
Applications of 19F-NMR in Fragment-Based Drug Discovery


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Screening Fragment Libraries
3. Characterizing Ligand Binding by 19F-NMR
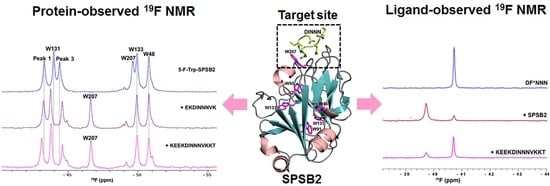
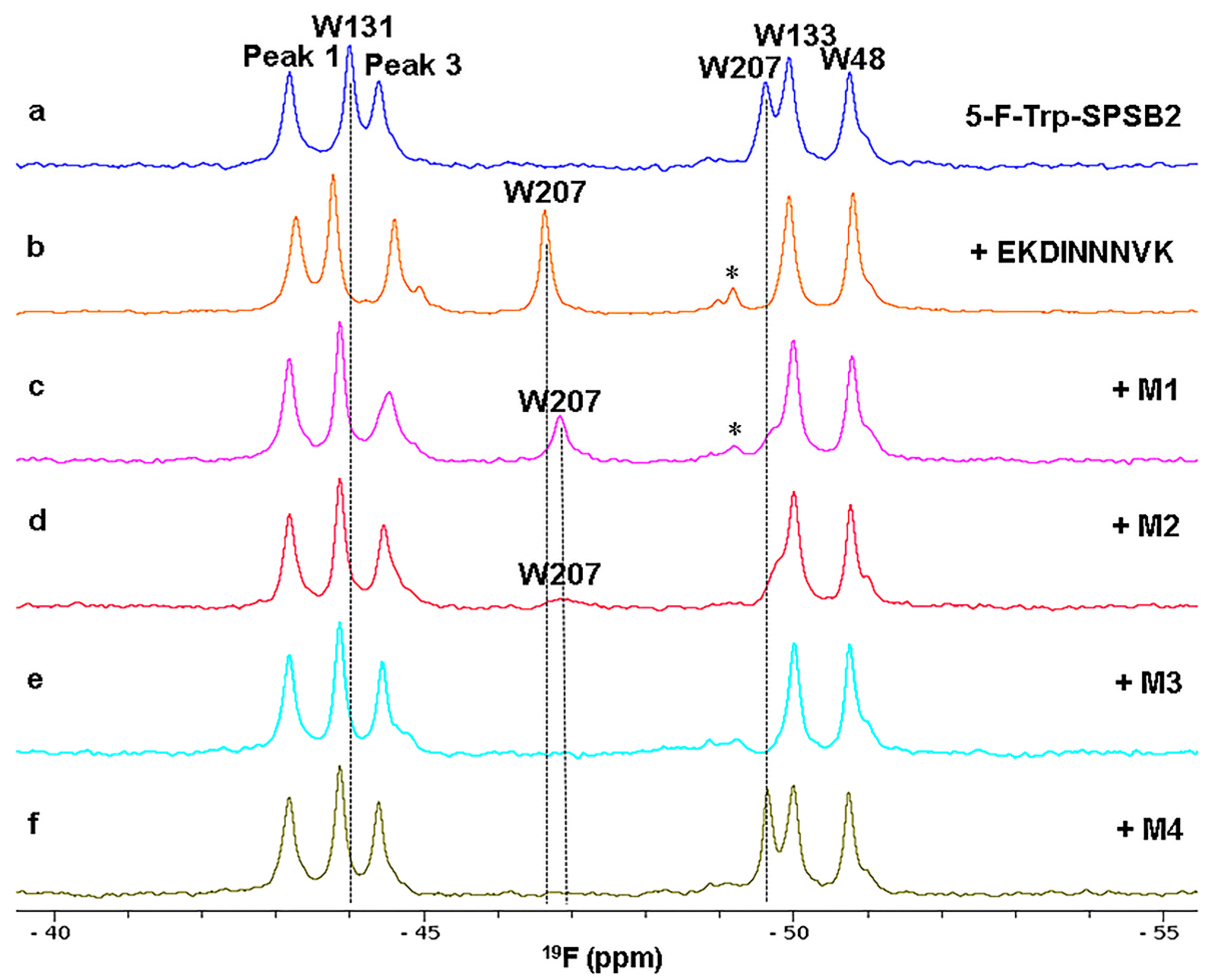
3.1. Ligand Binding to the SPRY Domain-Containing SOCS Box Protein 2 (SPSB2)
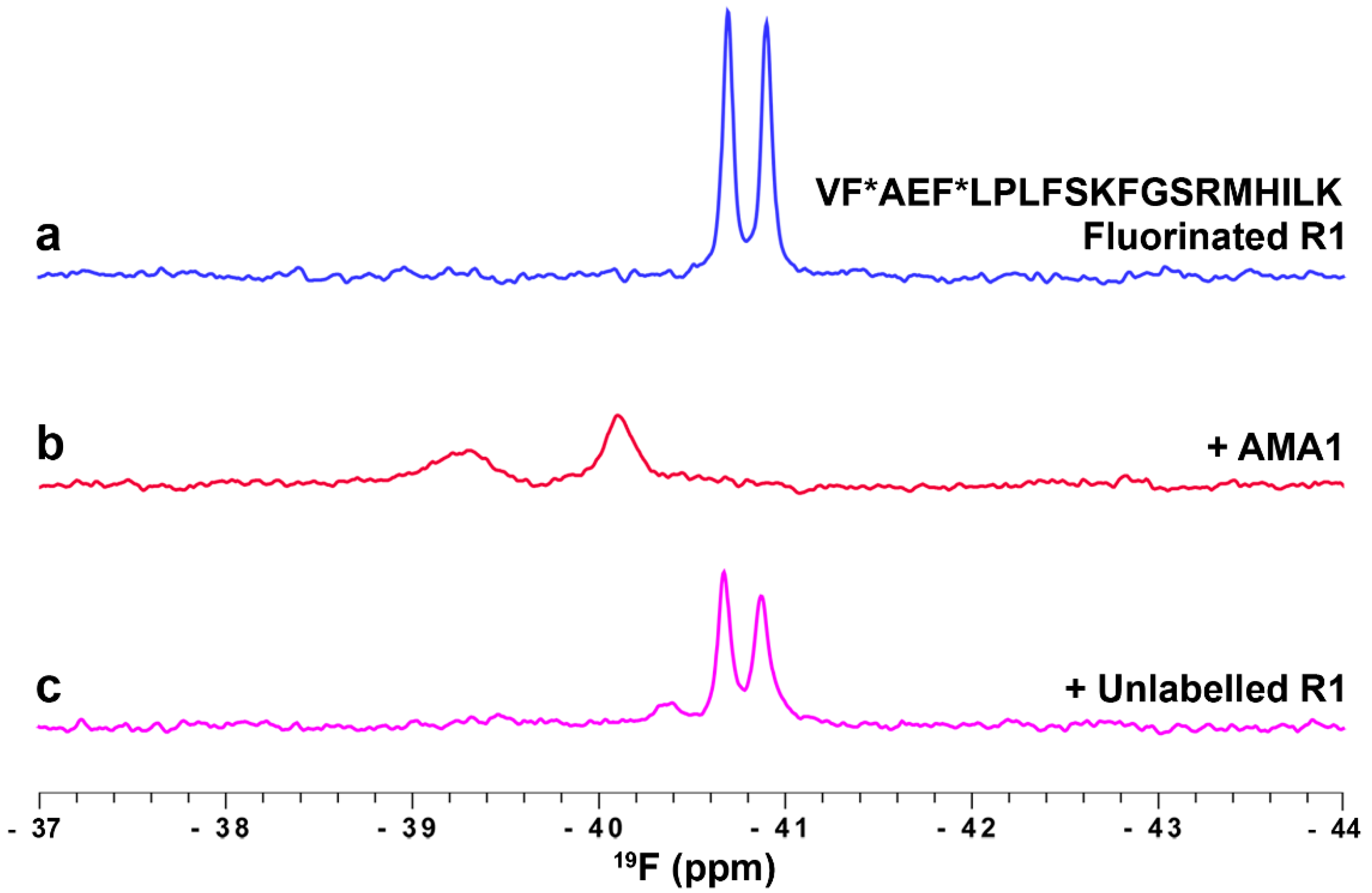
3.2. Ligand Binding to Apical Membrane Antigen 1 (AMA1)
3.3. Competitive Binding
3.3.1. Fluorinated Peptide Probe Targeting the iNOS Binding Site on SPSB2
3.3.2. Profiling the AMA1 Binding Site Using Fluorinated R1 Peptide
3.4. Ligand Binding Site Mapping
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Osborne, R. Fresh from the biotech pipeline—2012. Nat. Biotechnol. 2013, 31, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Cooper, M.A. Screening strategies to identify new antibiotics. Curr. Drug Targets 2012, 13, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Leeson, P.D.; St-Gallay, S.A. The influence of the ’organizational factor’ on compound quality in drug discovery. Nat. Rev. Drug Discov. 2011, 10, 749–765. [Google Scholar] [CrossRef] [PubMed]
- Practical Fragments. Available online: http://practicalfragments.blogspot.com.au/2015/01/fragments-in-clinic-2015-edition.html (accessed on 19 May 2016).
