
Microwave-Assisted Syntheses of Bioactive Seven-Membered, Macro-Sized Heterocycles and Their Fused Derivatives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
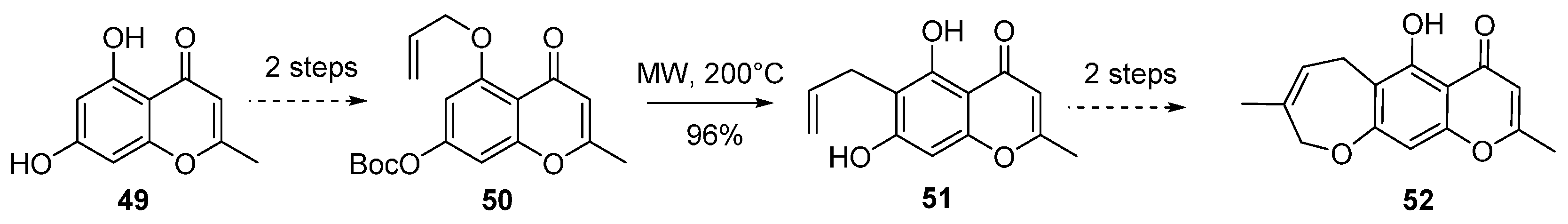{kind=link}
