
Rearrangements of Cycloalkenyl Aryl Ethers

 ,
,
Abstract
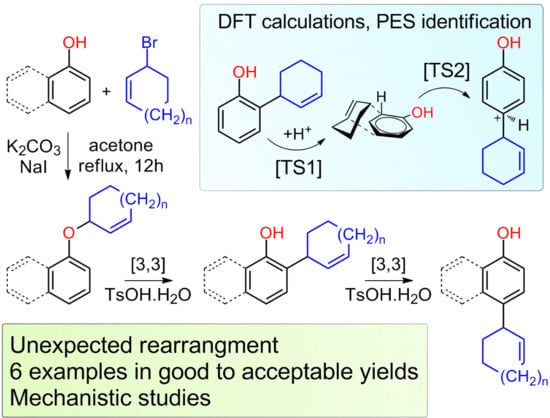
:
1. Introduction
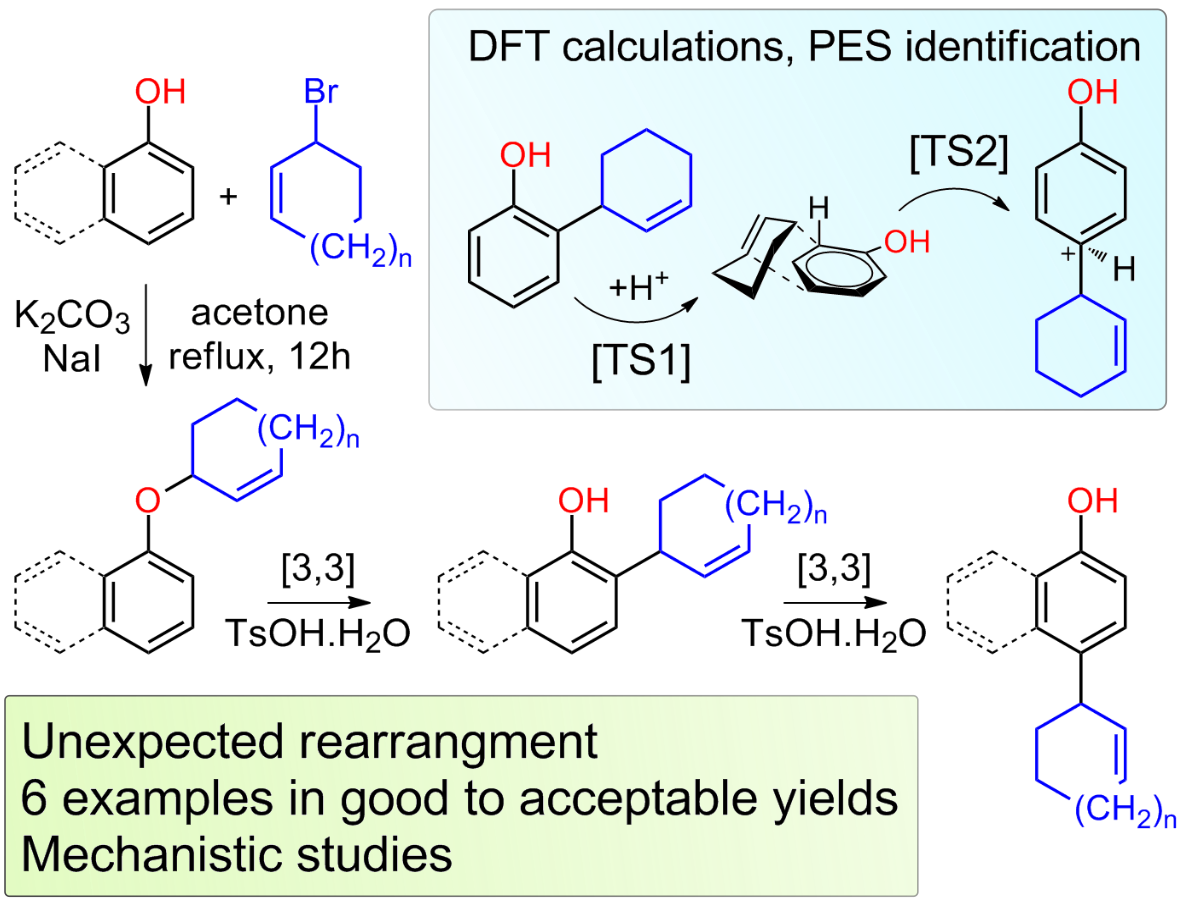
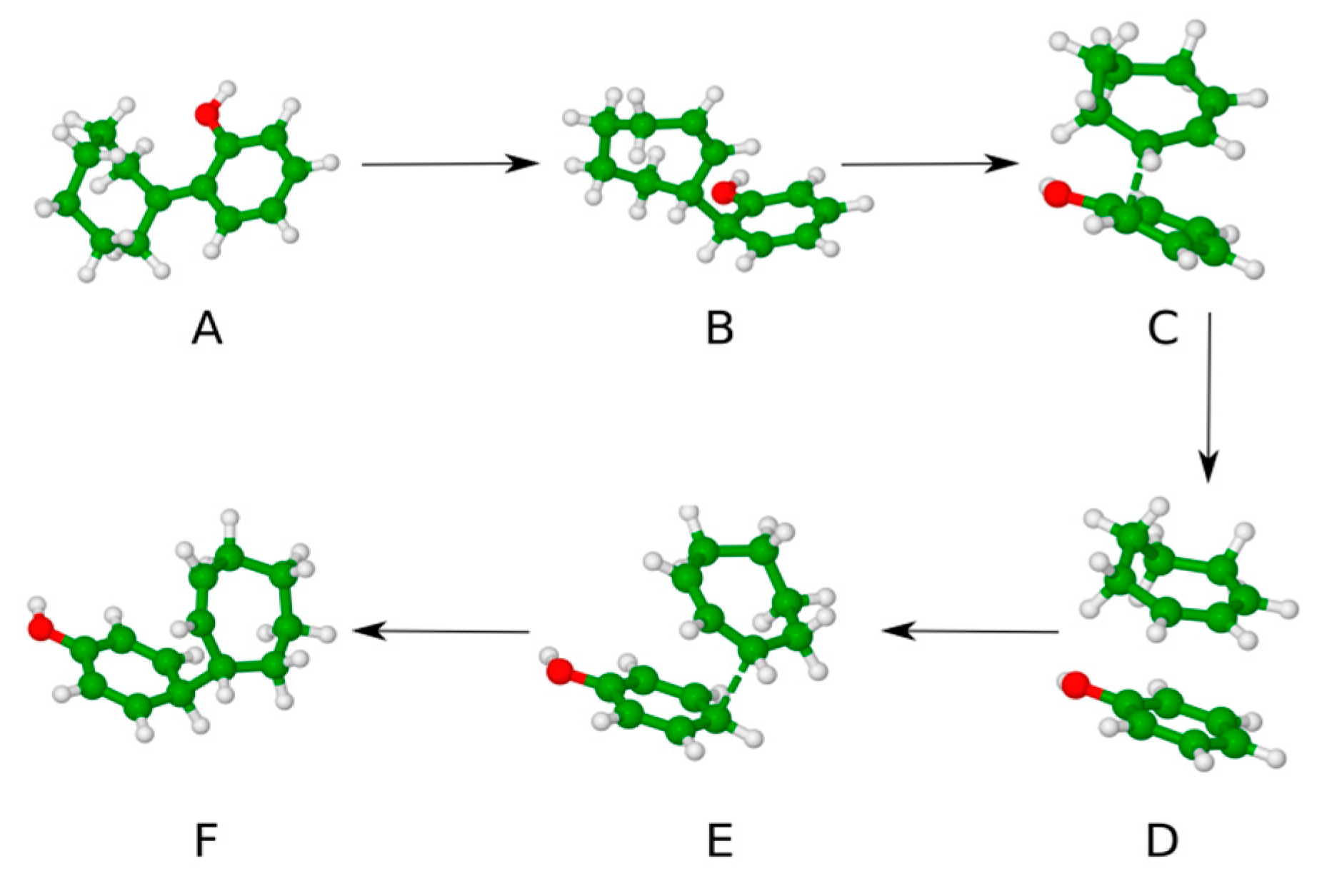
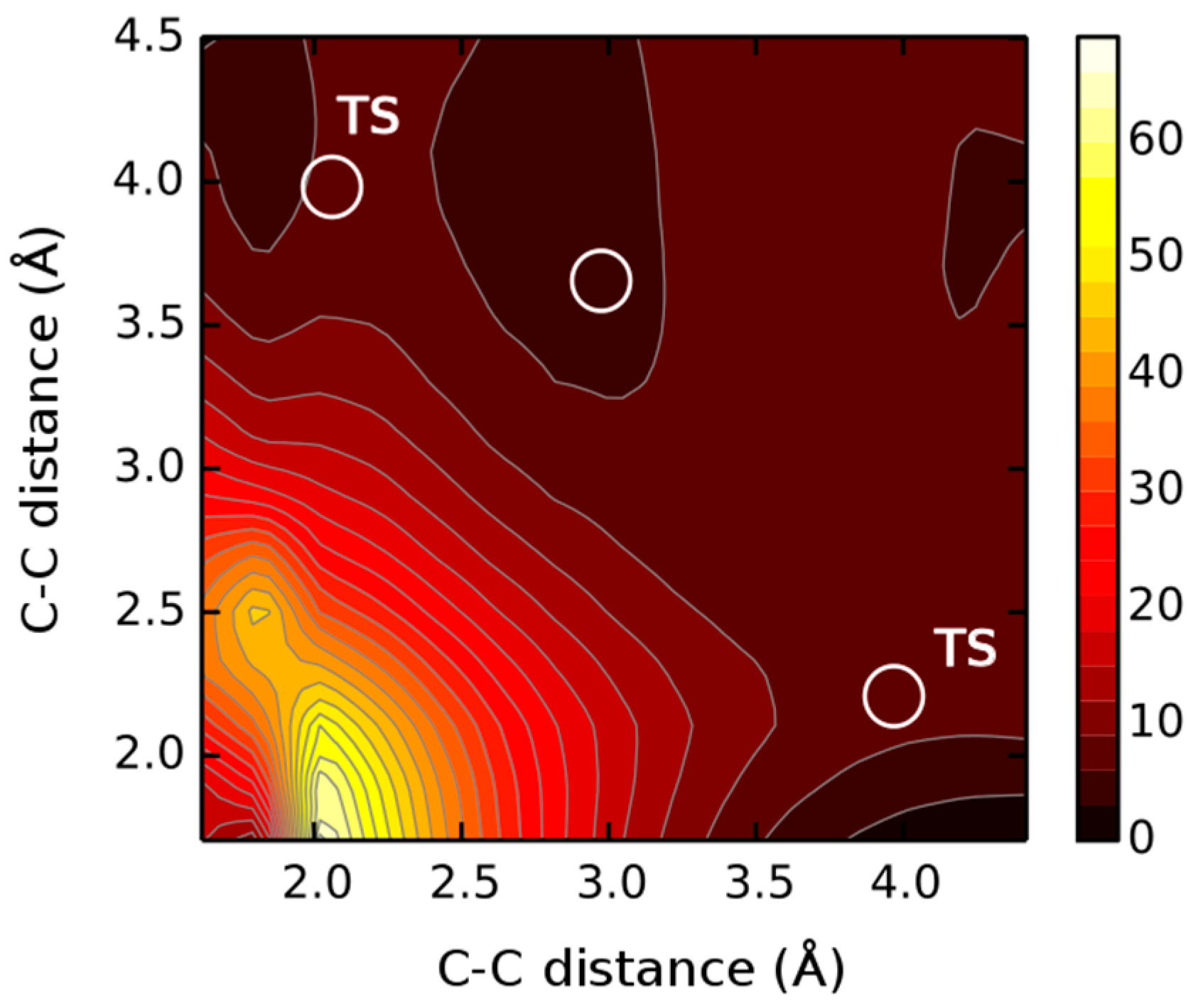
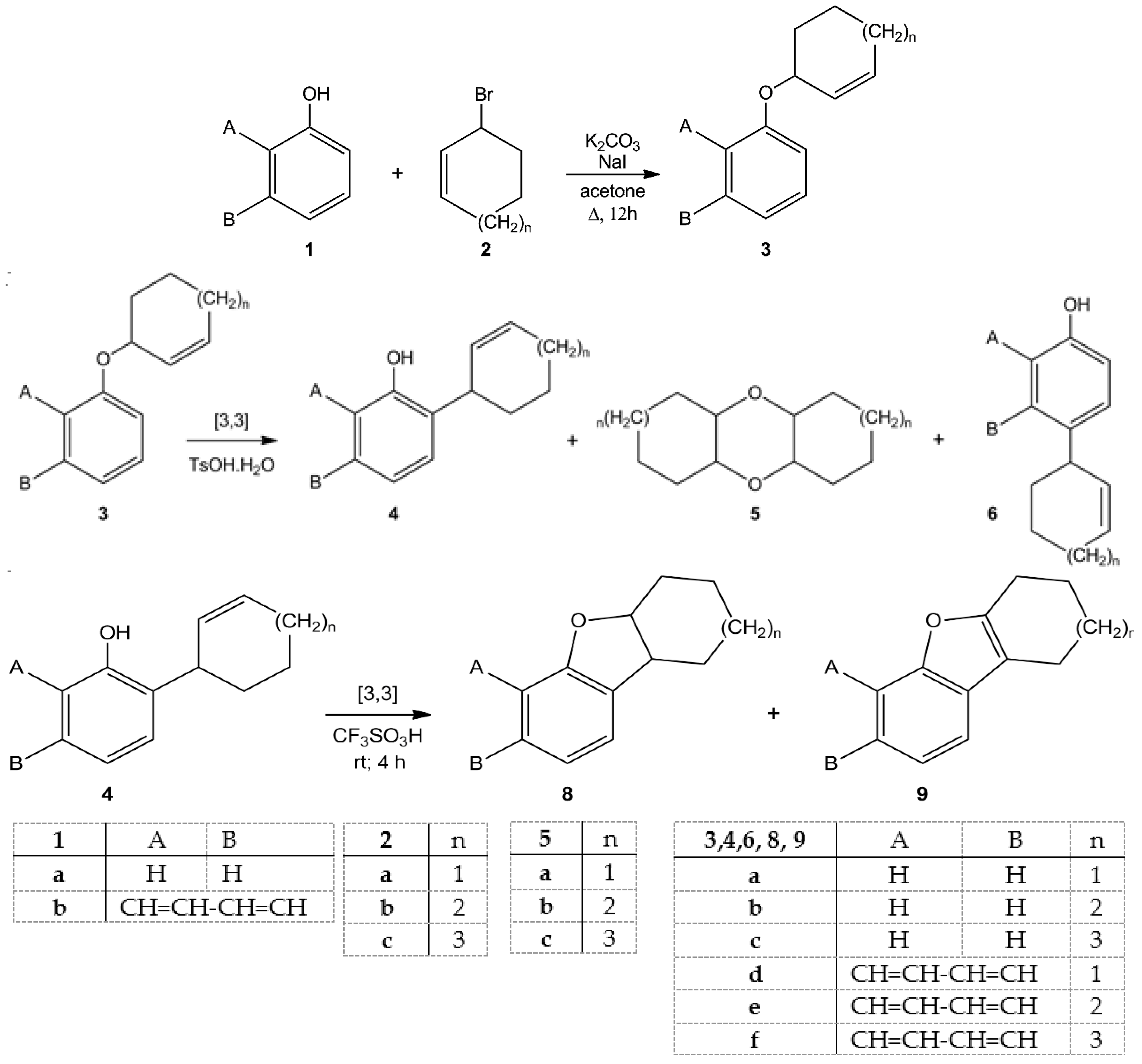
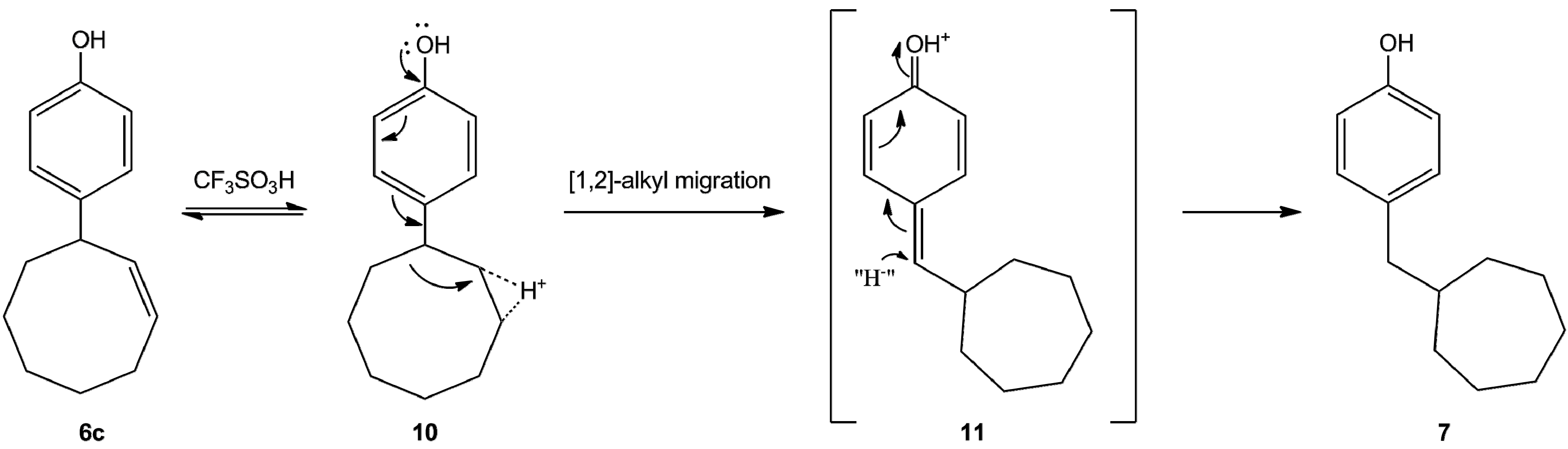
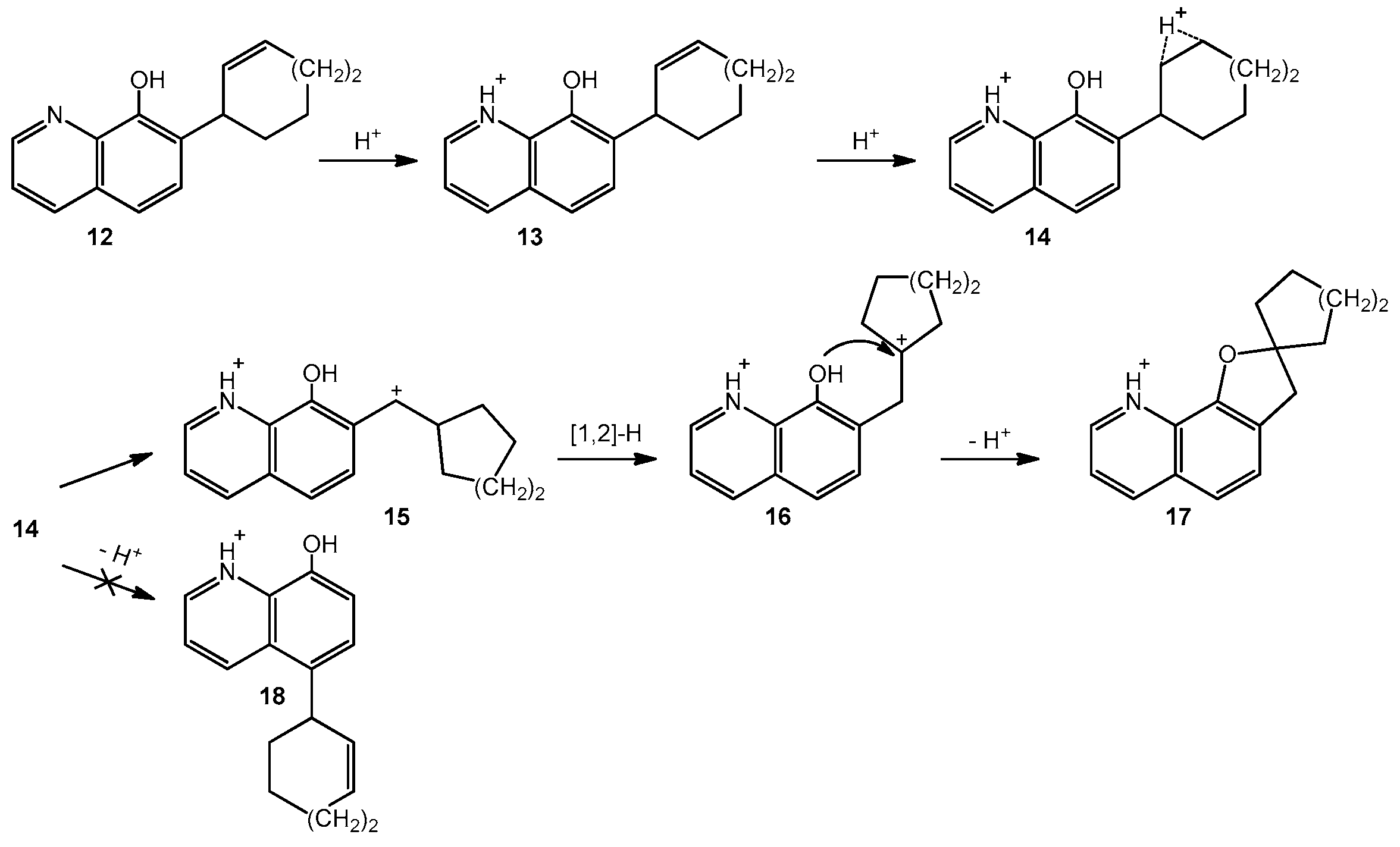
2. Results and Discussions
3. Experimental Section
3.1. General Information
3.2. Preparation of Ethers 3: General Procedure
3.3. Rearrangement of Ethers 3: General Procedure
3.4. General Procedures for Cyclization of Compounds 4a,b and 4d–f
4. Conclusions
Author Contributions
Conflicts of Interest
References and Notes
- Cicerale, S.; Lucas, L.; Keast, R. Biological activities of phenolic compounds present in virgin olive oil. Int. J. Mol. Sci. 2010, 11, 458–479. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Valentao, P.; Pereira, J.A.; Andrade, P.B. Fenolics: From chemistry to biology. Molecules 2009, 14, 2202–2211. [Google Scholar] [CrossRef]
