
Structures and Biogenesis of Fallaxosides D4, D5, D6 and D7, Trisulfated Non-Holostane Triterpene Glycosides from the Sea Cucumber Cucumaria fallax




Abstract
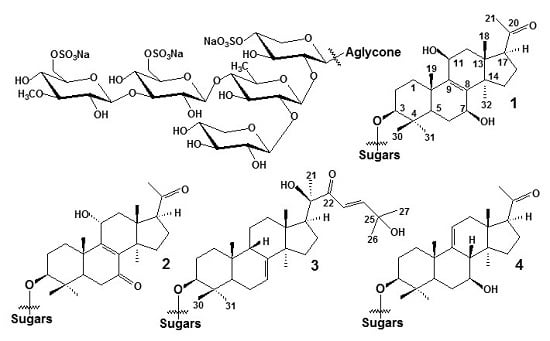
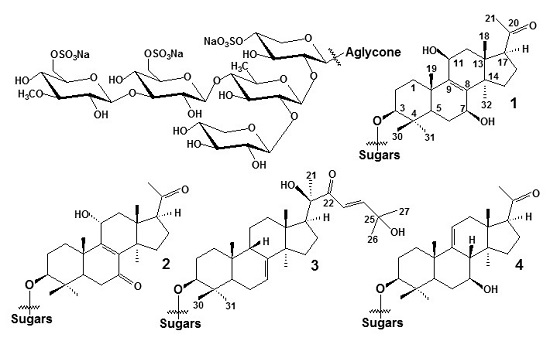
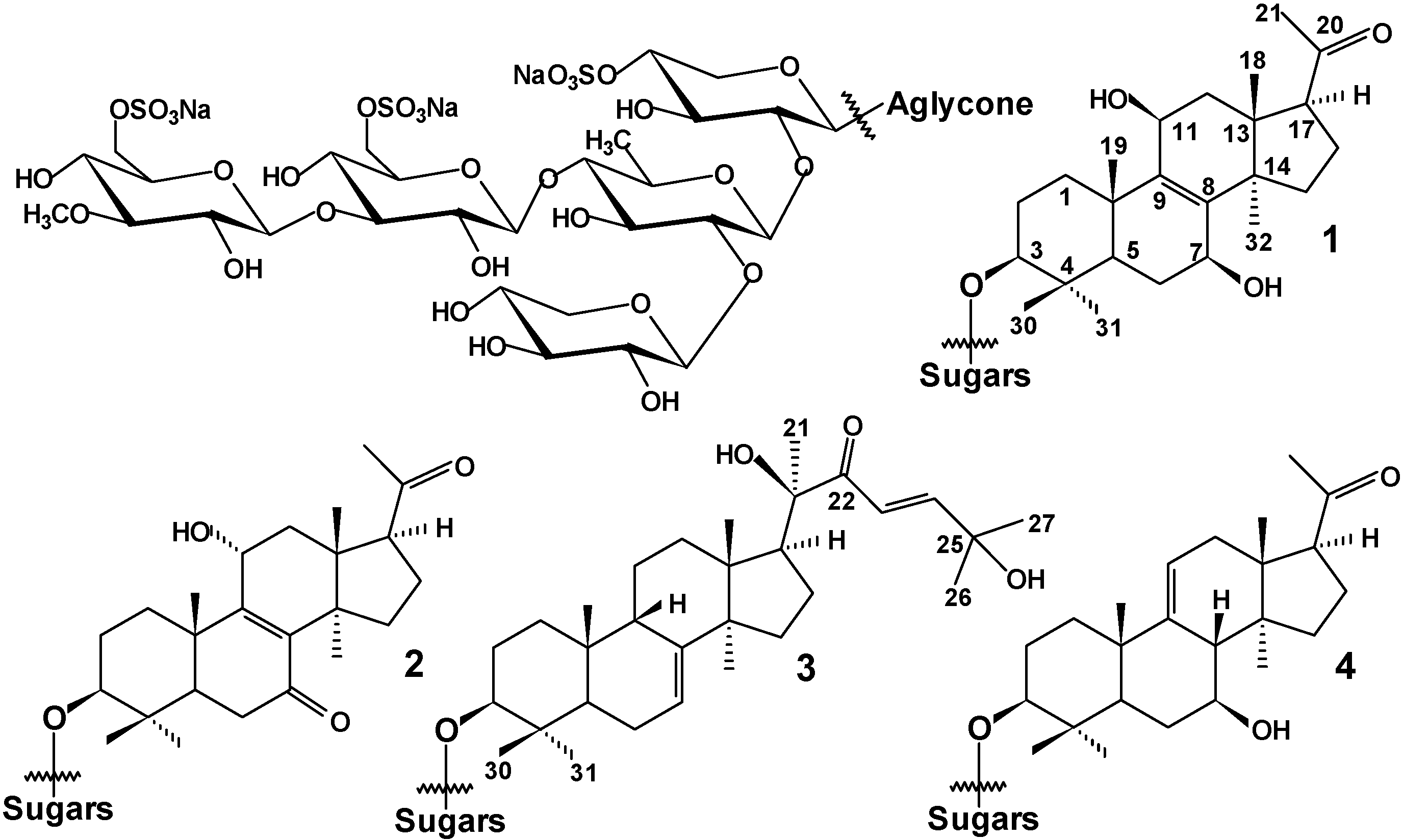
:
1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.4. Cell Culture
3.5. Cytotoxicity Activity
3.6. Hemolytic Activity
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stonik, V.A.; Kalinin, V.I.; Avilov, S.A. Toxins from the sea cucumbers (Holothuroids): Chemical structures, properties, taxonomic distribution, biosynthesis and evolution. J. Nat. Toxins 1999, 8, 235–248. [Google Scholar] [PubMed]
- Kalinin, V.I.; Kalinovskii, A.I.; Stonik, V.A. Onekotanogenin-A new triterpene genin from the holothurian Psolus fabricii. Chem. Nat. Comp. 1987, 23, 560–563. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Stonik, V.A.; Smirnov, A.V. Sea Cucumbers Triterpene Glycosides, the Recent Progress in Structural Elucidation and Chemotaxonomy. Phytochem. Rev. 2005, 4, 221–236. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Avilov, S.A.; Kalinovsky, A.I.; Dmitrenok, P.S.; Kalinin, V.I.; Morre, J.; Deinzer, M.L.; Woodward, C.; Collin, P.D. Glycosides from the North Atlantic sea cucumber Cucumaria frondosa V-Structures of five new minor trisulfated triterpene oligoglycosides, frondosides A7-1, A7-3, A7-4, and isofrondoside C. Can. J. Chem. 2007, 85, 626–636. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Kalinonovsky, A.I.; Avilov, S.A.; Andryjashenko, P.V.; Dmitrenok, P.S.; Kalinin, V.I.; Stonik, V.A. 3β-O-Glycosylated 16β-acetoxy-9β-H-lanosta-7,24-diene-3β,18,20β-triol, an intermediate metabolite from the sea cucumber Eupentacta fraudatrix and its biosynthetic significance. Biochem. Syst. Ecol. 2012, 44, 53–60. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Kalinovsky, A.I.; Avilov, S.A.; Andryjaschenko, P.V.; Dmitrenok, P.S.; Kalinin, V.I.; Martyyas, E.A.; Minin, K.V. Fallaxosides C1, C2, D1 and D2, Unusual Olygosulfated Triterpene Glycosides from the Sea Cucumber Cucumaria fallax (Cucumariidae, Dendrochirotida) and taxonomic status of this animal. Nat. Prod. Commun. 2016, 11, 939–945. [Google Scholar]