{kind=link}
{kind=link}
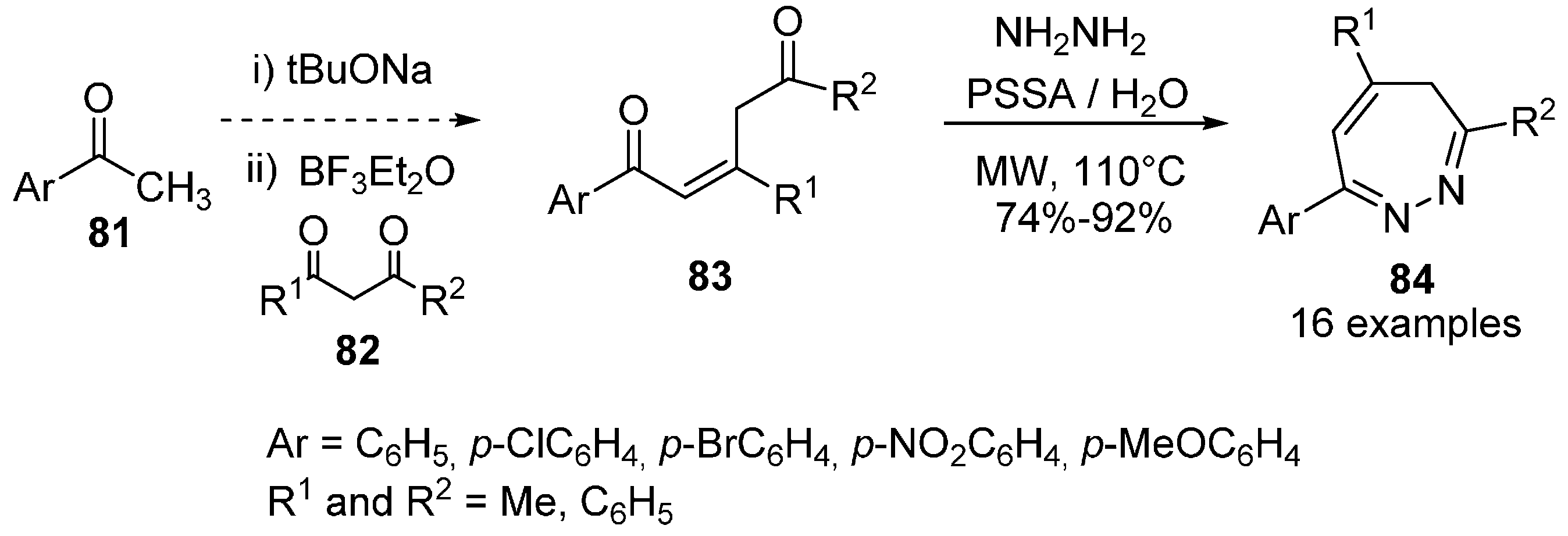{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
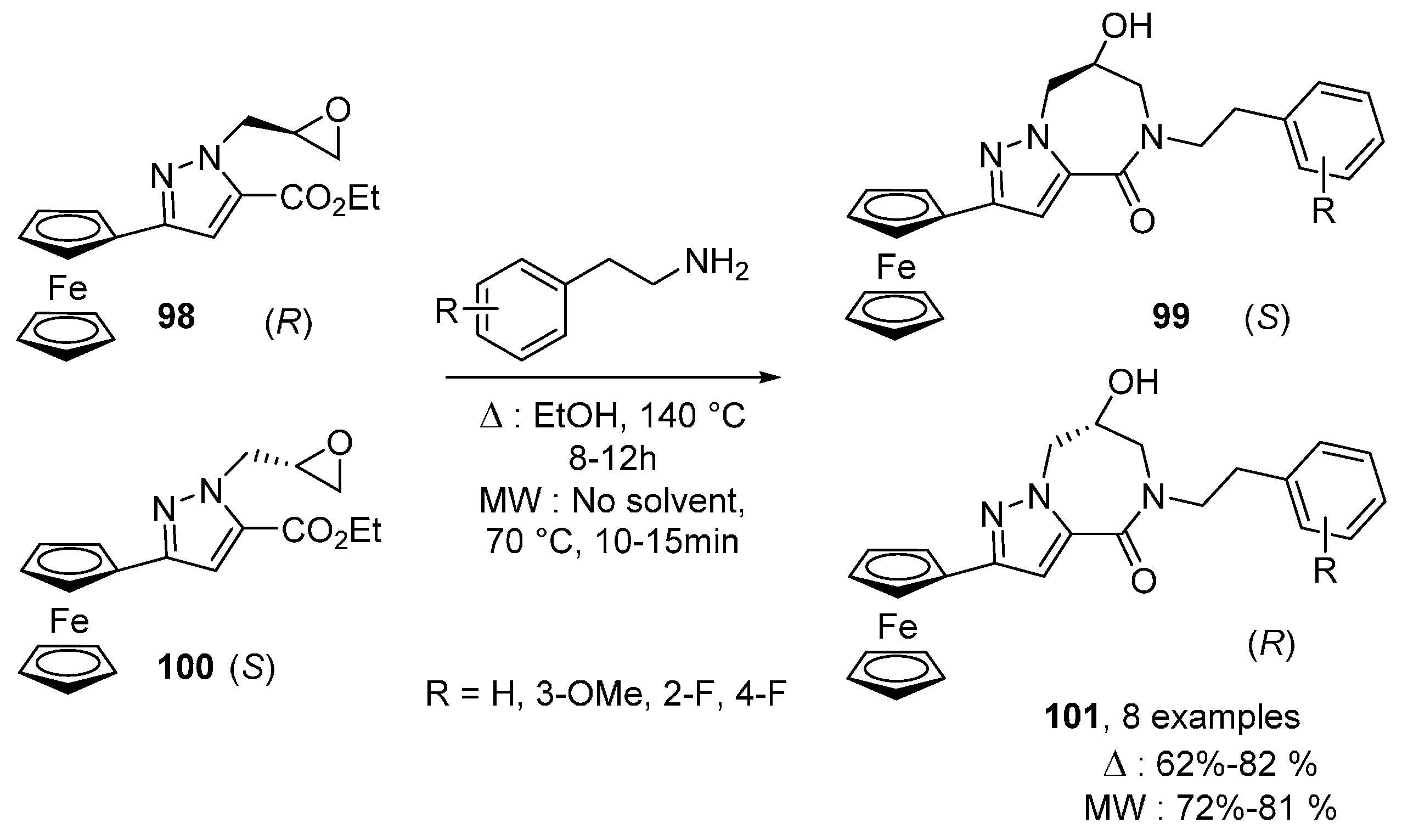{kind=link}
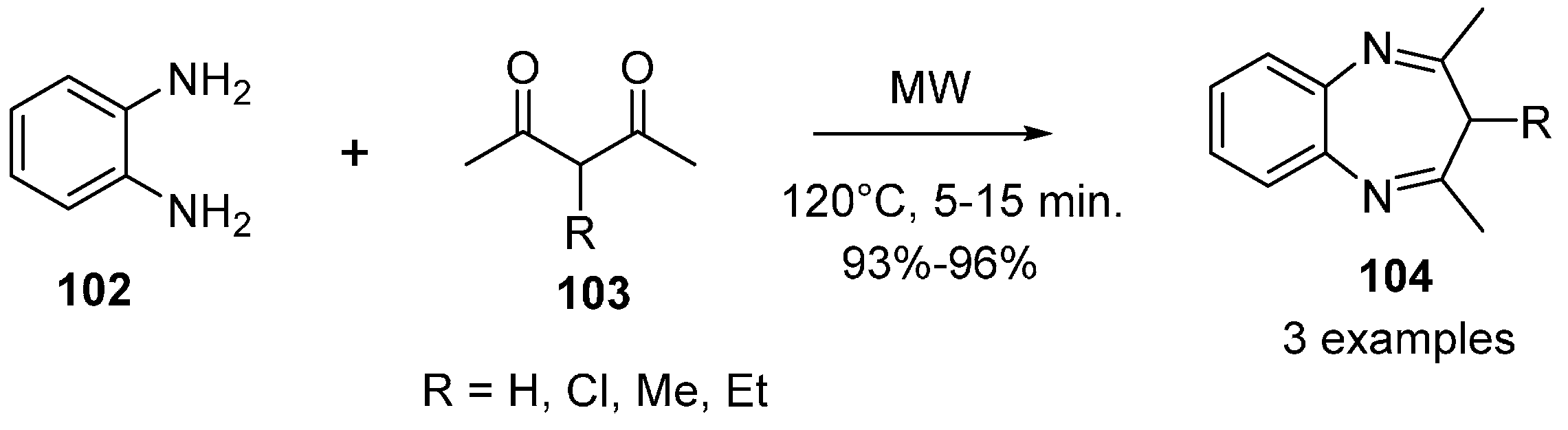{kind=link}