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Petros, A.M.; Dinges, J.; Augeri, D.J.; Baumeister, S.A.; Betebenner, D.A.; Bures, M.G.; Elmore, S.W.; Hajduk, P.J.; Joseph, M.K.; Landis, S.K.; et al. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J. Med. Chem. 2006, 49, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Verdonk, M.L.; Rees, D.C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Van Molle, I.; Thomann, A.; Buckley, D.L.; So, E.C.; Lang, S.; Crews, C.M.; Ciulli, A. Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1alpha protein-protein interface. Chem. Biol. 2012, 19, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.; Morton, C.J.; Simpson, J.S.; Scanlon, M.J. Design and evaluation of the performance of an NMR screening fragment library. Aust. J. Chem. 2013, 66, 1465–1472. [Google Scholar] [CrossRef]
- Scanlon, M.J.; Norton, R.S. Fragment-based drug discovery, an accessible approach to new therapeutics. Aust. Biochem. 2013, 44, 9–12. [Google Scholar]
- Patel, D.; Bauman, J.D.; Arnold, E. Advantages of crystallographic fragment screening: functional and mechanistic insights from a powerful platform for efficient drug discovery. Prog. Biophys. Mol. Biol. 2014, 116, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Oster, L.; Tapani, S.; Xue, Y.; Kack, H. Successful generation of structural information for fragment-based drug discovery. Drug Discov. Today 2015, 20, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fagerness, P.E.; Hadden, D.T.; Sarver, R.W.; Stockman, B.J. Fluorine-NMR experiments for high-throughput screening: Theoretical aspects, practical considerations, and range of applicability. J. Am. Chem. Soc. 2003, 125, 7696–7703. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Hommel, U.; Landrum, G.; Lewis, R.; Dalvit, C. Design and NMR-based screening of LEF, a library of chemical fragments with different local environment of fluorine. J. Am. Chem. Soc. 2009, 131, 12949–12959. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Dalvit, C. Fluorine local environment: From screening to drug design. Drug Discov. Today 2012, 17, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Lambruschini, C.; Veronesi, M.; Romeo, E.; Garau, G.; Bandiera, T.; Piomelli, D.; Scarpelli, R.; Dalvit, C. Development of fragment-based n-FABS NMR screening applied to the membrane enzyme FAAH. ChemBioChem 2013, 14, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.B.; Poppe, L.; Xia, X.; Cheng, A.C.; Sun, Y.; Michelsen, K.; Eastwood, H.; Schnier, P.D.; Nixey, T.; Zhong, W. Fragment based drug discovery: Practical implementation based on 19F-NMR spectroscopy. J. Med. Chem. 2012, 55, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.B.; Whittington, D.A.; Bartberger, M.D.; Sickmier, E.A.; Chen, K.; Cheng, Y.; Judd, T. Fragment linking approach using 19F-NMR spectroscopy to obtain highly potent and selective inhibitors of β-secretase. J. Med. Chem. 2016, 59, 3732–3749. [Google Scholar] [CrossRef] [PubMed]
- Gee, C.T.; Koleski, E.J.; Pomerantz, W.C. Fragment screening and druggability assessment for the CBP/p300 KIX domain through protein-observed 19F-NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2015, 54, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- Kitevski-LeBlanc, J.L.; Prosser, R.S. Current applications of 19F-NMR to studies of protein structure and dynamics. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 62, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Danielson, M.A.; Falke, J.J. Use of 19F-NMR to probe protein structure and conformational changes. Annu. Rev. Biophys. Biomol. Struct. 1996, 25, 163–195. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Lewis, R.S.; Curtis, J.M.; Zhan, Y.; Saunders, B.M.; Babon, J.J.; Kolesnik, T.B.; Low, A.; Masters, S.L.; Willson, T.A.; et al. The SPRY domain-containing SOCS box protein SPSB2 targets iNOS for proteasomal degradation. J. Cell Biol. 2010, 190, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S.; Kolesnik, T.B.; Kuang, Z.; D’Cruz, A.A.; Blewitt, M.E.; Masters, S.L.; Low, A.; Willson, T.; Norton, R.S.; Nicholson, S.E. TLR regulation of SPSB1 controls inducible nitric oxide synthase induction. J. Immunol. 2011, 187, 3798–3805. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Low, A.; Sharpe, T.D.; Uppenberg, J.; Yao, S.; Kuang, Z.; Savitsky, P.; Lewis, R.S.; Nicholson, S.E.; Norton, R.S.; et al. Structural basis for Par-4 recognition by the SPRY domain- and SOCS box-containing proteins SPSB1, SPSB2, and SPSB4. J. Mol. Biol. 2010, 401, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Yap, B.K.; Leung, E.W.; Yagi, H.; Galea, C.A.; Chhabra, S.; Chalmers, D.K.; Nicholson, S.E.; Thompson, P.E.; Norton, R.S. A potent cyclic peptide targeting SPSB2 protein as a potential anti-infective agent. J. Med. Chem. 2014, 57, 7006–7015. [Google Scholar] [CrossRef] [PubMed]
- Yap, B.K.; Harjani, J.R.; Leung, E.W.; Nicholson, S.E.; Scanlon, M.J.; Chalmers, D.K.; Thompson, P.E.; Baell, J.B.; Norton, R.S. Redox-stable cyclic peptide inhibitors of the SPSB2-iNOS interaction. FEBS Lett. 2016, 590, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.W.W.; Yagi, H.; Harjani, J.R.; Mulcair, M.D.; Scanlon, M.J.; Baell, J.B.; Norton, R.S. 19F-NMR as a probe of ligand interactions with the iNOS binding site of SPRY domain-containing SOCS box protein 2. Chem. Biol. Drug Des. 2014, 84, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Harjani, J.R.; Yap, B.K.; Leung, E.W.; Lucke, A.; Nicholson, S.E.; Scanlon, M.J.; Chalmers, D.K.; Thompson, P.E.; Norton, R.S.; et al. Design, synthesis, and characterization of cyclic peptidomimetics of the inducible nitric oxide synthase binding epitope that disrupt the protein-protein interaction involving SPRY domain-containing suppressor of cytokine signaling box protein (SPSB) 2 and inducible nitric oxide synthase. J. Med. Chem. 2016, in press. [Google Scholar]
- Rule, G.S.; Pratt, E.A.; Simplaceanu, V.; Ho, C. Nuclear magnetic resonance and molecular genetic studies of the membrane-bound D-lactate dehydrogenase of Escherichia coli. Biochemistry 1987, 26, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Pratt, E.A.; Rule, G.S. Membrane-bound D-lactate dehydrogenase of Escherichia coli: A model for protein interactions in membranes. Biochim. Biophys. Acta 1989, 988, 173–184. [Google Scholar] [CrossRef]
- Anderluh, G.; Razpotnik, A.; Podlesek, Z.; Macek, P.; Separovic, F.; Norton, R.S. Interaction of the eukaryotic pore-forming cytolysin equinatoxin II with model membranes: 19F-NMR studies. J. Mol. Biol. 2005, 347, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Crowley, P.B.; Kyne, C.; Monteith, W.B. Simple and inexpensive incorporation of 19F-tryptophan for protein NMR spectroscopy. Chem. Commun. (Camb.) 2012, 48, 10681–10683. [Google Scholar] [CrossRef] [PubMed]
- Arntson, K.E.; Pomerantz, W.C. Protein-observed fluorine NMR: A bioorthogonal approach for small molecule discovery. J. Med. Chem. 2015, 58, 5158–5171. [Google Scholar] [CrossRef] [PubMed]