- Miyamoto, T.; Togawa, K.; Higuchi, R.; Komori, T.; Sasaki, T. Structures of four new triterpenoid oligoglycosides: Ds-penaustrosides A, B, C and D from the sea cucumber Pentacta australis. J. Nat. Prod. 1992, 55, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Drozdova, O.A.; Avilov, S.A.; Kalinovskii, A.I.; Stonik, V.A.; Mil'grom, Y.M.; Rashkes, Y.V. Trisulfated glycosides from the holothurian Cucumaria japonica. Chem. Nat. Comp. 1993, 29, 309–313. [Google Scholar] [CrossRef]
- Ilyin, S.G.; Reshetnyak, M.V.; Afyatullov, S.S.; Stonik, V.A.; Elyakov, G.B. Crystallic and molecular structure of diacetate of holost-8(9)-en-3α,16β-diol. Proc. USSR Acad. Sci. 1985, 284, 356–359. [Google Scholar]
- Avilov, S.A.; Drozdova, O.A.; Kalinin, V.I.; Kalinovsky, A.I.; Stonik, V.A.; Gudimova, E.N.; Riguera, R.; Jimenez, C. Frondoside C. A new nonholostane triterpene glycoside from the sea cucumber Cucumaria frondosa: Structure and cytotoxicity of its desulfated derivative. Can. J. Chem. 1998, 76, 137–141. [Google Scholar]
- Stonik, V.A. Some terpenoid and steroid derivatives from echinoderms and sponges. Pure Appl. Chem. 1986, 58, 423–436. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Aminin, D.L.; Avilov, S.A.; Silchenko, A.S.; Stonik, V.A. Triterpene glycosides from sea cucucmbers (Holothurioidae, Echinodermata), biological activities and functions. In Studies in Natural Product Chemistry (Bioactive Natural Products); Atta-ur-Rahman, Ed.; Elsevier Science Publisher: Amsterdam, The Netherland, 2008; Volume 35, pp. 135–196. [Google Scholar]
- Makarieva, T.N.; Stonik, V.A.; Kapustina, I.I.; Boguslavsky, V.M.; Dmitrenok, P.S.; Kalinin, V.I.; Cordeiro, M.L.; Djerassi, C. Biogenetic studies on marine lipids. 42. Biosynthetic studies of steroid and triterpenoid metabolites in the sea cucumber Eupentacta fraudatrix. Steroids 1993, 58, 508–517. [Google Scholar] [CrossRef]