{kind=link}
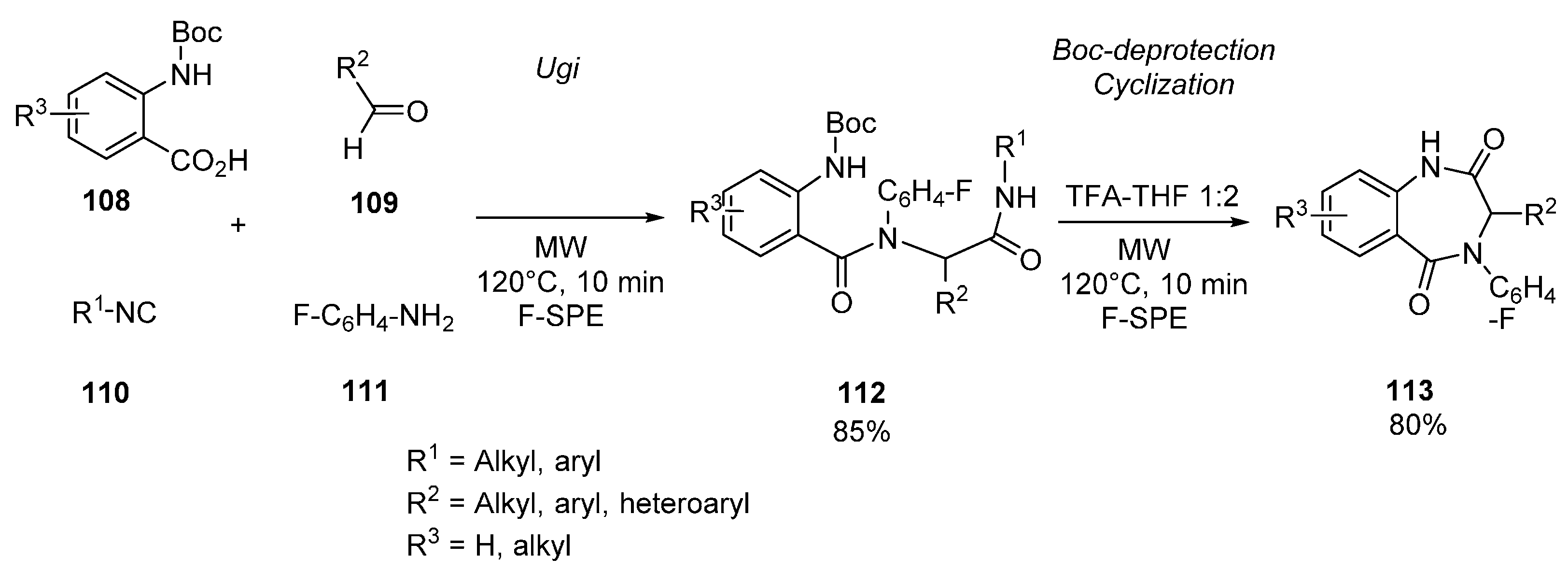{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
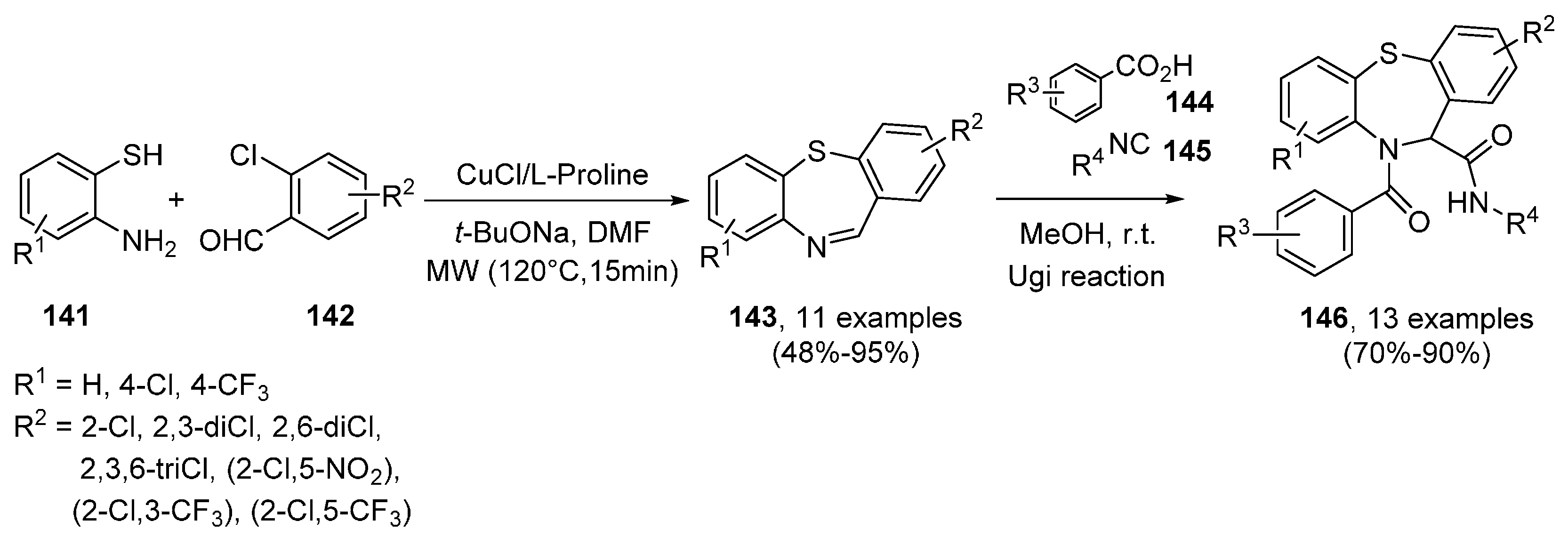{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