- Anders, R.F.; Adda, C.G.; Foley, M.; Norton, R.S. Recombinant protein vaccines against the asexual blood-stages of Plasmodium falciparum. Hum. Vaccin. 2010, 6, 1–15. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Anders, R.F.; Foley, M.; Norton, R.S. Apical membrane antigen 1 as an anti-malarial drug target. Curr. Top. Med. Chem. 2011, 11, 2039–2047. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.; MacRaild, C.A.; Riglar, D.T.; Chan, J.A.; Foley, M.; Baum, J.; Ralph, S.A.; Norton, R.S.; Cowman, A.F. Interaction between Plasmodium falciparum apical membrane antigen 1 and the rhoptry neck protein complex defines a key step in the erythrocyte invasion process of malaria parasites. J. Biol. Chem. 2010, 285, 14815–14822. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.L.; Arastu-Kapur, S.; Dubremetz, J.F.; Boothroyd, J.C. Plasmodium falciparum AMA1 binds a rhoptry neck protein homologous to TgRON4, a component of the moving junction in Toxoplasma gondii. Eukaryot. Cell 2006, 5, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Lamarque, M.; Besteiro, S.; Papoin, J.; Roques, M.; Vulliez-Le Normand, B.; Morlon-Guyot, J.; Dubremetz, J.F.; Fauquenoy, S.; Tomavo, S.; Faber, B.W.; et al. The RON2-AMA1 interaction is a critical step in moving junction-dependent invasion by apicomplexan parasites. PLoS Pathog. 2011, 7, e1001276. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.M.; Parisi, K.; Masciantonio, R.; Hoeck, J.; Casey, J.L.; Murphy, V.J.; Harris, K.S.; Batchelor, A.H.; Anders, R.F.; Foley, M. The most polymorphic residue on Plasmodium falciparum apical membrane antigen 1 determines binding of an invasion-inhibitory antibody. Infect. Immun. 2006, 74, 2628–2636. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.R.; Withers-Martinez, C.; Hackett, F.; Blackman, M.J. An inhibitory antibody blocks interactions between components of the malarial invasion machinery. PLoS Pathog. 2009, 5, e1000273. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Casey, J.L.; Coley, A.M.; Masciantonio, R.; Sabo, J.K.; Keizer, D.W.; Lee, E.F.; McMahon, A.; Norton, R.S.; Anders, R.F.; et al. Binding hot spot for invasion inhibitory molecules on Plasmodium falciparum apical membrane antigen 1. Infect. Immun. 2005, 73, 6981–6989. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, P.; Yasgar, A.; Luci, D.K.; Beatty, W.L.; Hu, X.; Andersen, J.; Narum, D.L.; Moch, J.K.; Sun, H.; Haynes, J.D.; et al. Disrupting malaria parasite AMA1-RON2 interaction with a small molecule prevents erythrocyte invasion. Nat. Commun. 2013, 4, 2261. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Becker, M.; Gupta, A.; Strike, P.; Murphy, V.J.; Anders, R.F.; Batchelor, A.H. Structure of AMA1 from Plasmodium falciparum reveals a clustering of polymorphisms that surround a conserved hydrophobic pocket. Proc. Natl. Acad. Sci. USA 2005, 102, 12736–12741. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.M.; Gupta, A.; Murphy, V.J.; Bai, T.; Kim, H.; Foley, M.; Anders, R.F.; Batchelor, A.H. Structure of the malaria antigen AMA1 in complex with a growth-inhibitory antibody. PLoS Pathog. 2007, 3, 1308–1319. [Google Scholar] [PubMed]
- Lim, S.S.; Yang, W.; Krishnarjuna, B.; Kannan Sivaraman, K.; Chandrashekaran, I.R.; Kass, I.; MacRaild, C.A.; Devine, S.M.; Debono, C.O.; Anders, R.F.; et al. Structure and dynamics of apical membrane antigen 1 from Plasmodium falciparum FVO. Biochemistry 2014, 53, 7310–7320. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, M.L.; Roques, M.; Lamarque, M.H.; Pugniere, M.; Douguet, D.; Crawford, J.; Lebrun, M.; Boulanger, M.J. Host cell invasion by apicomplexan parasites: Insights from the co-structure of AMA1 with a RON2 peptide. Science 2011, 333, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.S.; Debono, C.O.; MacRaild, C.A.; Chandrashekaran, I.R.; Dolezal, O.; Anders, R.F.; Simpson, J.S.; Scanlon, M.J.; Devine, S.M.; Scammells, P.J.; et al. Development of inhibitors of Plasmodium falciparum apical membrane antigen 1 based on fragment screening. Aust. J. Chem. 2013, 66, 1530–1536. [Google Scholar] [CrossRef]
