
Hydrolytic Metallo-Nanozymes: From Micelles and Vesicles to Gold Nanoparticles


Abstract
:1. Introduction
2. Surfactant-Based Hydrolytic “Metallonanosystems”
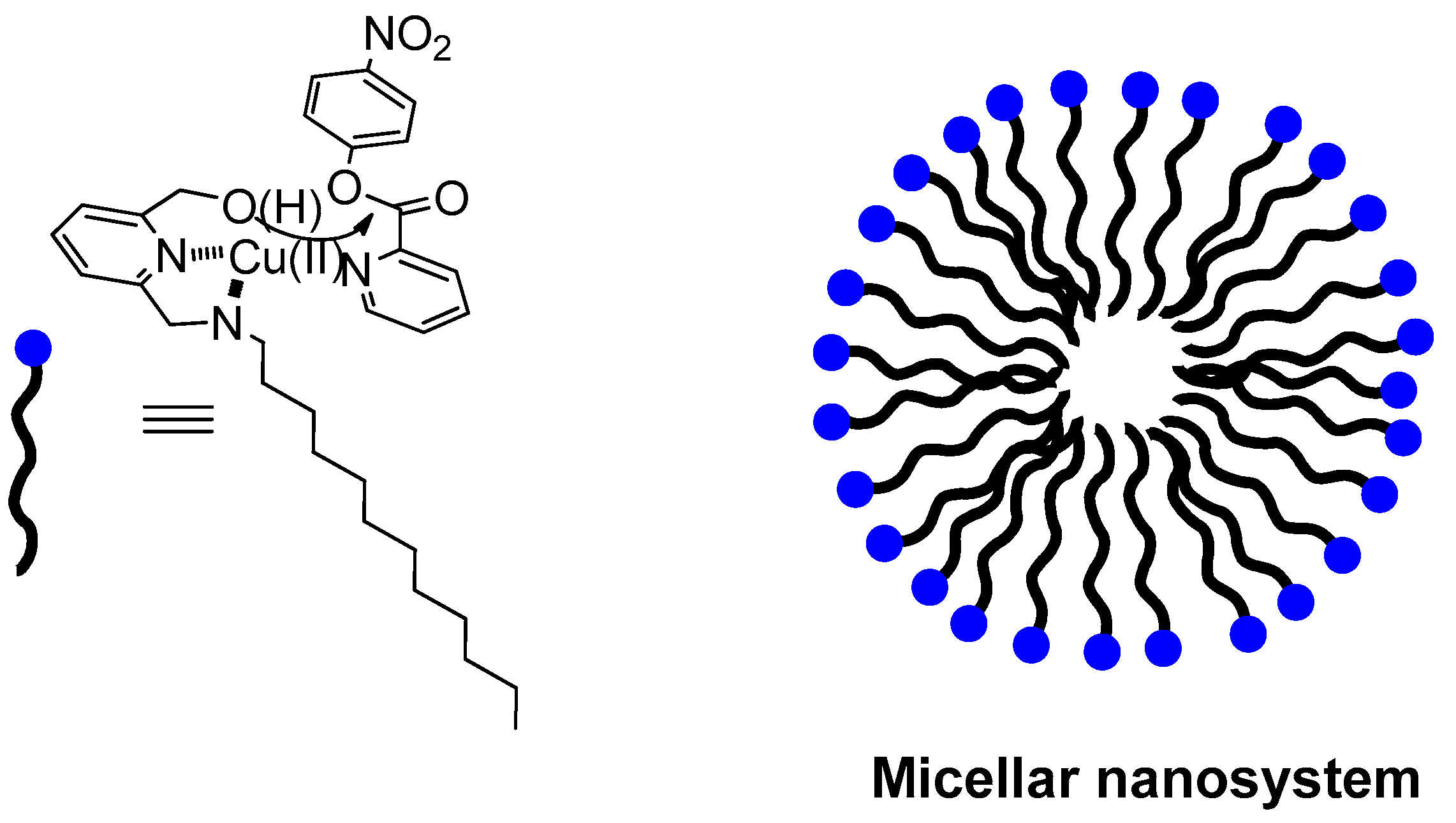
2.1. Metallomicelles
2.2. Metallovesicles
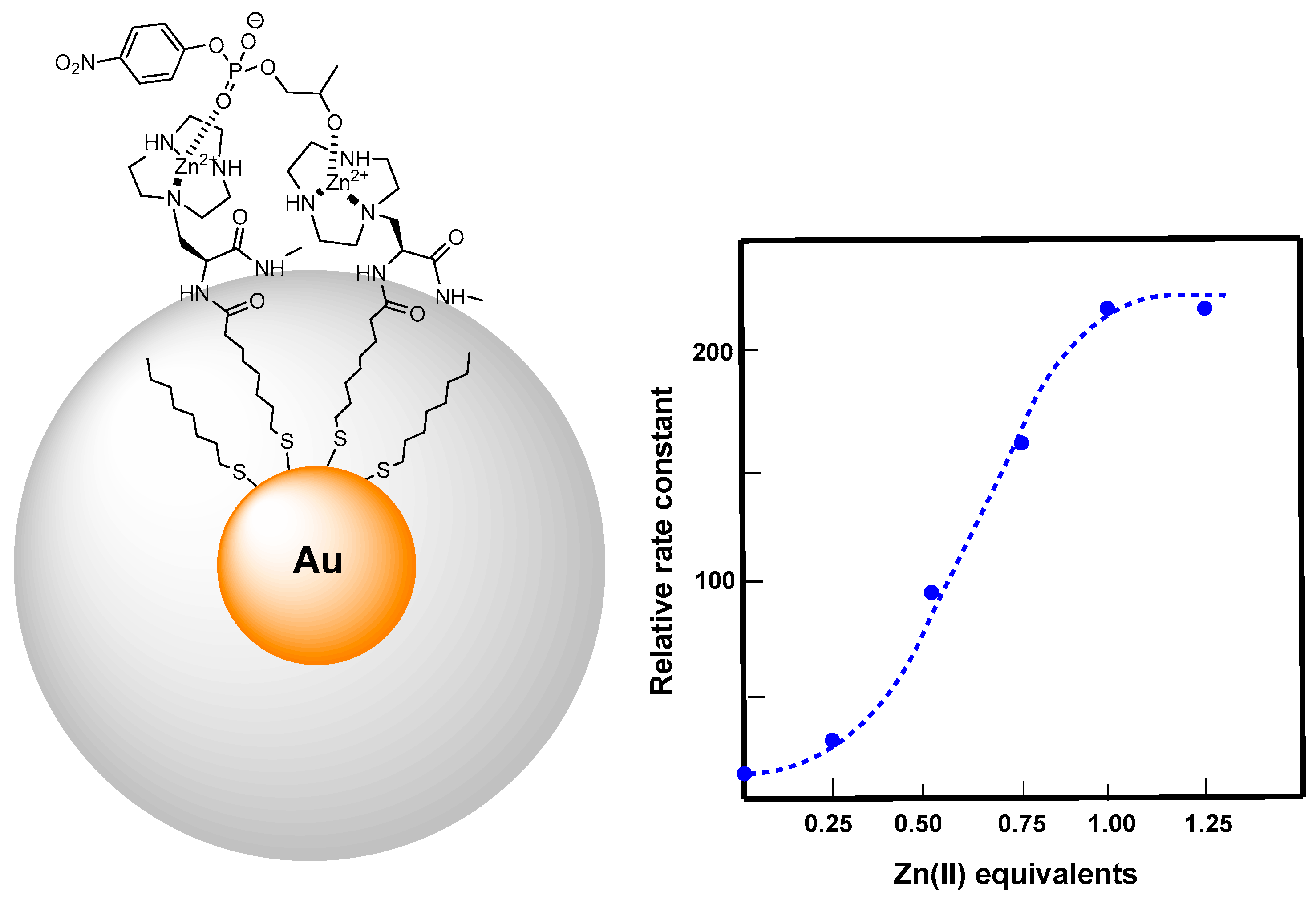
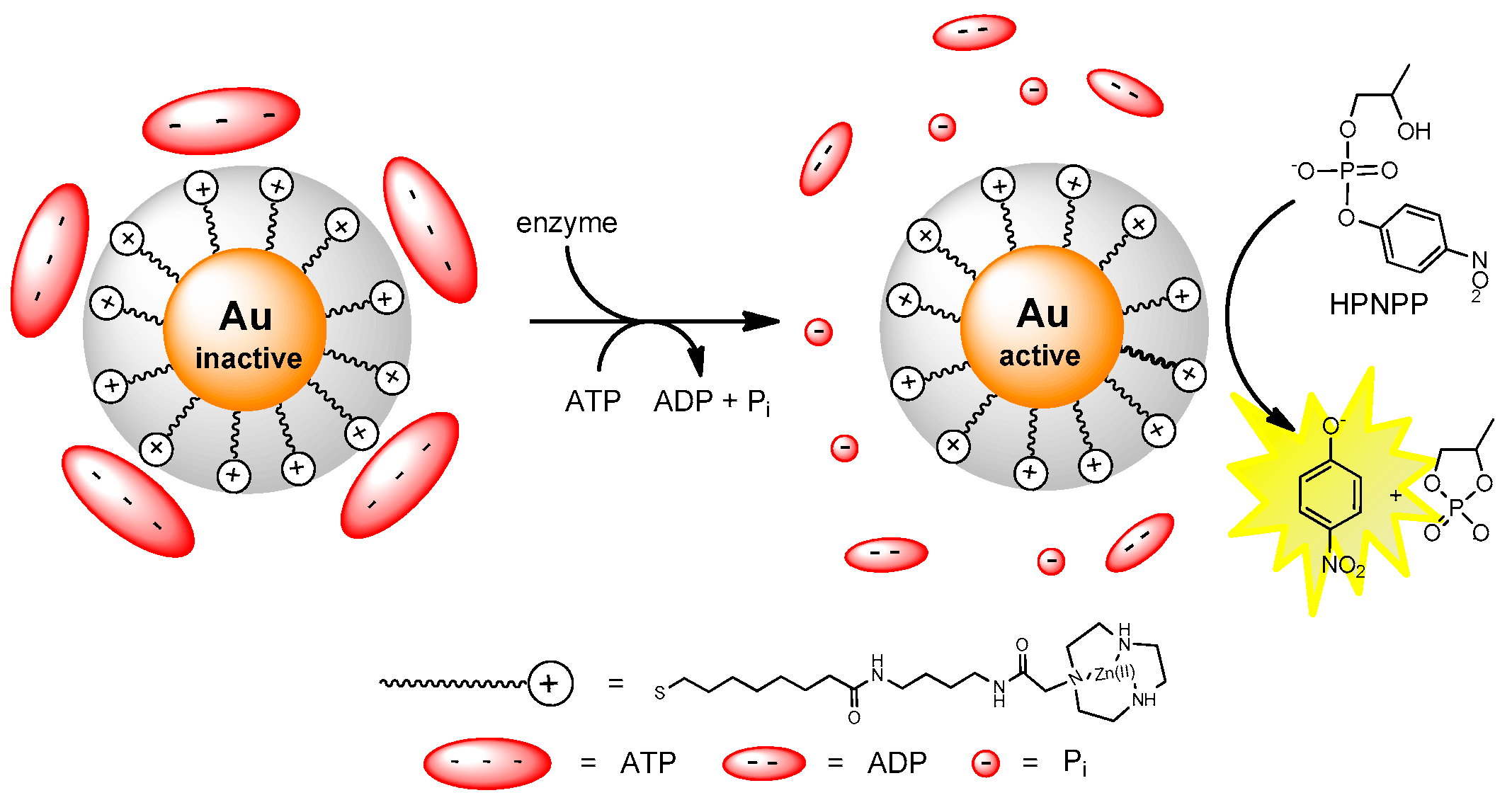
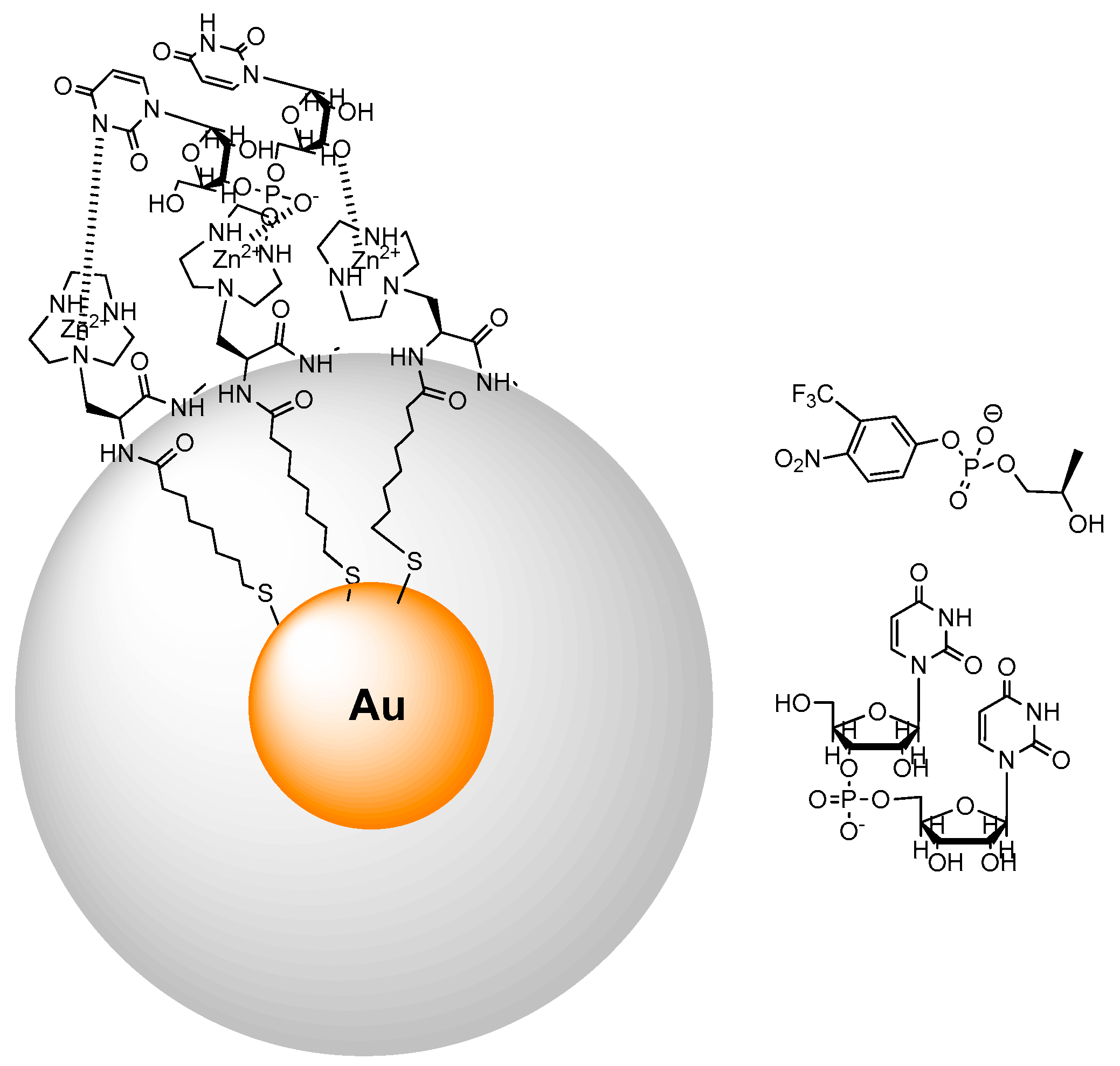
3. Metallonanoparticles
4. Comparison between the Nanosystems and the Quest for Cooperativity
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| PNPP | p-Nitrophenyl picolinate |
| DAAP | 4,4-(Dialkylamino)pyridine |
| PNPH | p-Nitrophenylhexanoate |
| PNPDPP | p-Nitrophenyl diphenyl phosphate |
| CTAB | Cetyltrimethylammonium bromide |
| AuNP | Gold nanoparticle |
| HPNPP | 2-Hydroxypropyl-p-nitrophenyl phosphate |
| BAPA | Bis-(2-amino-pyridinyl-6-methyl)amine |
| BNPP | Bis-p-nitrophenyl phosphate |
References
- Bhattacharya, S.; Kumari, N. Metallomicelles as potent catalysts for the ester hydrolysis reactions in water. Coord. Chem. Rev. 2009, 253, 2133–2149. [Google Scholar] [CrossRef]
- Mancin, F.; Scrimin, P.; Tecilla, P.; Tonellato, U. Amphiphilic metalloaggregates: Catalysis, transport, and sensing. Coord. Chem. Rev. 2009, 253, 2150–2165. [Google Scholar] [CrossRef]
- Zhang, J.; Meng, X.-G.; Zeng, X.-C.; Yu, X.Q. Metallomicellar supramolecular systems and their applications in catalytic reactions. Coord. Chem. Rev. 2009, 253, 2166–2177. [Google Scholar] [CrossRef]
- Desbouis, D.; Troitsky, I.P.; Belousoff, M.J.; Spiccia, L.; Graham, B. Copper(II), zinc(II) and nickel(II) complexes as nuclease mimetics. Coord. Chem. Rev. 2012, 256, 897–937. [Google Scholar] [CrossRef]
- Dong, Z.; Luo, Q.; Liu, J. Artificial enzymes based on supramolecular scaffolds. Chem. Soc. Rev. 2012, 41, 7890–7908. [Google Scholar] [CrossRef] [PubMed]
- La Sorella, G.; Strukul, G.; Scarso, A. Recent advances in catalysis in micellar media. Green Chem. 2015, 17, 644–683. [Google Scholar] [CrossRef]
- Moroi, Y.; Sugii, R.; Akine, C.; Matuura, R. Anionic surfactants with methylviologen and cupric ions as divalent cationic gegenion(II): Effect of alkyl chain length on solubility and micelle formation. J. Colloid Interface Sci. 1985, 108, 180–188. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Mei, G.C. Association phenomena. 7. Mixed chelate and comicellar catalysis of acetyl phosphate olysis reactions. J. Am. Chem. Soc. 1985, 107, 7964–7967. [Google Scholar] [CrossRef]
- Fornasier, R.; Milani, D.; Scrimin, P.; Tonellato, U. Functional micellar catalysis. Part 8. Catalysis of the hydrolysis of p-nitrophenylpicolinate by metal-chelating micelles containing copper(II) or zinc(II). Perkin Trans. J. Chem. Soc. 1986, 2, 233–237. [Google Scholar] [CrossRef]