- Prokofieva, N.G.; Likhatskaya, G.N.; Volkova, O.V.; Anisimov, M.M.; Kiseleva, M.I.; Ilyin, S.G.; Budina, T.A.; Pokhilo, N.D. The action of betulafolientetraol on the erythrocytes and model membranes. Biol. Membr. 1992, 9, 954–960. [Google Scholar]
- Taniyama, S.; Arakawa, O.; Terada, M.; Nishio, S.; Takatani, T.; Mahmud, Y.; Noguchi, T. Ostreopsis sp., a possible origin of palytoxin (PTX) in parrotfish Scarus ovifrons. Toxicon 2003, 42, 29–33. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.


{kind=link}
{kind=link}
| Atom | δC mult. a,b,c | δH mult. (J in Hz) d | HMBC | ROESY |
|---|---|---|---|---|
| Xyl1 (1→C-3) | ||||
| 1 | 104.7 CH | 4.76 d (6.9) | C: 3 | H-3, H-3, 5 Xyl1 |
| 2 | 81.3 CH | 3.97 t (8.6) | C: 1 Qui2; C: 1, 3 Xyl1 | H-1 Qui2; H-4 Xyl1 |
| 3 | 75.2 CH | 4.31 t (8.6) | C: 2, 4 Xyl1 | H-1, 5 Xyl1 |
| 4 | 76.1 CH | 4.98 dd (8.6, 13.8) | C: 3 Xyl1 | H-2 Xyl1 |
| 5 | 64.0 CH2 | 4.76 d (11.2) | C: 1, 3 Xyl1 | |
| 3.87 dd (8.6, 11.2) | H-1, 3 Xyl1 | |||
| Qui2 (1→2Xyl1) | ||||
| 1 | 102.0 CH | 5.20 d (7.8) | C: 2 Xyl1 | H-2 Xyl1; H-3, 5 Qui2 |
| 2 | 82.4 CH | 3.92 t (8.6) | C: 1 Xyl5; C: 1, 3 Qui2 | H-1 Xyl5; H-4 Qui2 |
| 3 | 75.2 CH | 3.98 t (8.6) | C: 2, 4 Qui2 | H-5 Qui2 |
| 4 | 86.3 CH | 3.43 t (8.6) | C: 1 Glc3; C: 3, 5, 6 Qui2 | H-1 Glc3; H-2, 6 Qui2 |
| 5 | 70.8 CH | 3.56 dd (6.0, 9.5) | H-1, 3, 6 Qui2 | |
| 6 | 17.8 CH3 | 1.55 d (6.0) | C: 4, 5 Qui2 | H-4, 5 Qui2 |
| Glc3 (1→4Qui2) | ||||
| 1 | 103.9 CH | 4.78 d (7.8) | C: 4 Qui2 | H-4 Qui2; H-5 Glc3 |
| 2 | 73.4 CH | 3.81 m | C: 1, 3 Glc3 | |
| 3 | 86.5 CH | 4.13 t (8.6) | C: 1 MeGlc4; C: 2, 4 Glc3 | H-1 MeGlc4; H-1 Glc3 |
| 4 | 69.1 CH | 3.80 t (8.6) | C: 5, 6 Glc3 | |
| 5 | 74.8 CH | 4.08 m | H-1 Glc3 | |
| 6 | 67.3 CH2 | 4.94 d (11.2) | ||
| 4.59 dd (6.9, 11.2) | C: 5 Glc3 | H-4 Glc3 | ||
| MeGlc4 (1→3Glc3) | ||||
| 1 | 104.7 CH | 5.15 d (8.4) | C: 3 Glc3 | H-3 Glc3; H-3, 5 MeGlc4 |
