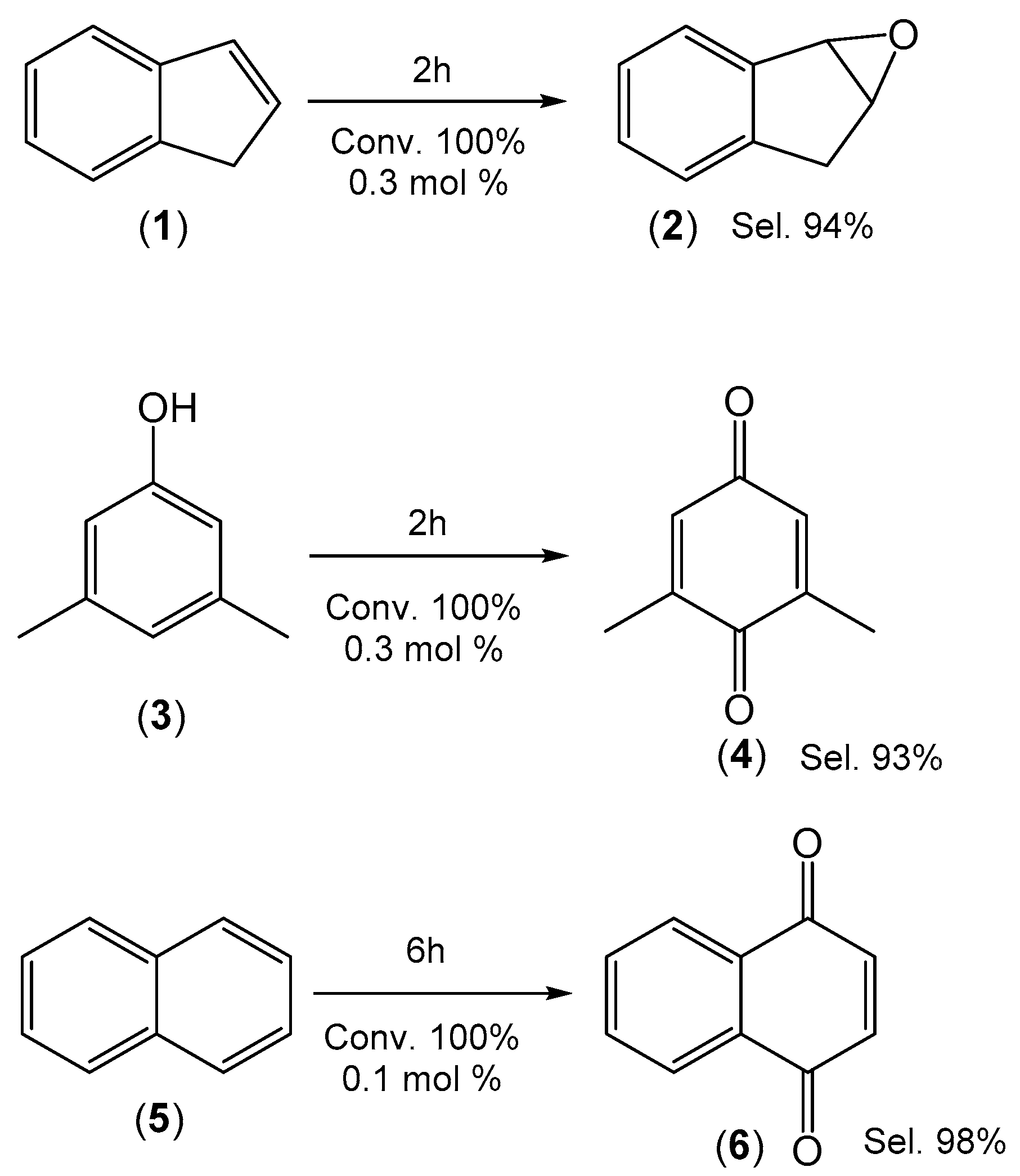
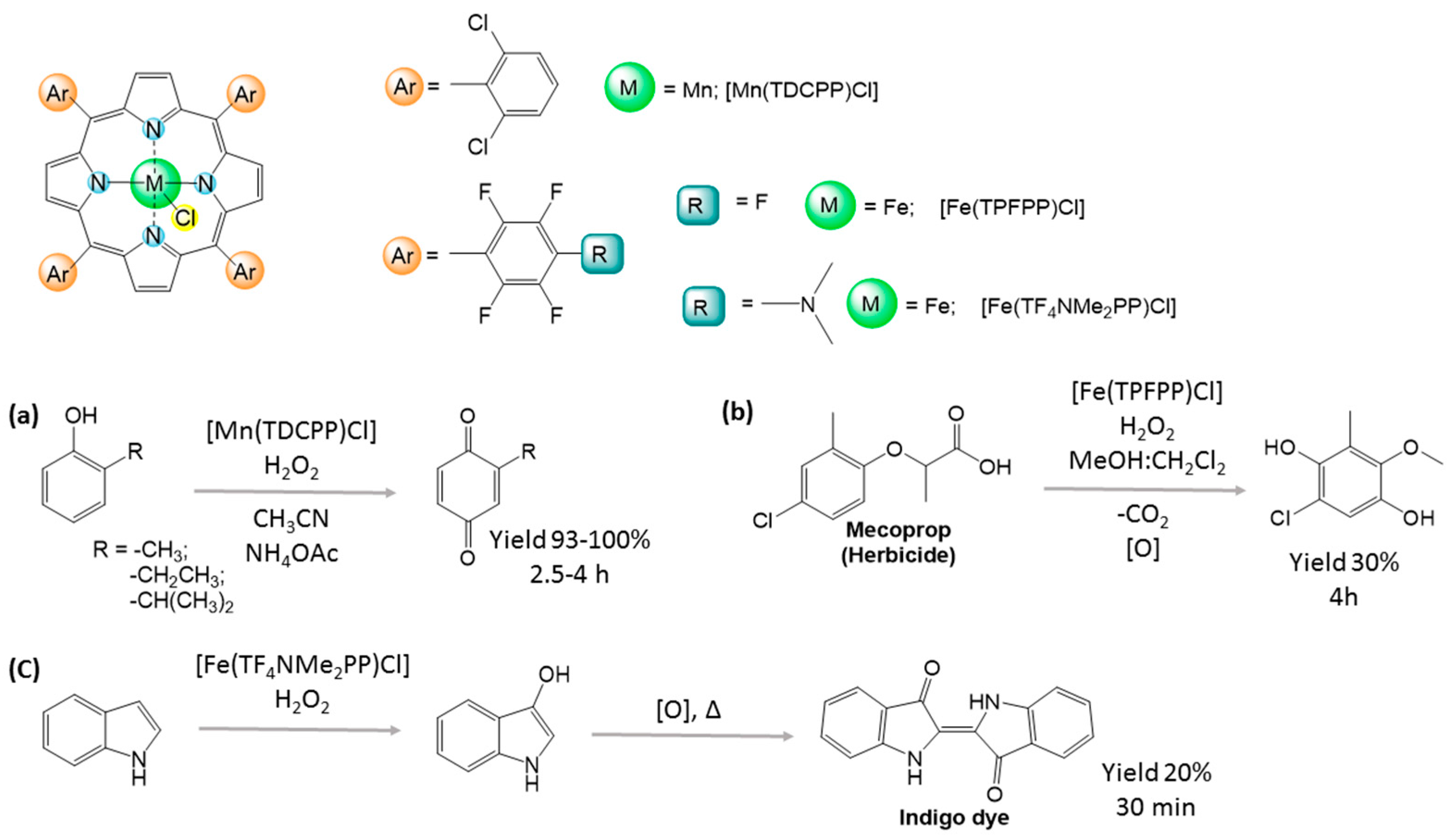
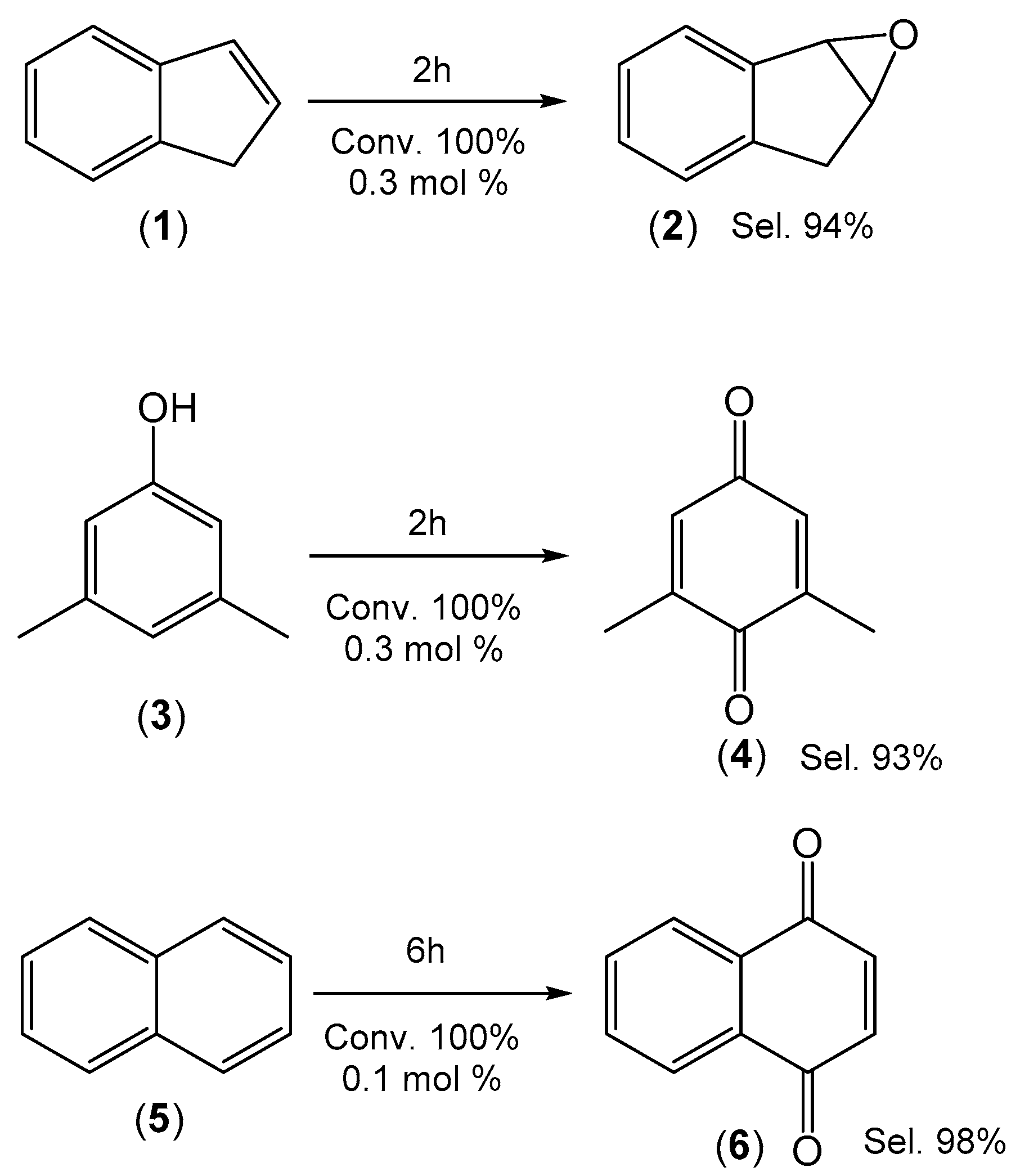
2.3. Green Oxidation of Aromatic Compounds Using [Fe(TPFPP)Cl]
3. Materials and Methods
3.1. Physical Measurements
3.2. Preparation of Metaloporphyrin Catalysts
3.3. Catalysis Experiments
3.4. Analytical Data for Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhupathiraju, N.V.S.D.K.; Rizvi, W.; Batteas, J.D.; Drain, C.M. Fluorinated porphyrinoids as efficient platforms for new photonic materials, sensors, and therapeutics. Org. Biomol. Chem. 2016, 14, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.-J.; Yun, K.-J.; Kang, M.-S.; Park, J.; Lee, K.-T.; Parka, S.B.; Shin, J.-H. Synthesis and in vitro photodynamic activities of water-soluble fluorinated tetrapyridylporphyrins as tumor photosensitizers. Bioorg. Med. Chem. Lett. 2007, 17, 2789–2794. [Google Scholar] [CrossRef] [PubMed]
- Milot, R.L.; Schmuttenmaer, C.A. Electron injection dynamics in high-potential porphyrin photoanodes. Acc. Chem. Res. 2015, 48, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Nakazono, T.; Parent, A.R.; Sakai, K. Improving singlet oxygen resistance during photochemical water oxidation by cobalt porphyrin catalysts. Chem. Eur. J. 2015, 21, 6723–6726. [Google Scholar] [CrossRef] [PubMed]
- Goslinski, T.; Piskorz, J. Fluorinated porphyrinoids and their biomedical applications. J. Photochem. Photobiol. C Photochem. Rev. 2011, 12, 304–321. [Google Scholar] [CrossRef]
- Costas, M. Selective C–H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. [Google Scholar] [CrossRef]
- Simões, M.M.Q.; Neves, C.M.B.; Pires, S.M.G.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mimicking P-450 processes and the use of metalloporphyrins. Pure Appl. Chem. 2013, 85, 1671–1681. [Google Scholar]
- Zucca, P.; Rescigno, A.; Rinaldi, A.C.; Sanjust, E. Biomimetic metalloporphines and metalloporphyrins as potential tools for delignification: Molecular mechanisms and application perspectives. J. Mol. Catal. A 2014, 388–389, 2–34. [Google Scholar] [CrossRef]