- Tagaki, W.; Ogino, K. Micellar models of zinc enzymes. Top. Curr. Chem. 1985, 128, 143–174. [Google Scholar]
- Gellman, S.H.; Petter, R.; Breslow, R. Catalytic hydrolysis of a phosphate triester by tetracoordinated zinc complexes. J. Am. Chem. Soc. 1986, 108, 2388–2394. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.M.; Gan, L.H.; Johnson, E.; Durst, D.H. Phosphate ester hydrolysis catalyzed by metallomicelles. J. Am. Chem. Soc. 1987, 109, 2800–2803. [Google Scholar] [CrossRef]
- Scrimin, P.; Tecilla, P.; Tonellato, U. Leaving group effect in the cleavage of picolinate esters catalyzed by hydroxy-functionalized metallomicelles. J. Org. Chem. 1994, 59, 18–24. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Snehalatha, K.; George, S.K. Synthesis of some copper(II)-chelating dialkylamino)pyridine amphiphiles and evaluation of their esterolytic capacities in cationic micellar media. J. Org. Chem. 1998, 63, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Snehalatha, K.; Kumar, V.P. Synthesis of new Cu(II)-chelating ligand amphiphiles and their esterolytic properties in cationic micelles. J. Org. Chem. 2003, 68, 2741–2747. [Google Scholar] [CrossRef] [PubMed]
- Ogino, K.; Kashihara, N.; Ueda, T.; Isaka, T.; Yoshida, T.; Tagaki, W. Hydrolytic metalloenzyme models. Metal ion dependent site-selective acylation of hydroxyl groups of bis-imidazole ligands catalyzed by Zn2+ and Cu2+ in the reaction with p-nitrophenyl 2-pyridinecarboxylate in a cationic surfactant micelle. Bull. Chem. Soc. Jpn. 1992, 65, 373–384. [Google Scholar] [CrossRef]
- Ogino, K.; Inoue, K.; Tagaki, W. Ester substrate PNIQ as a novel fluorescent probe for the study of acylation and deacylation steps in ester hydrolysis catalyzed by lipophilic imidazole·Zn2+ complexes in micelles. Tetrahedron Lett. 1992, 33, 4191–4194. [Google Scholar] [CrossRef]
- Weijnen, J.G.J.; Koudijs, A.; Schellekens, G.A.; Engbersen, J.F.J. Functionalised 1,10-phenanthroline metallocatalysts as models for hydrolytic metalloenzymes. Perkin Trans. J. Chem. Soc. 1992, 2, 829–834. [Google Scholar] [CrossRef]
- Scrimin, P.; Tecilla, P.; Tonellato, U.; Bunton, C.A. Nucleophilic Catalysis of Hydrolyses of Phosphate and Carboxylate Esters by Metallomicelles. Facts and Misconceptions. Coll. Surf. 1998, 144, 71–79. [Google Scholar] [CrossRef]
- Hampl, F.; Liska, F.; Mancin, F.; Tecilla, P.; Tonellato, U. Metallomicelles made of Ni(II) complexes of lipophilic 2-pyridineketoximes as powerful catalysts of the cleavage of carboxylic acid esters. Langmuir 1999, 15, 405–412. [Google Scholar] [CrossRef]