| 2 | 74.3 CH | 3.78 t (8.4) | C: 1 MeGlc4 | H-4 MeGlc4 |
| 3 | 86.3 CH | 3.64 t (9.3) | C: 2, 4 MeGlc4, OMe | H-1, 5 MeGlc4 |
| 4 | 69.8 CH | 4.01 m | C: 3, 5 MeGlc4 | H-2, 6 MeGlc4 |
| 5 | 75.5 CH | 4.01 m | C: 4, 6 MeGlc4 | H-1, 3 MeGlc4 |
| 6 | 67.0 CH2 | 4.92 brd (11.0) | C: 4, 5 MeGlc4 | |
| 4.75 brd (8.4) | C: 5 MeGlc4 | |||
| OMe | 60.4 CH3 | 3.80 s | C: 3 MeGlc4 | |
| Xyl5 (1→2Qui2) | ||||
| 1 | 105.1 CH | 5.22 d (7.6) | C: 2 Qui2 | H-2 Qui2; H-3, 5 Xyl5 |
| 2 | 74.8 CH | 3.91 t (7.6) | C: 1, 3 Xyl5 | |
| 3 | 76.3 CH | 4.07 t (8.4) | C: 2, 4 Xyl5 | H-1, 5 Xyl5 |
| 4 | 70.1 CH | 4.05 m | C: 3 Xyl5 | H-2 Xyl5 |
| 5 | 66.4 CH2 | 4.28 dd (5.1, 11.8) | C: 1, 4 Xyl5 | |
| 3.66 t (9.3) | C: 1, 3, 4 Xyl5 | H-1, 3 Xyl5 |
| Position | δC Mult. a | δH Mult. (J in Hz) | HMBC | ROESY |
|---|---|---|---|---|
| 1 | 34.9 CH2 | 2.47 m | H-11, H-19 | |
| 1.32 m | C: 18 | H-3, H-5, H-11 | ||
| 2 | 26.6 CH2 | 1.90 m | ||
| 1.79 m | H-19, H-30 | |||
| 3 | 88.6 CH | 3.13 dd (4.7, 12.2) | C: 30, 31, C: 1 Xyl1 | H-1, H-5, H-31, H-1 Xyl1 |
| 4 | 39.5 C | |||
| 5 | 51.4 CH | 1.07 dd (2.7, 15.6) | C: 4, 19, 30 | H-1, H-3, H-7, H-31 |
| 6 | 30.8 CH2 | 2.35 m | H-31 | |
| 1.99 m | C: 5, 7, 10 | H-19, H-30 | ||
| 7 | 67.7 CH | 4.55 t (8.1) | C: 8, 9 | H-5, H-15, H-32 |
| 8 | 139.9 C | |||
| 9 | 142.9 C | |||
| 10 | 38.3 C | |||
| 11 | 65.2 CH | 4.81 brd (6.8) | C: 8, 9, 13 | H-1 |
| 12 | 42.8 CH2 | 2.53 dd (7.5, 14.2) | C: 13, 18 | H-17, H-32 |
| 2.27 d (14.2) | C: 9, 11, 13, 14, 18 | H-18, H-21 | ||
| 13 | 45.2 C | |||
| 14 | 51.5 C | |||
| 15 | 32.0 CH2 | 2.71 brd (10.8) β | C: 14, 32 | H-18 |
| 1.60 brt (10.8) α | H-7, H-32 | |||
| 16 | 22.6 CH2 | 2.41 m | H-18 | |
| 1.68 m | H-32 | |||
| 17 | 58.9 CH | 2.83 t (8.8) | C: 12, 13, 16, 18, 20, 21 | H-12, H-21, H-32 |
| 18 | 19.7 CH3 | 1.23 s | C: 12, 13, 14, 17 | H-12, H-15, H-16, H-19, H-21 |
| 19 | 22.2 CH3 | 1.57 s | C: 1, 5, 9, 10 | H-1, H-2, H-5, H-6, H-18, H-30 |
| 20 | 211.8 C | |||
| 21 | 31.3 CH3 | 2.14 s | C: 17, 20 | |