- Kubota, R.; Asayama, S.; Kawakami, H. A bioinspired polymer-bound Mn-porphyrin as an artificial active center of catalase. Chem. Commun. 2014, 50, 15909–15912. [Google Scholar] [CrossRef] [PubMed]
- Mahammed, A.; Gross, Z. The importance of developing metal complexes with pronounced catalase-like activity. Catal. Sci. Technol. 2011, 1, 535–540. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Pereira, M.M.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mechanistic studies on metalloporphyrin epoxidation reactions with hydrogen peroxide: Evidence of two active species. J. Catal. 2005, 234, 76–87. [Google Scholar] [CrossRef]
- Pires, S.M.G.; Simões, M.M.Q.; Santos, I.C.M.S.; Rebelo, S.L.H.; Paz, F.A.A.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidation of organosulfur compounds using an iron(III) porphyrin complex: An environmentally safe and efficient approach. Appl. Catal. B: Environ. 2014, 160–161, 80–88. [Google Scholar] [CrossRef]
- Linhares, M.; Rebelo, S.L.H.; Simões, M.M.Q.; Silva, A.M.S.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Freire, C. Biomimetic oxidation of indole by Mn(III)porphyrins. Appl. Catal. A Gen. 2014, 470, 427–433. [Google Scholar] [CrossRef]
- Linhares, M.; Rebelo, S.L.H.; Biernacki, K.; Magalhães, A.L.; Freire, C. Biomimetic one-pot route to acridine epoxides. J. Org. Chem. 2015, 80, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Linhares, M.; Rebelo, S.L.H.; Neves, M.G.P.M.S.; Freire, C. Direct access to polycyclic peripheral diepoxy-meso-quinone derivatives from acene catalytic oxidation. RSC Adv. 2013, 3, 5350–5353. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Linhares, M.; Simões, M.M.Q.; Silva, A.M.S.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Freire, C. Catalytic production of indigo dye inspired on iron(III) porphyrin biosystems. J. Catal. 2014, 315, 33–40. [Google Scholar] [CrossRef]