- Mancin, F.; Tecilla, P.; Tonellato, U. Metallomicelles made of Ni(II) and Zn(II) complexes of 2-pyridinealdoxime-based ligands as catalysts of the cleavage of carboxylic acid esters. Langmuir 2000, 16, 227–233. [Google Scholar] [CrossRef]
- Kimura, E.; Hashimoto, H.; Koike, T. Hydrolysis of lipophilic esters catalyzed by a zinc(II) complex of a long alkyl-pendant macrocyclic tetraamine in micellar solution. J. Am. Chem. Soc. 1996, 118, 10963–10970. [Google Scholar] [CrossRef]
- Polyzos, A.; Hughes, A.B.; Christie, J.R. Catalysis of aryl ester hydrolysis in the presence of metallomicelles containing a copper(II) diethylenetriamine derivative. Langmuir 2007, 23, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.-G.; Jiang, X.; Gu, L.-N.; Hu, G. Gemini metallomicellar catalysis: Hydrolysis of p-nitrophenyl picolinate catalyzed by Cu(II) and Ni(II) complexes of macrocyclic ligands in gemini surfactant micelles. J. Mol. Catal. A Chem. 2007, 277, 15–20. [Google Scholar] [CrossRef]
- Du, J.; Jiang, B.; Kou, X.; Zeng, X.; Xiang, Q. Enhanced hydrolysis of carboxylic acid esters catalyzed by metallomicelles made of Cu(II) and Zn(II) complexes. J. Colloid Interface Sci. 2002, 256, 428–434. [Google Scholar] [CrossRef]
- Weijnen, J.G.J.; Engbersen, J.F.J. Catalytic hydrolysis of phosphate esters by metallocomplexes of 1,10-phenanthroline derivatives in micellar solution. Recl. Trav. Chim. Pays-Bas 1993, 112, 351–357. [Google Scholar] [CrossRef]
- Lim, Y.Y.; Tan, E.H.L.; Gan, L.H. Anionic effects on the hydrolysis of p-nitrophenyl diphenyl phosphate in surface active [Cu(R-tmed)X2] and [Cu(R-tmed)(acac)X] complexes where R-tmed = N,N,N′-Trimethyl-N′-alkylethylenediamine and X = halide or nitrate. J. Colloid Interface Sci. 1993, 157, 442–446. [Google Scholar] [CrossRef]
- Bunton, C.A.; Scrimin, P.; Tecilla, P. The source of catalysis of dephosphorylation of p-nitrophenyldiphenylphosphate by metallomicelles. Perkin Trans. 2 J. Chem. Soc. 1996, 419–425. [Google Scholar] [CrossRef]
- Scrimin, P.; Ghirlanda, G.; Tecilla, P.; Moss, R.A. Comparative reactivities of phosphate ester cleavages by metallomicelles. Langmuir 1996, 12, 6235–6241. [Google Scholar] [CrossRef]
- De Santi, G.; Scrimin, P.; Tonellato, U. Micellization triggers pseudo-intramolecular transacylation in Cu2+ complexes of hydrolytic metallomicelles. Tetrahedron Lett. 1990, 31, 4791–4794. [Google Scholar] [CrossRef]
- Fornasier, R.; Scrimin, P.; Tonellato, U.; Zanta, N. Highly enantioselective cleavage of α-amino acid p-nitrophenylesters by chiral metallomicelles. J. Chem. Soc. Chem. Commun. 1988, 716–718. [Google Scholar] [CrossRef]
