
Aza-Henry Reactions on C-Alkyl Substituted Aldimines



Abstract
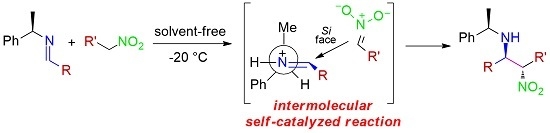
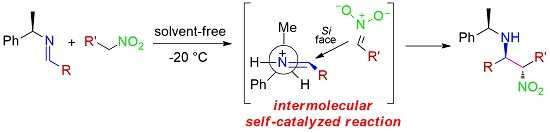
:
1. Introduction
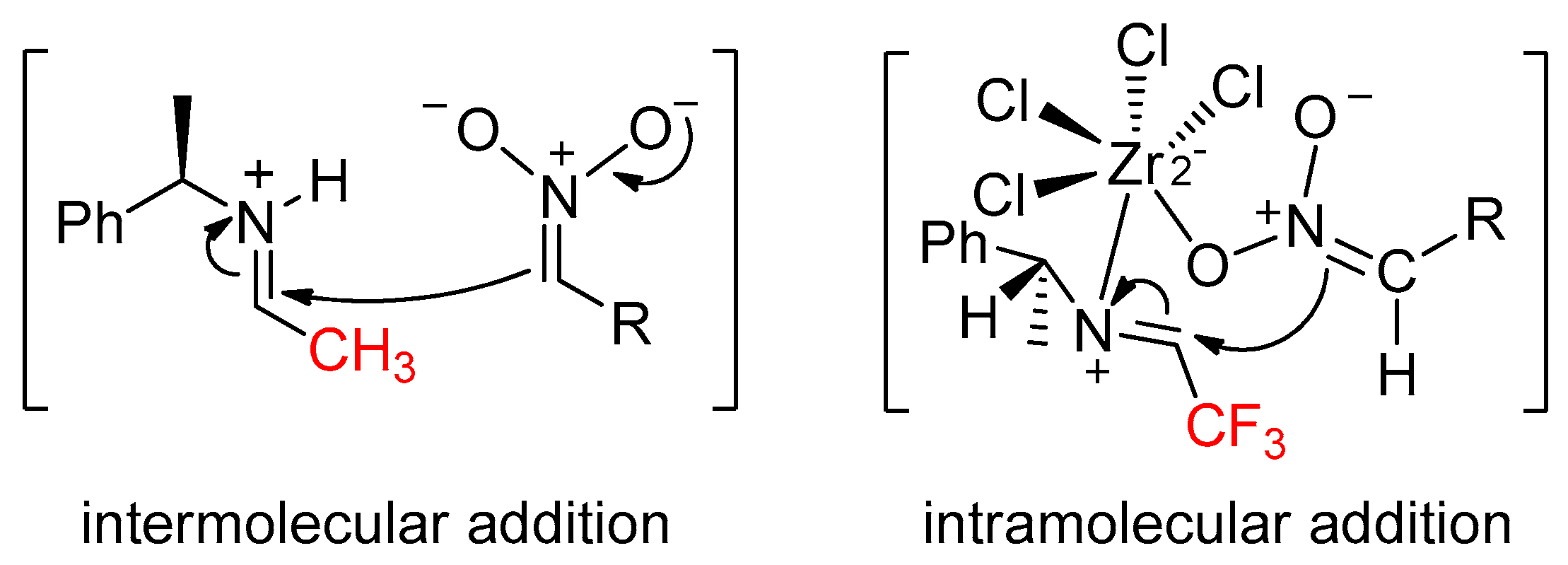
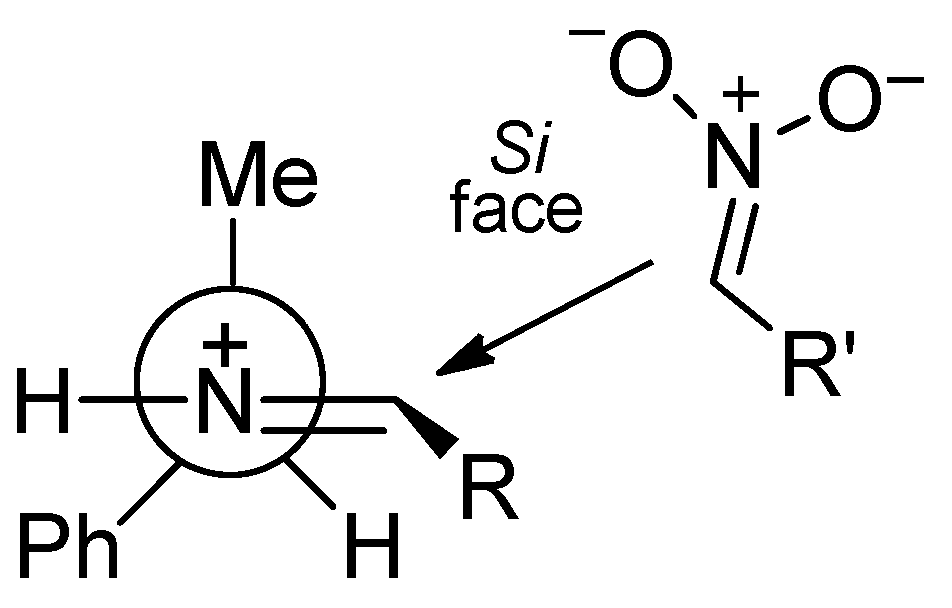
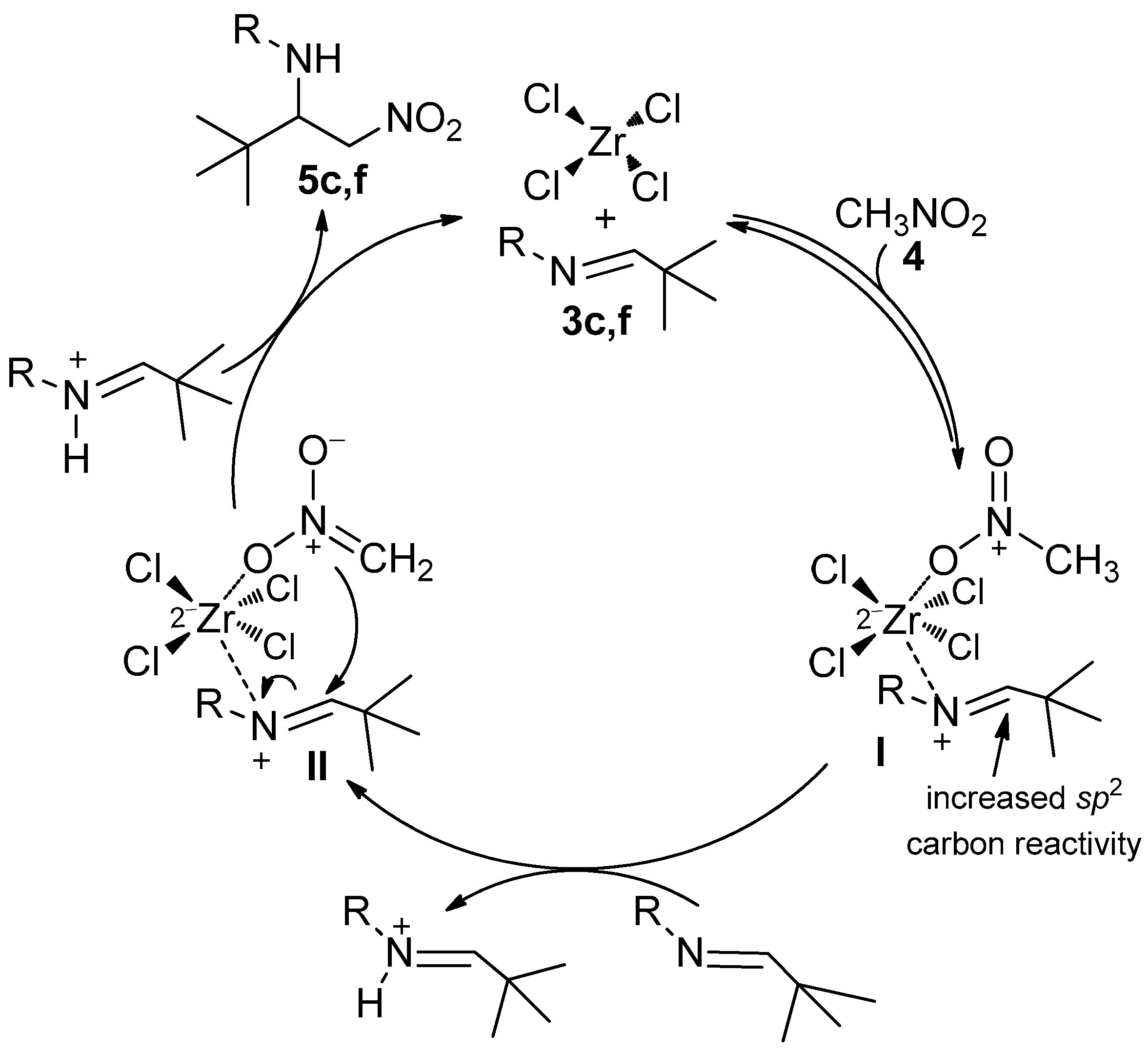
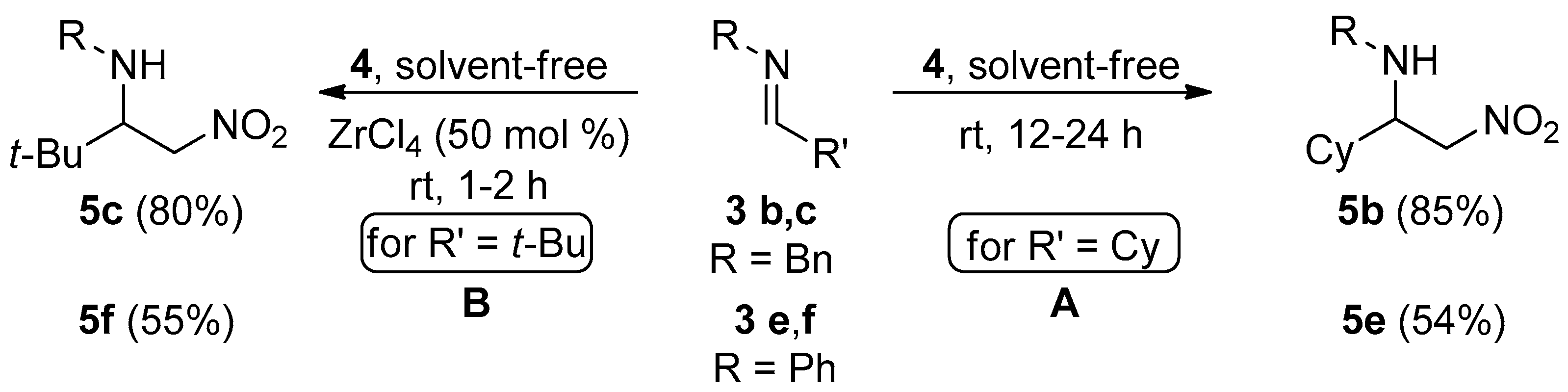
2. Results and Discussion
3. Experimental Section
3.1. General Procedure for the Synthesis of C-alkyl Imines (3a–i)
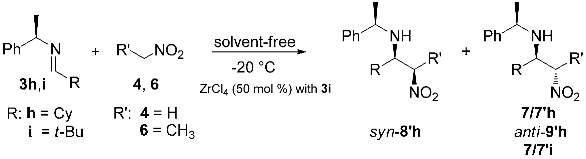
3.2. General Procedure for the Synthesis of C-alkyl β-nitro Amines
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Juhl, K.; Gathergood, N.; Jørgensen, K.A. Catalytic asymmetric direct Mannich reactions of carbonyl compounds with α-imino esters. Angew. Chem. Int. Ed. 2001, 40, 2995–2997. [Google Scholar] [CrossRef]
- Westermann, B. Asymmetric catalytic aza-Henry reactions leading to 1,2-diamines and 1,2-diaminocarboxylic acids. Angew. Chem. Int. Ed. 2003, 42, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Marques-Lopez, E.; Merino, P.; Tejero, T.; Herrera, R.P. Catalytic enantioselective aza-Henry reactions. Eur. J. Org. Chem. 2009, 2401–2420. [Google Scholar] [CrossRef]
- Nitabaru, T.; Kumagai, N.; Shibasaki, M. Catalytic Asymmetric Nitro-Mannich Reactions with a Yb/K Heterobimetallic Catalyst. Molecules 2010, 15, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Lucet, D.; le Gall, T.; Mioskowski, C. The Chemistry of Vicinal Diamines. Angew. Chem. Int. Ed. 1998, 37, 2580–2627. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. Recent synthetic developments in the nitro to carbonyl conversion (Nef reaction). Tetrahedron 2004, 60, 1017–1047. [Google Scholar] [CrossRef]