- Huang, W.Y.; Cai, Y.Z.; Zhang, Y. Natural phenolic compounds from medicinal herbs and dietary plants, potential use for cancer prevention. Nutr. Cancer 2010, 62, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Harbor, J.B. Biochemistry of Phenolic Compounds; Academic Press: London, UK, 1964. [Google Scholar]
- Vermeries, W.; Nicolson, R. Biochemistry of Phenolic Compounds; Springer: Heidelberg, Germany, 2006. [Google Scholar]
- Kadieva, M.G.; Oganesyan, F.T. Methods for the synthesis of benzofuran derivatives (Review). Chem. Heterocycl. Compd. 1997, 33, 1245–1258. [Google Scholar] [CrossRef]
- De Luca, I.; Nieddu, G.; Porcheddu, A.; Giacomelli, G. Some recent approaches to the synthesis of 2-substituted benzofurans. Curr. Med. Chem. 2009, 16, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Lee, T.W.; Wu, M.H. Synthesis of substituted dihydrobenzofurans and bis-dihydrobenzofurans. Heterocycles 2012, 85, 1607–1613. [Google Scholar] [CrossRef]
- Zhou, X.; Li, M.; Wang, X.B.; Wang, T.; Kong, Y. Synthesis of benzofuran derivatives via rearrangement and their inhibitory activity on acetylcholinesterase. Molecules 2010, 15, 8593–8601. [Google Scholar] [CrossRef] [PubMed]
- Khanam, H.; Uzzaman, S. Bioactive benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Kongkathip, N.; Hasitapan, K.; Pradiphal, N.; Kirtihara, K.; Jongkon, N.; Kongkathip, B. Synthesis of novel 2-(2-cyclopentyl)- and 2-(2-cyclohexyl) substituted 1-naphthol derivatives with anticyclooxygenase avtivity. Curr. Med. Chem. 2006, 13, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Kongkathip, B.; Sangma, C.; Kirtihara, K.; Luangkamin, S.; Hasitapan, K.; Jongkon, N.; Hannougbua, S.; Kongkathip, N. Inhibitory effect of 2-substituted-1-naphthol derivatives on cyclooxygenase I and II. Bioorg. Med. Chem. 2005, 13, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Orovecz, O.; Kovács, P.; Kaleta, Z.; Párkányi, L.; Szabó, É; Novák, L. Rearrangement of allyl aryl ethers; VI: Reaction of naphthols with cycloalkadienes. Synthesis 2003, 7, 1043–1048. [Google Scholar]
- Rehbein, J.; Hierseman, M. Mechanistic aspects of the aliphatic Claisen Rearrangement. In The Claisen Rearrangement, 1st ed; Horseman, M., Nubbemeyer, U., Eds.; Wiley-VCH: Winheim, Germany, 2007; pp. 525–557. [Google Scholar]
- Castro, A.M.M. Claisen rearrangement over the past nine decades. Chem. Rev. 2004, 104, 2939–3002. [Google Scholar] [CrossRef] [PubMed]
- Lutz, R.P. Catalysis of the Cope and Claisen rearrangements. Chem. Rev. 1984, 84, 205–247. [Google Scholar] [CrossRef]
- Rhoads, S.J.; Rauling, N.R. The Claisen and Cope rearrangements. Org. React. 1975, 22, 1–252. [Google Scholar]
- Nubbemeyer, U. Recent advances in asymmetric [3,3]-sigmatrop rearrangements. Synthesis 2003, 961–1008. [Google Scholar] [CrossRef]