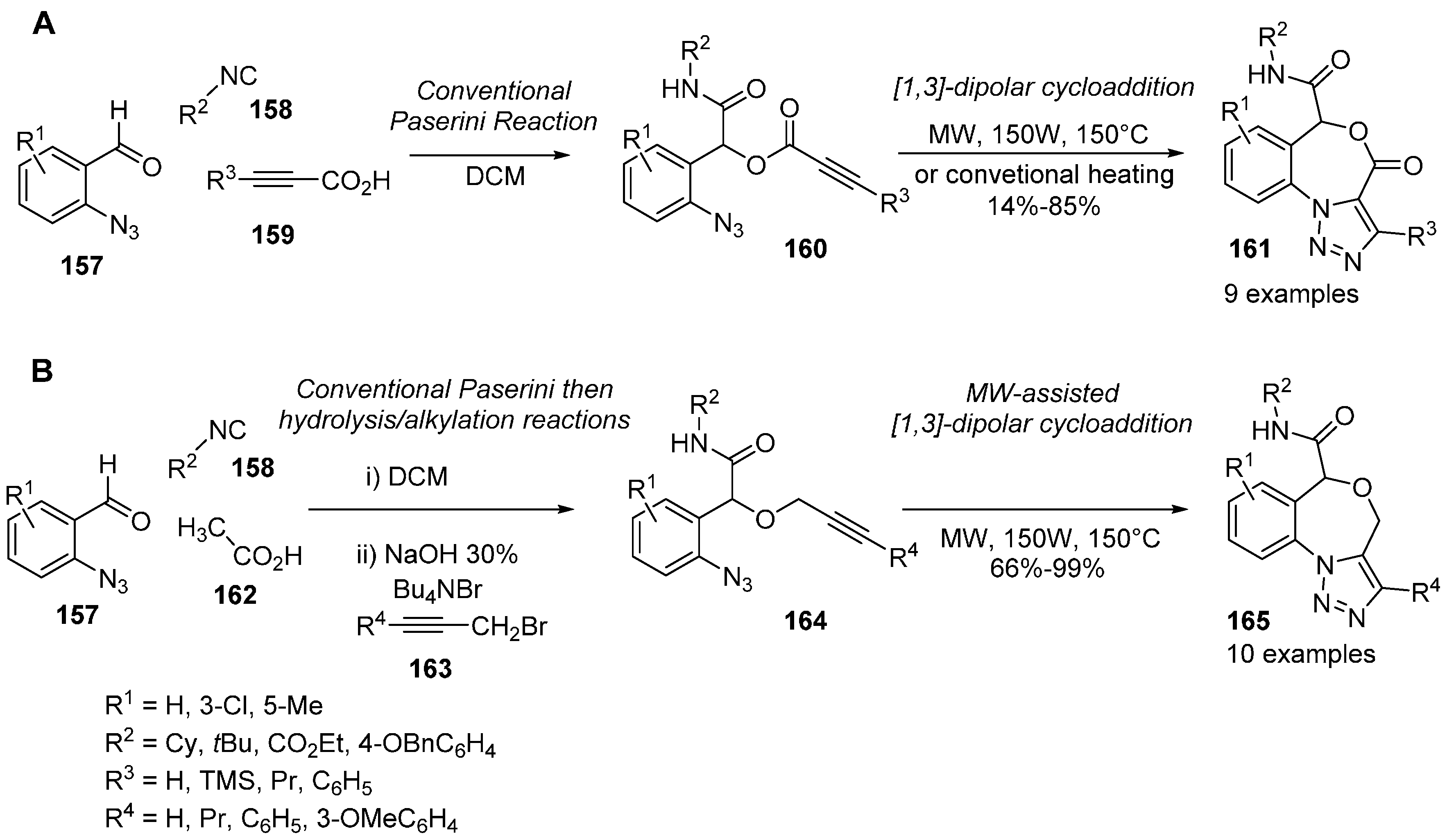{kind=link}
{kind=link}
{kind=link}
{kind=link}
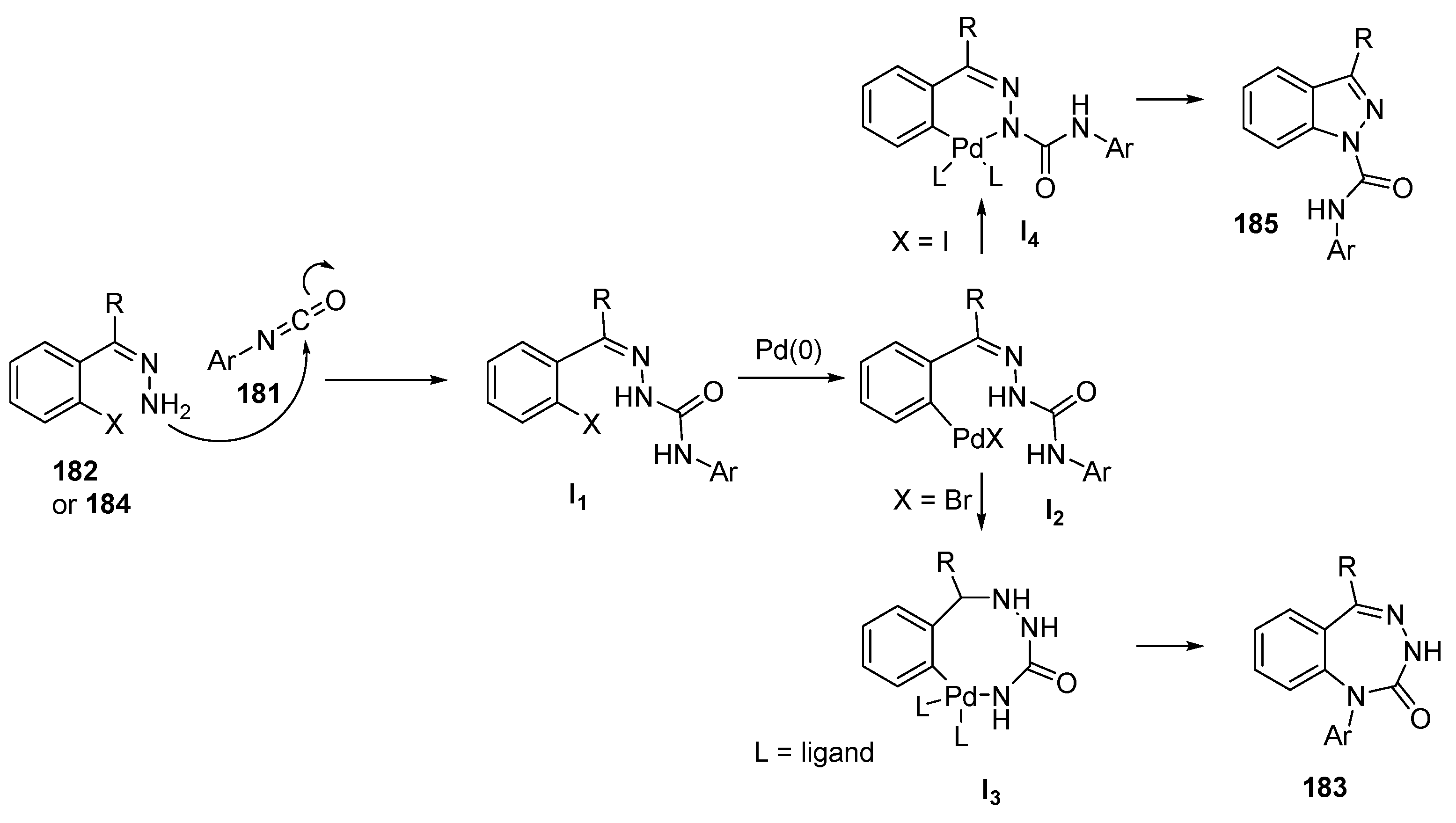{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthesis of Heterocyclic Compounds