- García Ruano, J.L.; López-Cantarero, J.; de Haro, T.; Alemán, J.; Cid, M.B. Preparation of α-amino ketones, β-amino hydroxylamines using asymmetric aza-Henry reactions of N-p-tolylsulfinylimines with nitroethane. Tetrahedron 2006, 62, 12197–12203. [Google Scholar] [CrossRef]
- Czekelius, C.; Carreira, E.M. Convenient Transformation of Optically Active Nitroalkanes into Chiral Aldoximes and Nitriles. Angew. Chem. Int. Ed. 2005, 44, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, S.; Pellacani, L.; Vergari, M.C. Fluorinated β-nitro amines by a selective ZrCl4-catalyzed aza-Henry reaction of (E)-trifluoromethyl aldimines. Org. Biomol. Chem. 2012, 10, 8207–8210. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, S.; Pelagalli, A.; Pellacani, L.; Sciubba, F.; Vergari, M.C. Trifluoromethyl-modified dipeptides by ZrCl4-promoted aza-Henry reactions. Amino Acids 2014, 46, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Parise, L.; Pelagalli, A.; Pellacani, L.; Sciubba, F.; Vergari, M.C.; Fioravanti, S. Ethyl Nitroacetate in Aza-Henry Addition on Trifluoromethyl Aldimines: A Solvent-Free Procedure To Obtain Chiral Trifluoromethyl α,β-Diamino Esters. J. Org. Chem. 2016, 81, 2864–2874. [Google Scholar] [CrossRef] [PubMed]