- Ghirlanda, G.; Scrimin, P.; Tecilla, P.; Toffoletti, A. Amphiphilic Cu(II) complexes modeled after the metal-complexation subunit of bleomycin antibiotics. Langmuir 1998, 14, 1646–1655. [Google Scholar] [CrossRef]
- Pengo, P.; Baltzer, L.; Pasquato, L.; Scrimin, P. Substrate modulation of the activity of an artificial nanoesterase made of peptide-functionalized gold nanoparticles. Angew Chem. Int. Ed. 2007, 46, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Pengo, P.; Polizzi, S.; Pasquato, L.; Scrimin, P. Carboxylate-imidazole cooperativity in dipeptide-functionalized gold nanoparticles with esterase-like activity. J. Am. Chem. Soc. 2005, 127, 1616–1617. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, J.G.J.; Koudijs, A.; Engbersen, J.F.J. Synthesis of chiral 1,10-phenanthroline ligands and the activity of metal-ion complexes in the enantioselective hydrolysis of N-protected amino acid esters. J. Org. Chem. 1992, 57, 7258–7265. [Google Scholar] [CrossRef]
- You, J.-S.; Yu, X.-Q.; Li, X.-S.; Yan, Q.-S.; Xie, R.-G. Enantioselective hydrolysis of long chain α-amino acid esters by chiral sulfur-containing macrocyclic metallomicelles. Tetrahedron Asym. 1998, 9, 1197–1203. [Google Scholar] [CrossRef]
- You, J.-S.; Yu, X.-Q.; Liu, K.; Tao, L.; Xiang, Q.-X.; Xie, R.-G. Synthesis of novel chiral lipophilic pyridyl-containing β-amino alcohol ligands and enantioselective hydrolysis of α-amino acid esters by chiral metallomicelles. Tetrahedron Asym. 1999, 10, 243–254. [Google Scholar] [CrossRef]
- You, J.-S.; Yu, X.-Q.; Su, X.-Y.; Wang, T.; Xiang, Q.-X.; Yang, M.; Xie, R.-G. Hydrolytic metalloenzyme models: Enantioselective hydrolysis of long chain α-amino acid esters by chiral metallomicelles composed of lipophilic l-histidinol. J. Mol. Catal. A Chem. 2003, 202, 17–22. [Google Scholar] [CrossRef]
- Gruber, B.; Kataev, E.; Aschenbrenner, J.; Stadlbauer, S.; König, B. Vesicles and micelles from amphiphilic zinc(II)–cyclen complexes as highly potent promoters of hydrolytic DNA cleavage. J. Am. Chem. Soc. 2011, 133, 20704–20707. [Google Scholar] [CrossRef] [PubMed]
- Poznik, M.; König, B. Cooperative hydrolysis of aryl esters on functionalized membrane surfaces and in micellar solutions. Org. Biomol. Chem. 2014, 12, 3175–3180. [Google Scholar] [CrossRef] [PubMed]
- Poznik, M.; Maitra, U.; König, B. The interface makes a difference: Lanthanide ion coated vesicles hydrolyze phosphodiesters. Org. Biomol. Chem. 2015, 13, 9789–9792. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.A.; Bhattacharya, S.; Scrimin, P.; Swarup, S. Surface-specific cleavage of a carbonate functionalized vesicular surfactant. J. Am. Chem. Soc. 1987, 109, 5740–5744. [Google Scholar] [CrossRef]