2.1. Seven-Membered Heterocycles
2.1.1. Seven-Membered Heterocycles with One Heteroatom
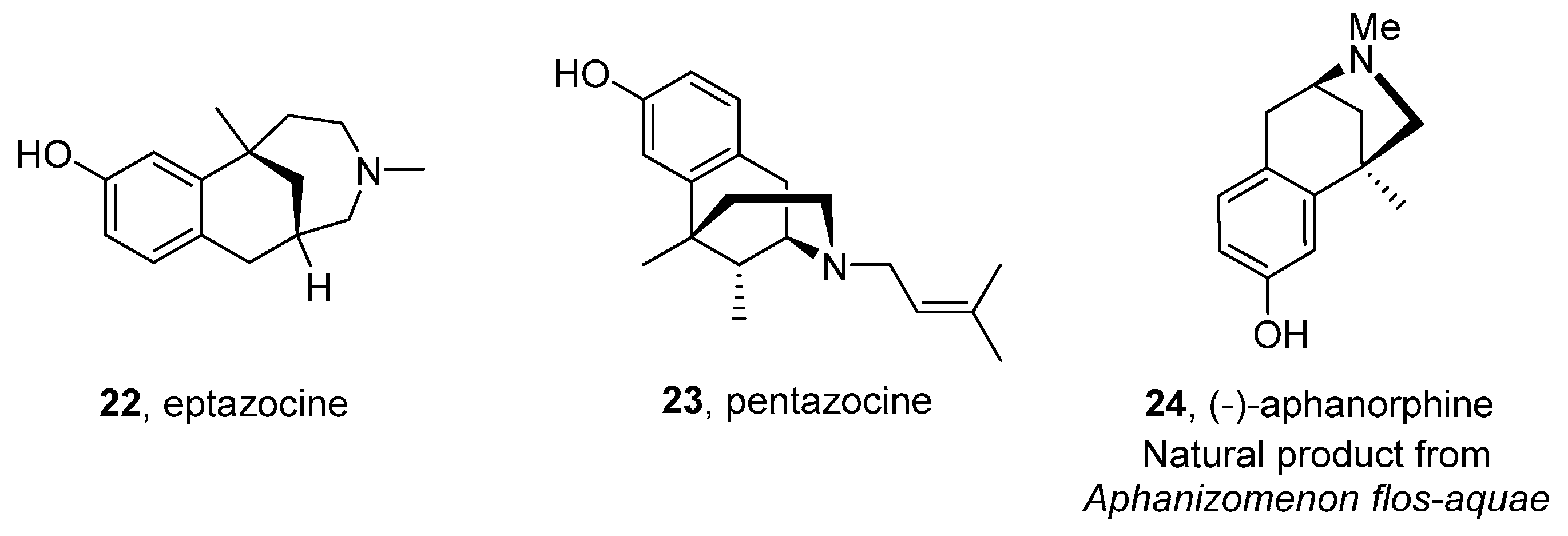
Benzazepines and Fused Analogues
2-Benzazepines and Their Derivatives