| 30 | 16.4 CH3 | 0.99 s | C: 3, 4, 5, 31 | H-2, H-6, H-19, H-31 |
| 31 | 27.9 CH3 | 1.15 s | C: 3, 4, 5, 30 | H-3, H-5, H-6, H-30 |
| 32 | 26.1 CH3 | 1.00 s | C: 8, 13, 14, 15 | H-7, H-12, H-15, H-16, H-17 |
| Position | δC Mult. a | δH Mult. (J in Hz) | HMBC | ROESY |
|---|---|---|---|---|
| 1 | 34.1 CH2 | 2.29 m | H-3, H-5 | |
| 2.01 m | C: 10 | H-11 | ||
| 2 | 26.5 CH2 | 2.00 m | C: 3 | H-19 |
| 1.77 m | H-19, H-30 | |||
| 3 | 87.8 CH | 3.13 dd (4.0; 11.6) | C: 4, 30, C: 1 Xyl1 | H-1, H-5, H-31, H-1 Xyl1 |
| 4 | 39.5 C | |||
| 5 | 50.5 CH | 1.83 dd (3.9; 13.5) | C: 4, 6, 10, 19, 30, 31 | H-1, H-3, H-31 |
| 6 | 36.9 CH2 | 2.56 m | C: 5, 7, 10 | |
| 2.50 m | C: 5, 7, 10 | H-19, H-30 | ||
| 7 | 201.4 C | |||
| 8 | 140.0 C | |||
| 9 | 163.1 C | |||
| 10 | 40.6 C | |||
| 11 | 64.1 CH | 4.70 dd (5.4; 8.9) | C: 8, 9, 10, 13 | H-1, H-18, H-19 |
| 12 | 43.5 CH2 | 2.58 dd (8.8; 13.1) | C: 9, 11, 13, 18 | H-18, H-21 |
| 2.40 dd (5.1; 13.4) | C: 11, 13, 17, 18 | H-17, H-32 | ||
| 13 | 48.5 C | |||
| 14 | 48.7 C | |||
| 15 | 33.4 CH2 | 2.30 m | C: 14, 17, 32 | H-32 |
| 1.86 m | H-18 | |||
| 16 | 22.1 CH2 | 2.27 m | H-18 | |
| 1.68 m | H-32 | |||
| 17 | 58.6 CH | 2.91 t (9.0) | C: 12, 13, 16, 18, 20 | H-12, H-21, H-32 |
| 18 | 18.6 CH3 | 0.58 s | C: 12, 13, 14, 17 | H-12, H-15, H-16, H-19, H-21 |
| 19 | 19.8 CH3 | 1.10 s | C: 1, 5, 9, 10 | H-2, H-6, H-18 |
| 20 | 211.7 C | |||
| 21 | 31.2 CH3 | 2.15 s | C: 17, 20 | H-17, H-18 |
| 30 | 16.0 CH3 | 1.01 s | C: 3, 4, 5, 31 | H-2, H-6 |
| 31 | 27.0 CH3 | 1.05 s | C: 3, 4, 5, 30 | H-3, H-5, H-6 |
| 32 | 25.0 CH3 | 1.40 s | C: 8, 13, 14, 15 | H-12, H-15, H-17 |
| Position | δC Mult. a | δH Mult. (J in Hz) | HMBC | ROESY |
|---|---|---|---|---|
| 1 | 35.5 CH2 | 1.32 m | H-11 | |
| 1.27 m | ||||
| 2 | 27.0 CH2 | 1.95 m | ||
| 1.78 m | H-19, H-30 | |||
| 3 | 88.9 CH | 3.14 dd (4.1, 11.6) | C: 30, C: 1 Xyl1 | H-5, H-31, H-1 Xyl1 |
| 4 | 39.5 C | |||
| 5 | 49.7 CH | 0.84 brd (12.7) | C: 1, 4, 30 | H-1, H-3, H-31 |