- Moss, R.A.; Swarup, S. Surface specific phosphate cleavage of a substrate functionalized vesicular surfactant. J. Am. Chem. Soc. 1986, 108, 5341–5342. [Google Scholar] [CrossRef]
- Moss, R.A.; Bhattacharya, S.; Chatterjee, S. Chemical differentiation of bilayer surfaces in functional dialkylammonium ion vesicles: Observation of surfactant flip-flop. J. Am. Chem. Soc. 1989, 111, 3680–3687. [Google Scholar] [CrossRef]
- Moss, R.A.; Ganguli, S.; Okumura, Y.; Fujita, T. Relation of surfactant monomer structure to flip-flop dynamics in surface-differentiated synthetic bilayer membranes. J. Am. Chem. Soc. 1990, 112, 6391–6392. [Google Scholar] [CrossRef]
- Scrimin, P.; Tecilla, P.; Tonellato, U. Cationic metallovesicles: Catalysis of the cleavage of p-nitrophenyl picolinate and control of Cu(II) permeation. J. Am. Chem. Soc. 1992, 114, 5086–5092. [Google Scholar] [CrossRef]
- Ghirlanda, G.; Scrimin, P.; Tecilla, P.; Tonellato, U. An hydrolytic reporter of Cu(II) availability in artificial liposomes. J. Org. Chem. 1993, 58, 3025–3029. [Google Scholar] [CrossRef]
- Scrimin, P.; Tecilla, P.; Moss, R.A.; Bracken, K. Control of permeation of lanthanide ions across phosphate-functionalized liposomal membranes. J. Am. Chem. Soc. 1998, 120, 1179–1185. [Google Scholar] [CrossRef]
- Scrimin, P.; Caruso, S.; Paggiarin, N.; Tecilla, P. Ln(III)-catalyzed cleavage of phosphate-functionalyzed synthetic lipids: Real time monitoring of vesicle decapsulation. Langmuir 2000, 16, 203–209. [Google Scholar] [CrossRef]
- Maiti, S.; Fortunati, I.; Ferrante, C.; Scrimin, P.; Prins, L.J. Dissipative self-assembly of vesicular nanoreactors. Nat. Chem. 2016, 8, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Cleij, M.C.; Scrimin, P.; Tecilla, P.; Tonellato, U. Chiral lipophilic ligands. 3. Control of enantioselectivity in Cu(II)-catalyzed cleavage of -amino acid esters by aggregate morphology. Langmuir 1996, 12, 2956–2960. [Google Scholar] [CrossRef]
- Pieters, G.; Prins, L.J. Catalytic self-assembled monolayers on gold nanoparticles. New J. Chem. 2012, 36, 1931–1939. [Google Scholar] [CrossRef]
- Pasquato, L.; Pengo, P.; Scrimin, P. Functional gold nanoparticles for recognition and catalysis. J. Mater. Chem. 2004, 14, 3481–3487. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, F.; Zhou, H.; Su, H. The structural and bonding evolution in cysteine-gold cluster complexes. Phys. Chem. Chem. Phys. 2013, 15, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.; Felluga, F.; Scrimin, P. A new ligand α-amino acid: (S)-2-Amino-3-[1-(1,4,7-triazacyclononane)]propanoic acid. Tetrahedron Lett. 1998, 39, 7159–7162. [Google Scholar] [CrossRef]