3-Benzazepines and Their Derivatives
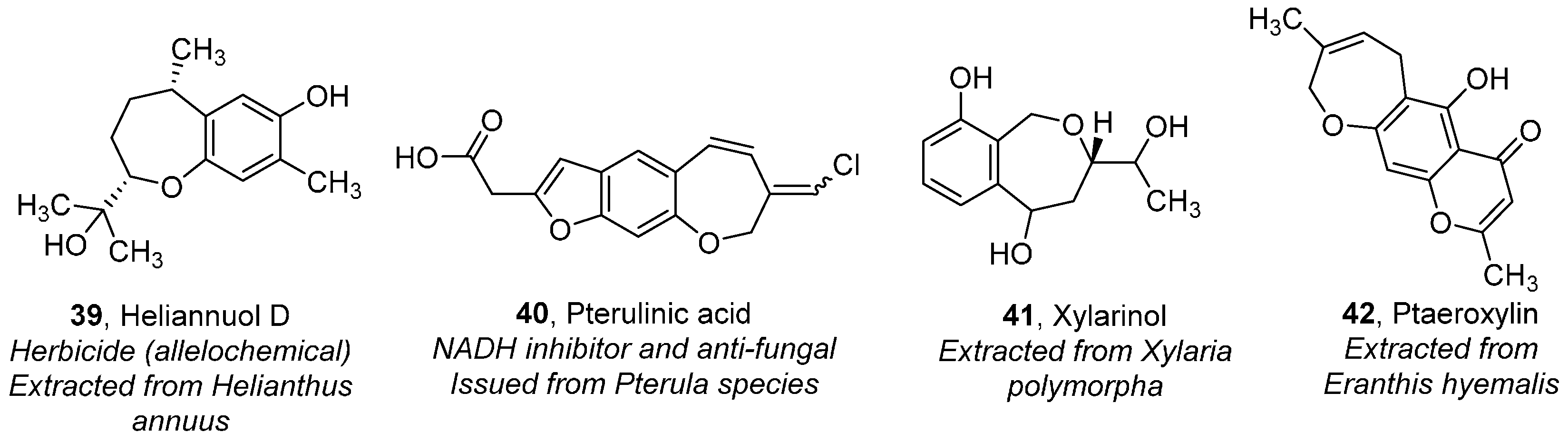
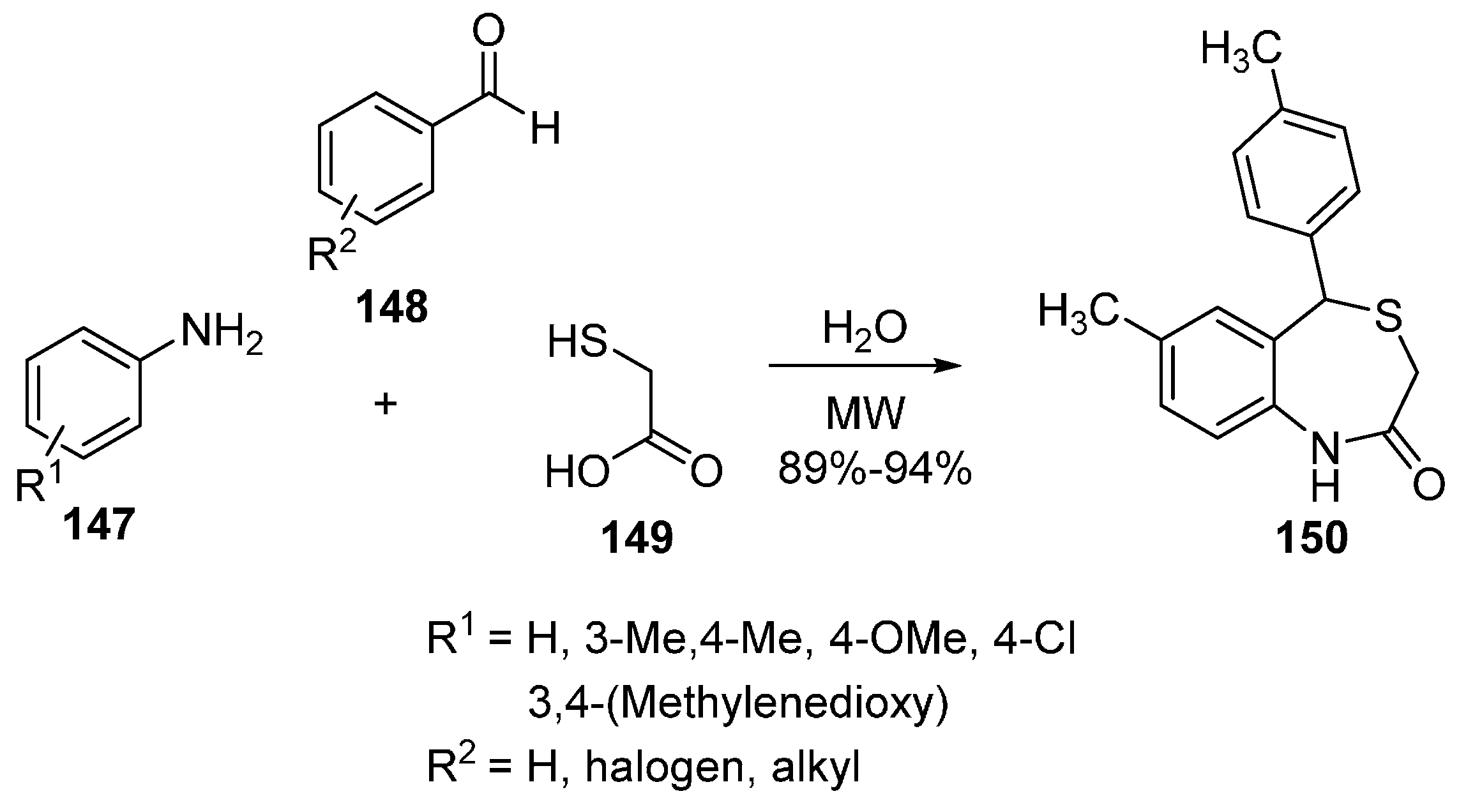
Oxepanes, Thiepanes and Their Derivatives
2.1.2. Seven-Membered Heterocycles with Two Heteroatoms