- Liu, W.; Huang, X.; Cheng, M.J.; Nielsen, R.J.; Goddard, W.A., III; Groves, J.T. Oxidative aliphatic C-H fluorination with fluoride ion catalyzed by a manganese porphyrin. Science 2012, 337, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.M.Q.; De Paula, R.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Metalloporphyrins in the biomimetic oxidative valorization of natural and other organic substrates. J. Porphyr. Phthal. 2009, 13, 589–596. [Google Scholar] [CrossRef]
- Rocha, M.; Rebelo, S.L.H.; Freire, C. Enantioselective arene epoxidation by Jacobsen catalyst: Role of protic solvent and co-catalyst on the activation of hydrogen peroxide. Appl. Catal. A Gen. 2013, 460–461, 116–123. [Google Scholar] [CrossRef]
- Silva, G.; Pires, S.M.G.; Silva, V.L.M.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Rebelo, S.L.H.; Silva, A.M.S.; Cavaleiro, J.A.S. A green and sustainable method for the oxidation of 3-dihydrobenzo[c]thiophenes using metalloporphyrin complexes. Catal. Commun. 2014, 56, 68–71. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidation of alkylaromatics with hydrogen peroxide catalysed by manganese(III) porphyrins in the presence of ammonium acetate. J. Mol. Catal. A Chem. 2003, 201, 9–22. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Pereira, M.M.; Monsanto, P.V.; Burrows, H.D. Catalytic oxidative degradation of s-triazine and phenoxyalkanoic acid based herbicides with metalloporphyrins and hydrogen peroxide: Identification of two distinct reaction schemes. J. Mol. Catal. A Chem. 2009, 297, 35–43. [Google Scholar] [CrossRef]
- Gonsalves, A.M.A.R.; Varejão, J.M.T.B.; Pereira, M.M. Some new aspects related related to the synthesis of mesosubstituted porphyrins. J. Heterocycl. Chem. 1991, 28, 635–640. [Google Scholar] [CrossRef]
- Johnstone, R.A.W.; Nunes, M.L.P.G.; Pereira, M.M.; Gonsalves, A.M.A.R.; Serra, A.C. Improved synthesis of 5,10,15,20-tetrakisaryl- and tetrakisalkylporphyrins. Heterocycles 1996, 43, 1423–1437. [Google Scholar]