- Baer, H.H.; Urbas, L. The Chemistry of the Nitro and Nitroso Groups, Part. 2; Patai, S., Ed.; Interscience: New York, NY, USA, 1970; p. 117. [Google Scholar]
- Zhang, X-J.; Lai, T-B.; Kong, R.Y-C. Biology of Fluoro-Organic Compounds. Top. Curr. Chem. 2012, 308, 365–404. [Google Scholar]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.; Petrozza, A.; Cavallo, G.; Lanzani, G.; Matteucci, F.; Bruce, D.W.; Houbenov, N.; Metrangolo, P.; Resnati, G. Anisotropic ionic conductivity in fluorinated ionic liquid crystals suitable for optoelectronic applications. J. Mater. Chem. A 2013, 1, 6572–6578. [Google Scholar] [CrossRef]
- Hird, M. Fluorinated liquid crystals—Properties and applications. Chem. Soc. Rev. 2007, 36, 2070–2095. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Verma, R.; Shreeve, J.M. Review of ionic liquids with fluorine-containing anions. J. Fluor. Chem. 2006, 127, 159–176. [Google Scholar] [CrossRef]
- Zimmer, L.E.; Sparr, C.; Gilmour, R. Fluorine Conformational Effects in Organocatalysis: An Emerging Strategy for Molecular Design. Angew. Chem. Int. Ed. 2011, 50, 11860–11871. [Google Scholar] [CrossRef] [PubMed]
- Hunter, L. Organo-fluorine chemistry II. Beilstein. J. Org. Chem. 2010, 6, No. 38. [Google Scholar] [CrossRef]
- Resnati, G.; Soloshonok, V.A. Fluoroorganic Chemistry: Synthetic challenges and biomedical rewards, Tetrahedron Symposia-in-Print n. 58. Tetrahedron 1996, 52, 1–330. [Google Scholar]
- Carroccia, L.; Fioravanti, S.; Pellacani, L.; Tardella, P.A. Solvent-Free Stereoselective Synthesis of (E)-Trifluoromethyl Imines and Hydrazones. Synthesis 2010, 4096–4100. [Google Scholar]