- Manea, F.; Bodar-Houillon, F.; Pasquato, L.; Scrimin, P. Nanozymes: Gold-nanoparticle-based transphosphorylation catalysts. Angew. Chem. Int. Ed. 2004, 43, 6165–6169. [Google Scholar] [CrossRef] [PubMed]
- Diez-Castellnou, M.; Mancin, F.; Scrimin, P. Efficient phosphodiester cleaving nanozymes resulting from multivalency and local medium polarity control. J. Am. Chem. Soc. 2014, 136, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.Q.; Mareque-Rivas, J.C.; de Rosales, R.T.M.; Williams, N.H. A highly reactive mononuclear Zn(II) complex for phosphodiester cleavage. J. Am. Chem. Soc. 2005, 127, 13470–13471. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.Q.; Mareque-Rivas, J.C.; Williams, N.H. Comparing a mononuclear Zn(II) complex with hydrogen bond donors with a dinuclear Zn(II) complex for catalysing phosphate ester cleavage. Chem. Commun. 2006, 1845–1847. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.Q.; Natale, D.; Prabaharan, R.; Mareque-Rivas, J.C.; Williams, N.H. Efficient phosphodiester binding and cleavage by a Zn(II) complex combining hydrogen-bonding interactions and double Lewis acid activation. Angew. Chem. Int. Ed. 2006, 45, 7056–7059. [Google Scholar] [CrossRef] [PubMed]
- Linjalathi, H.; Feng, G.Q.; Mareque-Rivas, J.C.; Mikkola, S.; Williams, N.H. Cleavage and isomerization of UpU promoted by dinuclear metal ion complexes. J. Am. Chem. Soc. 2008, 130, 4232–4233. [Google Scholar] [CrossRef] [PubMed]
- Livieri, M.; Mancin, F.; Saielli, G.; Chin, J.; Tonellato, U. Mimicking enzymes: Cooperation between organic functional groups and metal ions in the cleavage of phosphate diesters. Chem. Eur. J. 2007, 13, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Livieri, M.; Mancin, F.; Tonellato, U.; Chin, J. Multiple functional group cooperation in phosphate diester cleavage promoted by Zn(II) complexes. Chem. Commun. 2004, 2862–2863. [Google Scholar] [CrossRef] [PubMed]
- Tirel, E.Y.; Williams, N.H. Enhancing phosphate diester cleavage by a zinc complex through controlling nucleophile coordination. Chem. Eur. J. 2015, 21, 7053–7056. [Google Scholar] [CrossRef] [PubMed]
- Tirel, E.Y.; Bellamy, Z.; Adams, H.; Lebrun, V.; Duarte, F.; Williams, N.H. Catalytic zinc complexes for phosphate diester hydrolysis. Angew. Chem. Int. Ed. 2014, 53, 8246–8250. [Google Scholar] [CrossRef] [PubMed]