- Ivakura, I.; Kaneko, Y.; Hayashi, S.; Yabushita, A.; Kobayashi, T. The reaction mechanism of Claisen rearrangement obtained by transition state spectroscopy and single direct-dynamic trajectory. Molecules 2013, 18, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- In the mass spectrum of one reaction product 1,2,3,4,56,6a,10,11, 14a-dodecahydrodicycloocta[b,e][1,4]dioxin was observed (mass number 246.1620). This compounds may be formed from the expected hexadecahydro compound (5, n = 3). This reaction may be the hydride source. Unfortunately, so far we have been unable to isolate this compound in pure state.
- Pisanenko, D.A.; Smimov-Zamkov, Y.I.; Srebrodolsky, Y.I. Investigated the reaction of the more reactive 3-(4-methoxyphenyl)cyclopentene and phenol, and 3-(4-ethoxyphenyl)cyclopentene in the presence of BF3·H3PO4 at 60–85 °C, in CCl4. When 0.3 eq. of catalyst was used at 60 °C they observed the formation of arylcycloalkylation products (1,3- and 1,1-diarylcyclopentanes and 1,4-diaryl1,3-cyclopentadienes). Increasing the temperature intensified the process of ionic hydrogenation and isomerization of 1,3- and 1,1-diarylcyclopentanes and promoted the formation of 1,4-diaryl-1,3-cyclopentadienes. Ukrainskii Khimicheskii Zhurnal (Russian Ed.) 2007, 73, 118–127. [Google Scholar]
- The referees recommended to perform the acid-catalyzed reaction with [(4-alkyl-2-cyclohexen-1-yl)oxy]benzene. We have prepared the [(4-methyl-2-cyclohex-1-yl)oxy]benzene. The rearrangement reaction was carried out between 20 °C and 40 °C, and we obtained only 2-(6-methyl-2-cyclohexen-1-yl)phenol as a mixture of cis and trans isomers (ratio 2:3). We repeated the reaction in the presence of the reactive anisole and we have also isolated the above products, and no “cross reaction” was observed. We consider these results as an evidence for the intramolecular reaction.
- Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013.