- Adler, A.D.; Longo, F.R.; Kampas, F.; Kim, J. On the preparation of metallopophyrins. J. Inorg. Nucl. Chem. 1970, 32, 2443–2445. [Google Scholar] [CrossRef]
- Kadish, K.M.; Han, B.C.; Franzen, M.M.; Araullo-McAdams, C. Syntheses and spectroscopic characterization of (T(p-Me2N)F4PP)H2 and (T(p-Me2N)F4PP)M where T(p-Me2N)F4PP = the dianion of meso-tetrakis(o,o,m,m-tetrafluoro-p-(dimethylamino)phenyl)porphyrin and M = cobalt(II), copper(II), or nickel(II). Structures of (T(p-Me2N)F4PP)Co and meso-tetrakis(pentafluorophenyl)porphinatocobalt(II), (TF5PP)Co. J. Am. Chem. Soc. 1990, 112, 8364–8368. [Google Scholar]
- Samaroo, D.; Soll, C.E.; Todaro, L.J.; Drain, C.M. Efficient microwave-assisted synthesis of amine-substituted tetrakis(pentafluorophenyl)porphyrin. Org. Lett. 2006, 8, 4985–4988. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.; Andrade, M.; Pereira, C.; Pereira, A.M.; Rebelo, S.L.H.; Araújo, J.P.; Pires, J.; Carvalho, A.P.; Freire, C. Alkene epoxidation by manganese(III) complexes immobilized onto nanostructured carbon CMK-3. Catal. Today 2013, 203, 103–110. [Google Scholar] [CrossRef]
- Lipińska, M.E.; Rebelo, S.L.H.; Freire, C. Iron(III) porphyrin anchored onto organosilylated multi-walled carbon nanotubes as active catalyst for epoxidation reactions in eco-compatible conditions. J. Mater. Sci. 2014, 49, 1494–1505. [Google Scholar] [CrossRef]
- De Paula, R.; Faustino, M.A.F.; Pinto, D.C.G.A.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Kinetic study of meso-tetraphenylporphyrin synthesis under microwave irradiation. J. Heterocycl. Chem. 2008, 4, 453–459. [Google Scholar] [CrossRef]
- Chandrasekharam, M.; Rao, C.S.; Singh, S.P.; Kantam, M.L.; Reddy, M.R.; Reddy, P.Y.; Toru, T. Microwave-assisted synthesis of metalloporphyrazines. Tetrahedron Lett. 2007, 48, 2627–2630. [Google Scholar] [CrossRef]