- Manea, F.; Bindoli, C.; Polizzi, S.; Lay, L.; Scrimin, P. Expeditious synthesis of water-soluble, monolayer-protected gold nanoparticles of controlled size and monolayer composition. Langmuir 2008, 24, 4120–4124. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, R.; Selvestrel, F.; Lombardo, V.; Sissi, C.; Polizzi, S.; Mancin, F.; Tonellato, U.; Scrimin, P. Phosphate diester and DNA hydrolysis by a multivalent, nanoparticle-based catalyst. J. Am. Chem. Soc. 2008, 130, 15744–15745. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, R.; Scrimin, P.; Mancin, F. Phosphate diesters cleavage mediated by Ce(IV) complexes self-assembled on gold nanoparticles. Org. Biomol. Chem. 2010, 8, 2622–2626. [Google Scholar] [CrossRef] [PubMed]
- Franklin, S.J. Lanthanide-mediated DNA hydrolysis. Curr. Opin. Chem. Biol. 2001, 5, 201–208. [Google Scholar] [CrossRef]
- Pieters, G.; Cazzolaro, A.; Bonomi, R.; Prins, L.J. Self-assembly and selective exchange of oligoanions on the surface of monolayer protected Au nanoparticles in water. Chem. Commun. 2012, 48, 1916–1918. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, R.; Cazzolaro, A.; Sansone, A.; Scrimin, P.; Prins, L.J. Detection of enzyme activity through catalytic signal amplification with functionalized gold nanoparticles. Angew. Chem. Int. Ed. 2011, 50, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Scrimin, P.; Prins, L.J. Sensing through signal amplification. Chem. Soc. Rev. 2011, 40, 4488–4505. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.-Y.; Pezzato, C.; Scrimin, P.; Prins, L.J. Chiral nanozymes-gold nanoparticle-based transphosphorylation catalysts capable of enantiomeric discrimination. Chem. Eur. J. 2016, 22, 7028–7032. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.; Felluga, F.; Tecilla, P.; Formaggio, F.; Crisma, M.; Toniolo, C.; Scrimin, P. A bimetallic helical heptapeptide as a transphosphorylation catalyst in water. J. Am. Chem. Soc. 1999, 121, 6948–6949. [Google Scholar] [CrossRef]
- Scarso, A.; Scheffer, U.; Göbel, M.; Broxterman, Q.B.; Kaptein, B.; Formaggio, F.; Toniolo, C.; Scrimin, P. A peptide template as an allosteric supramolecular catalyst for the cleavage of phosphate esters. Proc. Natl. Acad. Sci. USA 2002, 99, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Manea, F.; Fiammengo, R.; Prins, L.J.; Pasquato, L.; Scrimin, P. Metallodendrimers as transphosphorylation catalysts. J. Am. Chem. Soc. 2007, 129, 6982–6983. [Google Scholar] [CrossRef] [PubMed]
- Zaupa, G.; Mora, C.; Bonomi, R.; Prins, L.J.; Scrimin, P. Catalytic self-assembled monolayers on Au nanoparticles: The source of catalysis of a transphosphorylation reaction. Chem. Eur. J. 2011, 17, 4879–4889. [Google Scholar] [CrossRef] [PubMed]
- Zaramella, D.; Scrimin, P.; Prins, L.J. Self-assembly of a catalytic multivalent peptide-nanoparticle complex. J. Am. Chem. Soc. 2012, 134, 8396–8399. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.R. Speed limits for artificial ribonucleases. Comm. Inorg. Chem. 2008, 29, 169–188. [Google Scholar] [CrossRef]