- Chai, J.D.; Gordon, H. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Krenske, E.H.; Davison, E.C.; Forbes, I.T.; Warner, J.A.; Smith, A.L.; Holmes, A.B.; Houk, K.N. Reverse Cope elimination of hydroxylamines and alkenes or alkynes. Theoretical investigation of tether length and substituent effects. J. Am. Chem. Soc. 2012, 134, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Törincsi, M.; Kolonits, P.; Novák, L. Synthesis and further rearrangements of 7-(2-cycloalken-1-yl)-8-quinolins. Monatsch. Chem. 2014, 145, 993–999. [Google Scholar] [CrossRef]
- Blomquist, A.T.; Kwlatek, J. Addition of ketene to cyclic conjugated dienes. J. Am. Chem. Soc. 1951, 73, 2098–2100. [Google Scholar] [CrossRef]
- Menurath, M.; Gauducheau, M. α-(2-Hydroxy-3-oxocycloheptane) propinlactone. Ric. Sci. Rend. Sez. A 1961, 1, 95–96. [Google Scholar]
- Bond, C.W.; Cresswell, A.J.; Davies, S.G.; Fletcher, A.M.; Kurasawa, W.; Lee, J.A.; Roberts, P.M. Ammonium-directed oxidation of cyclic allylic and homoallylic amines. J. Org. Chem. 2009, 74, 6735–6748. [Google Scholar] [CrossRef] [PubMed]
- Gaux, C.; Lhoste, P.; Simon, D.; Masden, A. Palladium(II)-catalyzed phenoxycarbonylation of allylic carbonates. J. Organomet. Chem. 1996, 511, 139–143. [Google Scholar] [CrossRef]
- Nautiyal, P.; Rastogi, S.N. Prepared by the reaction of phenol and 1-bromocycloheptene. Synthesis of 2-aryloxy-8-oxabicyclo[5.1.0]octanes. Ind. J. Chem. Sect. B 1979, 17B, 159–161. [Google Scholar]
- Wisser, M.S.; Harrity, J.P.A.; Joseph, P.A.; Hoveyda, A.H. Zirconium-catalyzed kinetic resolution of cyclic allylic ethers. JACS 1996, 118, 3779–3780. [Google Scholar]
- Cornforth, J.W.; Hughes, G.K.; Lions, F. Prepared by the reaction of phenol and 1,2-dibromocyclohexene. The pyrrolysis of phenyl cyclohexenyl ether. J. Proc. Soc. NSW 1938, 71, 323–329. [Google Scholar]
- Leo, E.A.; Delgado, J.; Domingo, L.R.; Espinos, A.M.; Miranda, M.A.; Tormos, R. Photogeneration of o-quinone methides from o-cycloalkenylphenols. J. Org. Chem. 2003, 68, 9643–9647. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yingiong, C.; Shaupeng, L.; Xin, C.; Han, J.; Jia, H.; Xiang, W.; Decal, Z.; Zhou, G.C. Synthesis and inhibitory evaluation of cyclohexen-2-yl and cyclohexyl-substituted phenols and quinones to endothelial cell and cancer cells. Eur. J. Med. Chem. 2010, 45, 2147–2153. [Google Scholar]