- Fioravanti, S.; Olivieri, L.; Pellacani, L.; Tardella, P.A. In the aziridination reactions with ethyl nosyloxycarbamate unfluorinated imines were able to deprotonate in situ the aminating agent, acting both as base and substrate: A Novel Approach to Chiral Spirodiaziridines. Tetrahedron Lett. 1998, 39, 6391–6392. [Google Scholar] [CrossRef]
- Carroccia, L.; Fioravanti, S.; Pellacani, L.; Sadun, C.; Tardella, P.A. Synthesis of optically active trifluoromethyl substituted diaziridines and oxaziridines. Tetrahedron 2011, 67, 5375–5381. [Google Scholar] [CrossRef]
- O′Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Champagne, P.A.; Desroches, J.; Paquin, J.-F. Organic Fluorine as a Hydrogen-Bond Acceptor: Recent Examples and Applications. Synthesis 2015, 47, 306–322. [Google Scholar]
- Claridge, T.D.W. High-Resolution NMR Techniques in Organic Chemistry, 2nd ed.; Elsevier Science: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Davies, A.G.; Kennedy, J.D. Organometallic reactions. Part XIX. Some reactions of aldehydes with aminotin compounds. J. Chem. Soc. C 1971, 68–73. [Google Scholar] [CrossRef]
- Hegedus, L.S.; Imwinkelried, R.; Sargent, M.A.; Dvorak, D.; Satoh, Y. Synthesis of optically active β-lactams by the photolytic reaction of imines with optically active chromium carbene complexes. J. Am. Chem. Soc. 1990, 112, 1109–1117. [Google Scholar] [CrossRef]