- Bunn, S.E.; Liu, C.T.; Lu, Z.-L.; Neverov, A.A.; Brown, R.S. The dinuclear Zn(II) complex catalyzed cyclization of a series of 2-hydroxypropyl aryl phosphate RNA models: Progressive change in mechanism from rate-limiting PO bond cleavage to substrate binding. J. Am. Chem. Soc. 2007, 129, 16238–16248. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, E. Nanomaterials with enzyme-like characteristics (nanozymes): Next-generation artificial enzymes. Chem. Soc. Rev. 2013, 42, 6060–6093. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, H.-B.; Ji, L.-N.; Mao, Z.-W. Insights into metalloenzyme microenvironments: Biomimetic metal complexes with a functional second coordination sphere. Chem. Soc. Rev. 2013, 42, 8360–8375. [Google Scholar] [CrossRef] [PubMed]
- Avenier, F.; Hollfelder, F. Combining medium effects and cofactor catalysis: Metal-coordinated synzymes accelerate phosphate transfer by 108. Chem. Eur. J. 2009, 15, 12371–12380. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.; Tecilla, P.; Baltzer, L.; Scrimin, P. De novo metallonucleases based on helix-loop-helix motifs. Chem. Eur. J. 2004, 10, 4163–4170. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.T.; Cangelosi, V.M.; Zastrow, M.L.; Tegoni, M.; Plegaria, J.S.; Tebo, A.G.; Mocny, C.S.; Ruckthong, L.; Qayyum, H.; Pecoraro, V.L. Protein Design: Toward Functional Metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
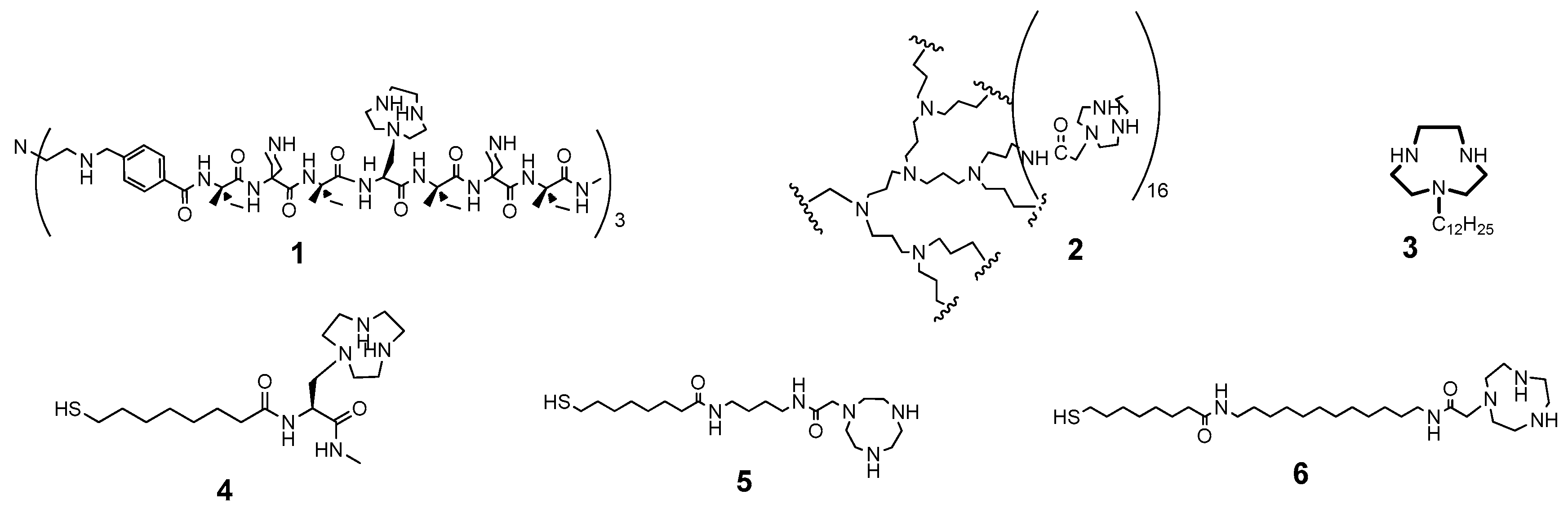{kind=link}
{kind=link}
| Ligand 1 | System | k2 or kcat/Km (s−1M−1) 2 | Kinetic Advantage | References |
|---|---|---|---|---|
| 1,4,7-Triazacyclononane | Monomer | 0.007 | 1 | [56,74] |
| 1 | Monomolecular trispeptide | 0.033 | 5 | [75] |
| 2 | Monomolecular dendrimer | 0.64 | 91 | [76] |
| 3 | Micelle | 0.028 | 4 | [56] |
| 4 | AuNP | 4.4 | 629 | [56] |
| 5 | AuNP | 21.7 | 3100 | [77] |
| 6 | AuNP | 638 | 91,143 | [57] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancin, F.; Prins, L.J.; Pengo, P.; Pasquato, L.; Tecilla, P.; Scrimin, P. Hydrolytic Metallo-Nanozymes: From Micelles and Vesicles to Gold Nanoparticles. Molecules 2016, 21, 1014. https://doi.org/10.3390/molecules21081014
Mancin F, Prins LJ, Pengo P, Pasquato L, Tecilla P, Scrimin P. Hydrolytic Metallo-Nanozymes: From Micelles and Vesicles to Gold Nanoparticles. Molecules. 2016; 21(8):1014. https://doi.org/10.3390/molecules21081014
Chicago/Turabian StyleMancin, Fabrizio, Leonard J. Prins, Paolo Pengo, Lucia Pasquato, Paolo Tecilla, and Paolo Scrimin. 2016. "Hydrolytic Metallo-Nanozymes: From Micelles and Vesicles to Gold Nanoparticles" Molecules 21, no. 8: 1014. https://doi.org/10.3390/molecules21081014