- Nashimura, T.; Ohtaka, S.; Kimura, A.; Hayama, H.; Haseba, Y.; Yasuhiro, H.; Uemura, S. Prepared by the alkylation of 1-naphthol with cyclohexanone using cation-exchanged montmorillonite. Metal cation-exchanged montmorillonite (Mn + mont)-catalyzed reductive alkylation of phenol and 1-naphthol with cyclohexanones. Appl. Catal. A Gen. 2000, 194–195, 415–425. [Google Scholar]
- Kurono, N.; Honda, E.; Komatsu, F.; Orito, K.; Tokuda, M. Regioselective synthesis of substituted 1-indalol, 2,3-dihydrobenzofurans and 2,3-dihydroindoles by electrochemical radical cyclization using an arene mediator. Tetrahedron 2004, 60, 1791–1801. [Google Scholar] [CrossRef]
- Bachelot, J.P.; Caubere, P. Prepared by the condensation of cyclic ketone enolates with benzofuranes. Aryne and SNAr reactions of polyhalobenzenes. 6. Synthesis of benzofurans. J. Org. Chem. 1982, 47, 234–238. [Google Scholar] [CrossRef]
- Laan, J.A.M.; Giesen, F.L.L.; Ward, J.P. Cycloalkylation of PhOH with cyclic dienes in the presence of aluminum phenoxide. Chem. Ind. 1989, 354–355. [Google Scholar]
- Sample Availability: Samples of the compounds 3–8 are available from the authors.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Materials | Temperature (°C) | Reaction Time (h) | Product(s) (Yields %) |
|---|---|---|---|---|
| 1 | 3a | 20 | 24 | 4a (28); 6a (22); 5a (12) |
| 2 | 3b | 50 | 12 | 4b (26); 6b (20); 5b (14) |
| 3 | 3c | 50 | 22 | 4c (47) |
| 4 | 3d | 20 | 24 | 4d (37); 6d (14); 5a (14) |
| 5 | 3e | 20 | 40 | 4e (22); 6e (15); 5b (16) |
| 6 | 3f | 50 | 8 | 4f (47) |
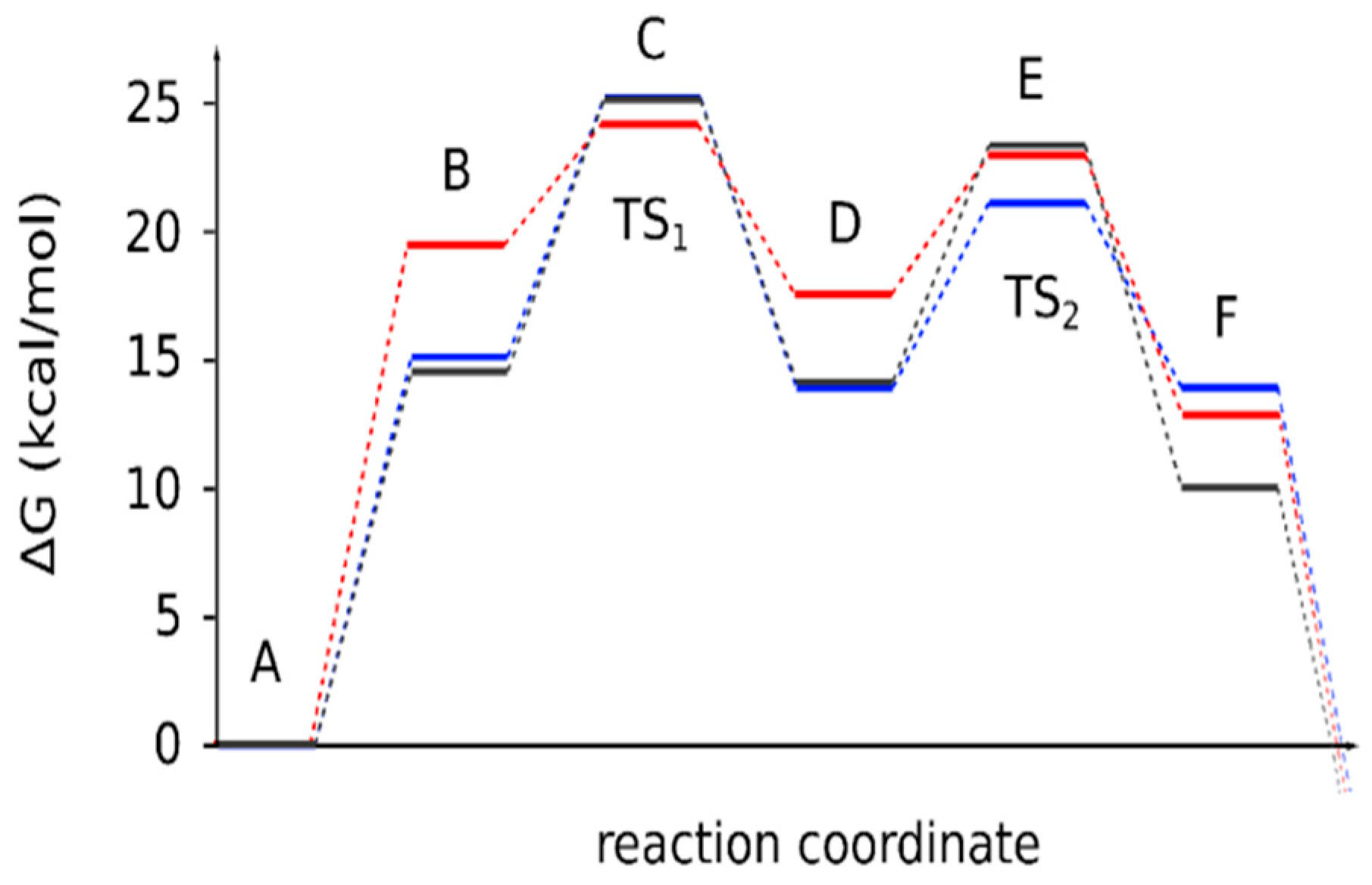
| Reaction Stages | 4a | 4b | 4c |
|---|---|---|---|
| A | 0 | 0 | 0 |
| B | 14.5 | 15.1 | 19.4 |
| C (TS1) | 25.1 | 25.2 | 24.1 |
| D | 14.1 | 13.9 | 17.5 |
| E (TS2) | 23.2 | 21.1 | 22.9 |
| F | 10.0 | 13.9 | 12.8 |
| ΔGr | −0.9 | −1.5 | −5.6 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Törincsi, M.; Nagy, M.; Bihari, T.; Stirling, A.; Kolonits, P.; Novak, L. Rearrangements of Cycloalkenyl Aryl Ethers. Molecules 2016, 21, 503. https://doi.org/10.3390/molecules21040503
Törincsi M, Nagy M, Bihari T, Stirling A, Kolonits P, Novak L. Rearrangements of Cycloalkenyl Aryl Ethers. Molecules. 2016; 21(4):503. https://doi.org/10.3390/molecules21040503
Chicago/Turabian StyleTörincsi, Mercedesz, Melinda Nagy, Tamás Bihari, András Stirling, Pál Kolonits, and Lajos Novak. 2016. "Rearrangements of Cycloalkenyl Aryl Ethers" Molecules 21, no. 4: 503. https://doi.org/10.3390/molecules21040503