- Yamasaki, S.; Fujii, K.; Wada, R.; Kanai, M.; Shibasaki, M. A General Catalytic Allylation Using Allyltrimethoxysilane. J. Am. Chem. Soc. 2002, 124, 6536–6537. [Google Scholar] [CrossRef] [PubMed]
- Cossío, F.P.; Alonso, C.; Lecea, B.; Ayerbe, M.; Rubiales, G.; Palacios, F. Mechanism and Stereoselectivity of the Aza-Wittig Reaction between Phosphazenes and Aldehydes. J. Org. Chem. 2006, 71, 2839–2847. [Google Scholar] [CrossRef] [PubMed]
- Makioka, Y.; Shindo, T.; Taniguchi, Y.; Takaki, K.; Fujiwara, Y. Ytterbium(III) Triflate Catalyzed Synthesis of Quinoline Derivatives from N-Arylaldimines and Vinyl Ethers. Synthesis 1995, 801–804. [Google Scholar] [CrossRef]
- Abrams, W.R.; Kallen, R.G. Equilibriums and kinetics of N-hydroxymethylamine formation from aromatic exocyclic amines and formaldehyde. Effects of nucleophilicity and catalyst strength upon mechanisms of catalysis of carbinolamine formation. J. Am. Chem. Soc. 1976, 98, 7777–7789. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Harada, K. Asymmetric syntheses of amino acids by addition of cyanide to the Schiff bases in the presence of cyanide-modified hemin-copolymer. Tetrahedron Lett. 1989, 30, 4535–4538. [Google Scholar] [CrossRef]
- Alvaro, G.; Pacioni, P.; Savoia, D. Addition of Organozincate Reagents to Imines Derived from (S)-1-Phenylethylamine and Ethyl (S)-Valinate—Synthesis of (S)-1-(2-Pyridyl)Alkylamines. Chem. Eur. J. 1997, 3, 726–731. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds not available from the authors.