| 6 | 23.1 CH2 | 1.92 m | H-31 | |
| 1.83 m | H-19, H-30 | |||
| 7 | 122.2 CH | 5.59 m | C: 9, 14 | H-15, H-32 |
| 8 | 149.7 C | |||
| 9 | 48.1 CH | 2.28 brd (12.8) | H-18, H-19 | |
| 10 | 35.6 C | |||
| 11 | 22.8 CH2 | 1.63 m | ||
| 1.38 m | H-32 | |||
| 12 | 34.8 CH2 | 1.96 m | H-17, H-21 | |
| 1.74 m | H-18 | |||
| 13 | 45.3 C | |||
| 14 | 52.9 C | |||
| 15 | 33.4 CH2 | 1.59 m | H-18 | |
| 1.50 m | H-7, H-32 | |||
| 16 | 22.2 CH2 | 1.93 m | H-23 | |
| 1.54 m | H-17, H-32 | |||
| 17 | 52.8 CH | 2.39 t (8.8) | C: 14, 16, 18 | H-21, H-32 |
| 18 | 24.8 CH3 | 1.28 s | C: 12, 14, 17 | H-9, H-12, H-16, H-21 |
| 19 | 24.5 CH3 | 0.93 s | C: 1, 5, 9, 10 | H-2, H-6, H-9 |
| 20 | 80.5 C | |||
| 21 | 24.5 CH3 | 1.62 s | C: 17, 20, 22 | H-12, H-17, H-18 |
| 22 | 205.3 C | |||
| 23 | 119.9 C | 7.41 d (15.5) | C: 22, 24, 25 | H-17, H-21, H-26, H-27, H-16 |
| 24 | 156.5 C | 7.40 d (15.5) | C: 22, 23, 25 | |
| 25 | 70.5 C | |||
| 26 | 29.3 CH3 | 1.47 s | C: 24, 25, 27 | |
| 27 | 29.2 CH3 | 1.48 s | C: 24, 25, 26 | |
| 30 | 17.4 CH3 | 1.02 s | C: 3, 4, 5, 31 | H-2, H-6 |
| 31 | 28.7 CH3 | 1.17 s | C: 3, 4, 5, 30 | H-3, H-5, H-6 |
| 32 | 30.7 CH3 | 1.03 s | C: 8, 13, 14, 15 | H-7, H-11, H-12, H-15, H-17 |
| Position | δC Mult. a | δH Mult. (J in Hz) | HMBC | ROESY |
|---|---|---|---|---|
| 1 | 36.4 CH2 | 1.60 m | H-11, H-19 | |
| 1.24 m | H-3, H-5, H-11 | |||
| 2 | 26.7 CH2 | 2.00 m | ||
| 1.77 m | H-19, H-30 | |||
| 3 | 88.5 CH | 3.10 dd (4.1; 11.8) | H-1, H-5, H-31, H-1 Xyl1 | |
| 4 | 39.5 C | |||
| 5 | 49.3 CH | 0.86 brd (13.7) | H-1, H-3, H-7, H-31 | |
| 6 | 31.8 CH2 | 2.15 m | H-31 | |
| 1.79 m | ||||
| 7 | 71.6 CH | 3.83 m | H-5, H-32 | |
| 8 | 49.8 CH | 2.38 m | H-15, H-18, H-19 | |
| 9 | 147.6 C | |||
| 10 | 38.9 C | |||
| 11 | 116.0 CH | 5.27 m | C: 10, 13 | H-1 |
| 12 | 35.8 CH2 | 2.34 m | H-32 | |
| 1.94 m | H-18 | |||
| 13 | 46.8 C | |||
| 14 | 47.3 C | |||
| 15 | 36.6 CH2 | 2.05 m | H-32 | |
| 1.94 m | ||||
| 16 | 22.5 CH2 | 2.31 m | H-18 | |
| 1.62 m | H-32 | |||