Seven-Membered Heterocycles with Two Nitrogen atoms
Diazepines
Diazepines Fused with Five-Membered Cycles
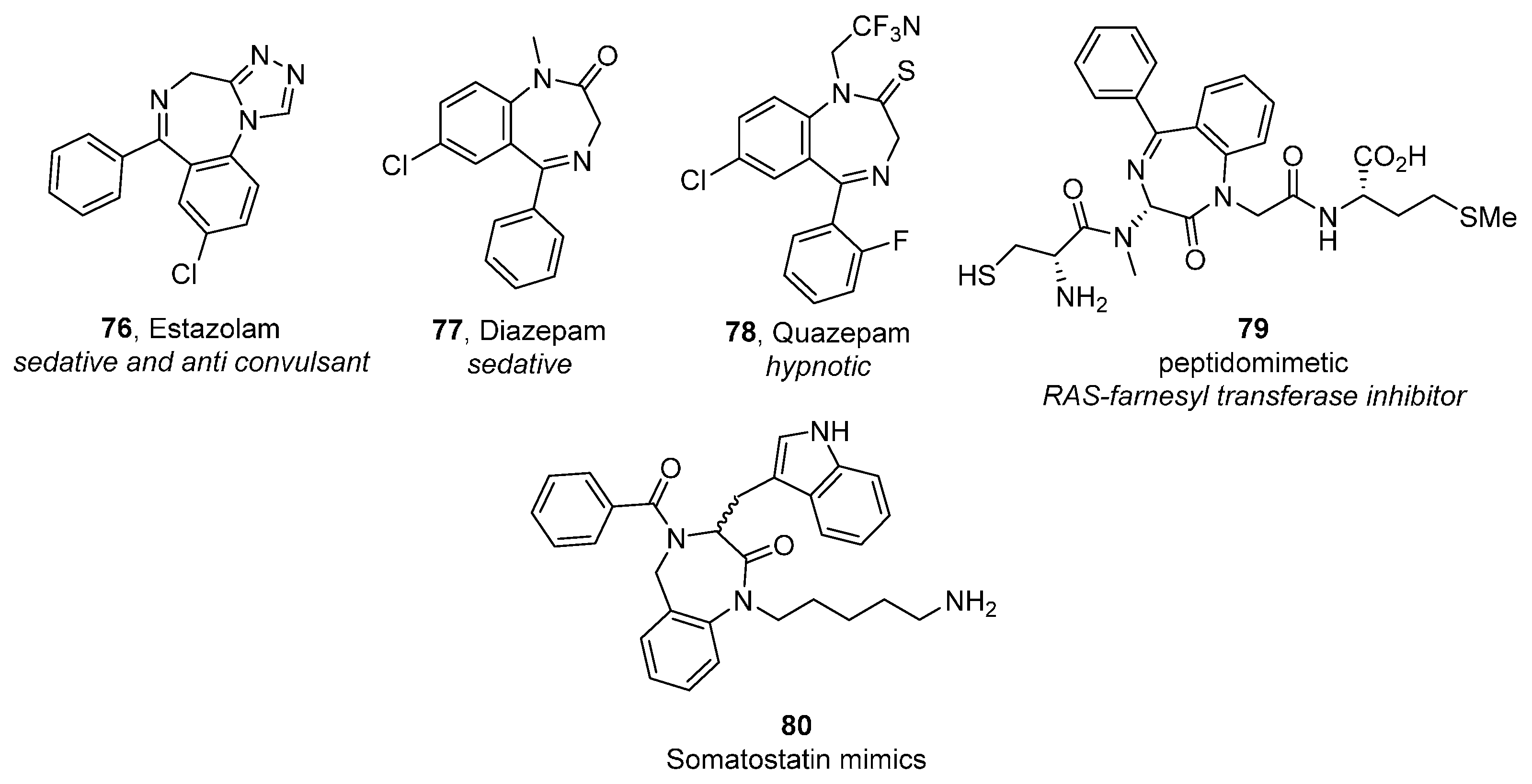
Benzodiazepines and Their Fused Analogues
Seven-Membered Heterocycles with Two Distinct Heteroatoms