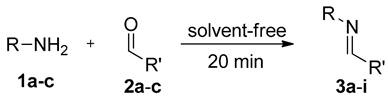
| Entry | 1 | R | 2 | R′ | Product 3 | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | a | Bn | a | Me | a | 85 |
| 2 | b | Cy | b | 90 | ||
| 3 | b | t-Bu | c | 95 | ||
| 4 | b | Ph | a | Me | d | 78 |
| 5 | b | Cy | e | 80 | ||
| 6 | c | t-Bu | f | 83 | ||
| 7 | c |  | a | Me | g | 80 |
| 8 | b | Cy | h | 88 | ||
| 9 | c | t-Bu | i | 95 |

| Entry | R | R′ | Product | Time (h) | Yield (%) b | syn/anti a | Dr a |
|---|---|---|---|---|---|---|---|
| 1 | CH3 | H | 7/7′g | 1 | 90 | − | 3:7 |
| 2 | CF3 c | 7/7′j | 3 | 80 | − | 8:2 | |
| 3 | CH3 | CH3 | syn-8/8′g; anti-9/9′g | 8 | 84 | 1:1 | 2:8 |
| 4 | CF3 c | syn-8/8′j; anti-9/9′j | 18 | 70 | 3:7 | 7.2:2.8 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
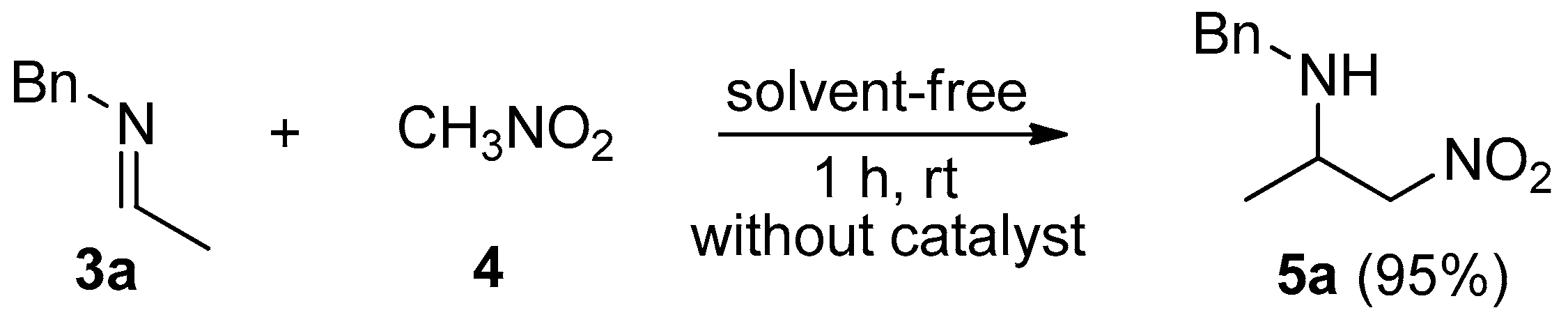{kind=link}
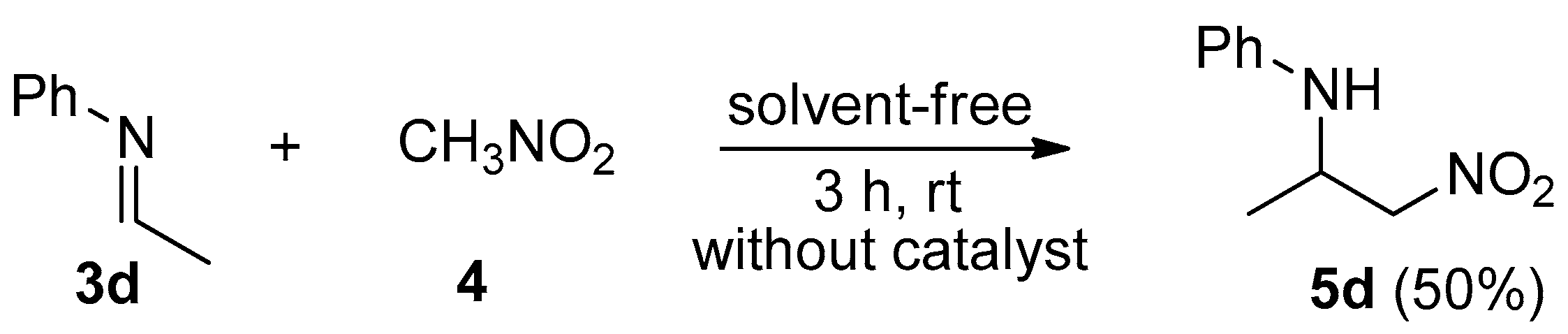{kind=link}
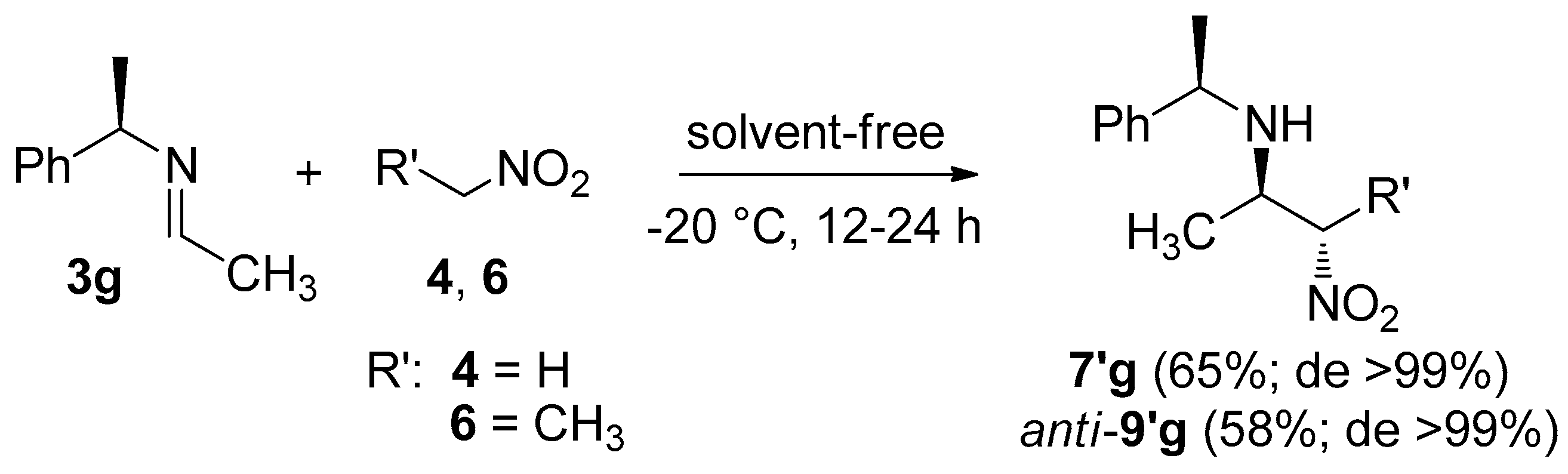{kind=link}
{kind=link}
| Entry | R | R' | Product | Time (h) | Yield (%) b | syn/anti a | Dr a |
|---|---|---|---|---|---|---|---|