| 17 | 59.3 CH | 2.92 t (9.0) | C: 13, 18 | H-12, H-32 |
| 18 | 16.4 CH3 | 0.59 s | C: 12, 13, 17 | H-8, H-12, H-19 |
| 19 | 21.8 CH3 | 1.00 s | C: 5, 9, 10 | H-1, H-2, H-8, H-18 |
| 20 | 212.5 C | |||
| 21 | 31.2 CH3 | 2.18 s | C: 17, 20 | |
| 30 | 16.6 CH3 | 0.94 s | C: 3, 4, 5, 31 | H-2, H-31 |
| 31 | 27.9 CH3 | 1.14 s | C: 3, 4, 5, 30 | H-3, H-5, H-6, H-30 |
| 32 | 18.3 CH3 | 0.99 s | C: 8, 13, 15 | H-7, H-15, H-16, H-17 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silchenko, A.S.; Kalinovsky, A.I.; Avilov, S.A.; Andryjaschenko, P.V.; Dmitrenok, P.S.; Kalinin, V.I.; Chingizova, E.A.; Minin, K.V.; Stonik, V.A. Structures and Biogenesis of Fallaxosides D4, D5, D6 and D7, Trisulfated Non-Holostane Triterpene Glycosides from the Sea Cucumber Cucumaria fallax. Molecules 2016, 21, 939. https://doi.org/10.3390/molecules21070939
Silchenko AS, Kalinovsky AI, Avilov SA, Andryjaschenko PV, Dmitrenok PS, Kalinin VI, Chingizova EA, Minin KV, Stonik VA. Structures and Biogenesis of Fallaxosides D4, D5, D6 and D7, Trisulfated Non-Holostane Triterpene Glycosides from the Sea Cucumber Cucumaria fallax. Molecules. 2016; 21(7):939. https://doi.org/10.3390/molecules21070939
Chicago/Turabian StyleSilchenko, Alexandra S., Anatoly I. Kalinovsky, Sergey A. Avilov, Pelageya V. Andryjaschenko, Pavel S. Dmitrenok, Vladimir I. Kalinin, Ekaterina A. Chingizova, Kirill V. Minin, and Valentin A. Stonik. 2016. "Structures and Biogenesis of Fallaxosides D4, D5, D6 and D7, Trisulfated Non-Holostane Triterpene Glycosides from the Sea Cucumber Cucumaria fallax" Molecules 21, no. 7: 939. https://doi.org/10.3390/molecules21070939
APA StyleSilchenko, A. S., Kalinovsky, A. I., Avilov, S. A., Andryjaschenko, P. V., Dmitrenok, P. S., Kalinin, V. I., Chingizova, E. A., Minin, K. V., & Stonik, V. A. (2016). Structures and Biogenesis of Fallaxosides D4, D5, D6 and D7, Trisulfated Non-Holostane Triterpene Glycosides from the Sea Cucumber Cucumaria fallax. Molecules, 21(7), 939. https://doi.org/10.3390/molecules21070939