- Ge, X.; MacRaild, C.A.; Devine, S.M.; Debono, C.O.; Wang, G.; Scammells, P.J.; Scanlon, M.J.; Anders, R.F.; Foley, M.; Norton, R.S. Ligand-induced conformational change of Plasmodium falciparum AMA1 detected using 19F NMR. J. Med. Chem. 2014, 57, 6419–6427. [Google Scholar] [CrossRef] [PubMed]
- Krishnarjuna, B.; Lim, S.S.; Devine, S.M.; Debono, C.O.; Lam, R.; Chandrashekaran, I.R.; Jaipuria, G.; Yagi, H.; Atreya, H.S.; Scanlon, M.J.; et al. Solution NMR characterization of apical membrane antigen 1 and small molecule interactions as a basis for designing new antimalarials. J. Mol. Recog. 2016, 29, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Devine, S.M.; Mulcair, M.D.; Debono, C.O.; Leung, E.W.; Nissink, J.W.; Lim, S.S.; Chandrashekaran, I.R.; Vazirani, M.; Mohanty, B.; Simpson, J.S.; et al. Promiscuous 2-aminothiazoles (PrATs): A frequent hitting scaffold. J. Med. Chem. 2015, 58, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C. Theoretical analysis of the competition ligand-based NMR experiments and selected applications to fragment screening and binding constant measurements. Concepts Magn. Reson. A 2008, 32, 341–372. [Google Scholar] [CrossRef]
- Richards, K.L.; Rowe, M.L.; Hudson, P.B.; Williamson, R.A.; Howard, M.J. Combined ligand-observe 19F and protein-observe 15N,1H-HSQC NMR suggests phenylalanine as the key d-somatostatin residue recognized by human protein disulfide isomerase. Sci. Rep. 2016, 6, 19518. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.F.; Yao, S.; Sabo, J.K.; Fairlie, W.D.; Stevenson, R.A.; Harris, K.S.; Anders, R.F.; Foley, M.; Norton, R.S. Peptide inhibitors of the malaria surface protein, apical membrane antigen 1: Identification of key binding residues. Biopolymers 2011, 95, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; MacRaild, C.A.; Mohanty, B.; Mobli, M.; Cowieson, N.P.; Anders, R.F.; Simpson, J.S.; McGowan, S.; Norton, R.S.; Scanlon, M.J. Molecular insights into the interaction between Plasmodium falciparum apical membrane antigen 1 and an invasion-inhibitory peptide. PLoS ONE 2014, 9, e109674. [Google Scholar] [CrossRef] [PubMed]
- Vulliez-Le Normand, B.; Tonkin, M.L.; Lamarque, M.H.; Langer, S.; Hoos, S.; Roques, M.; Saul, F.A.; Faber, B.W.; Bentley, G.A.; Boulanger, M.J.; et al. Structural and functional insights into the malaria parasite moving junction complex. PLoS Pathog. 2012, 8, e1002755. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.A.V.; MacRaild, C.A.; Seow, J.; Krishnarjuna, B.; Drinkwater, N.; Rouet, R.; Anders, R.F.; Christ, D.; McGowan, S.; Norton, R.S. Structural basis for epitope masking and strain specificity of a conserved epitope in an intrinsically disordered malaria vaccine candidate. Sci. Rep. 2015, 5, 10103. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norton, R.S.; Leung, E.W.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of 19F-NMR in Fragment-Based Drug Discovery. Molecules 2016, 21, 860. https://doi.org/10.3390/molecules21070860
Norton RS, Leung EWW, Chandrashekaran IR, MacRaild CA. Applications of 19F-NMR in Fragment-Based Drug Discovery. Molecules. 2016; 21(7):860. https://doi.org/10.3390/molecules21070860
Chicago/Turabian StyleNorton, Raymond S., Eleanor W. W. Leung, Indu R. Chandrashekaran, and Christopher A. MacRaild. 2016. "Applications of 19F-NMR in Fragment-Based Drug Discovery" Molecules 21, no. 7: 860. https://doi.org/10.3390/molecules21070860