| 1 | Cy | H | 7/7′h | 24 | 45 | − | 99:1 |
| 2 | CH3 | syn-8/8′h; anti-9/9′h | 48 | 56 | 1:1 | 99:1 | |
| 3 | t-Bu c | H | 7/7′i | 8 | 55 | − | 99:1 |
| 4 | CH3 | − | − | − | − | − |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelagalli, A.; Pellacani, L.; Scandozza, E.; Fioravanti, S. Aza-Henry Reactions on C-Alkyl Substituted Aldimines. Molecules 2016, 21, 723. https://doi.org/10.3390/molecules21060723
Pelagalli A, Pellacani L, Scandozza E, Fioravanti S. Aza-Henry Reactions on C-Alkyl Substituted Aldimines. Molecules. 2016; 21(6):723. https://doi.org/10.3390/molecules21060723
Chicago/Turabian StylePelagalli, Alessia, Lucio Pellacani, Elia Scandozza, and Stefania Fioravanti. 2016. "Aza-Henry Reactions on C-Alkyl Substituted Aldimines" Molecules 21, no. 6: 723. https://doi.org/10.3390/molecules21060723
APA StylePelagalli, A., Pellacani, L., Scandozza, E., & Fioravanti, S. (2016). Aza-Henry Reactions on C-Alkyl Substituted Aldimines. Molecules, 21(6), 723. https://doi.org/10.3390/molecules21060723