- Boscencu, R. Microwave synthesis under solvent-free conditions and spectral studies of some mesoporphyrinic complexes. Molecules 2012, 17, 5592–5603. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.N.; Aguiar, A.; Balula, S.S.; Silva, A.M.G.; Rangel, M. Characterization of a μ-oxo-bridged diiron porphyrin by ESI-LTQ-Orbitrap-MS. J. Mass Spectrom. 2014, 49, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Juillard, J. Dimethylformamide: Purification, tests for purity and physical properties. Pure Appl. Chem. 1977, 49, 885–892. [Google Scholar]
- Rebelo, S.L.H.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. An efficient approach for aromatic epoxidation using hydrogen peroxide and Mn(III) porphyrins. Chem. Commun. 2004, 7, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Vatsis, K.P.; Coon, M.J. Ipso-Substitution by Cytochrome P450 with Conversion of p-hydroxybenzene derivatives to hydroquinone: Evidence for hydroperoxo-iron as the active oxygen species. Arch. Biochem. Biophys. 2002, 397, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds H2TPFPP and [Fe(TFPPP)Cl] are available from the authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Heating | Solvent | Temperature | Time | FeCl2:H2TPFPP a |
|---|---|---|---|---|---|
| 1 | conventional | DMF, Py | 155 °C | 48 h | 200 |
| 2 | conventional | DMF, Py | 120 °C | 48 h | 200 |
| 3 | microwave | DMF | 120 °C | 24 h | 100 |
| 4 | microwave | CH3CN | 120 °C b | 3 h b | 20 |
| 5 | microwave | DMF | 160 °C b | 3 h b | 20 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rebelo, S.L.H.; Silva, A.M.N.; Medforth, C.J.; Freire, C. Iron(III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions. Molecules 2016, 21, 481. https://doi.org/10.3390/molecules21040481
Rebelo SLH, Silva AMN, Medforth CJ, Freire C. Iron(III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions. Molecules. 2016; 21(4):481. https://doi.org/10.3390/molecules21040481
Chicago/Turabian StyleRebelo, Susana L. H., André M. N. Silva, Craig J. Medforth, and Cristina Freire. 2016. "Iron(III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions" Molecules 21, no. 4: 481. https://doi.org/10.3390/molecules21040481