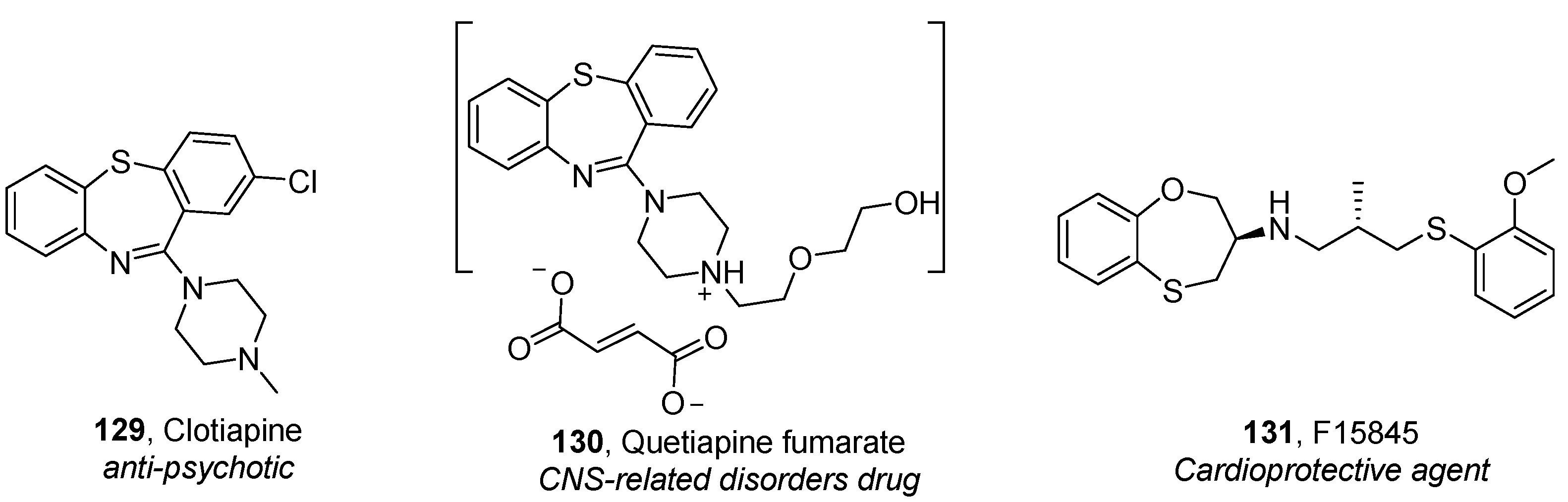
Benzothiazepines, Benzothiepines and Their Derivatives
Oxazepines and Their Derivatives
2.1.3. Seven-Membered Heterocycles with Three Heteroatoms
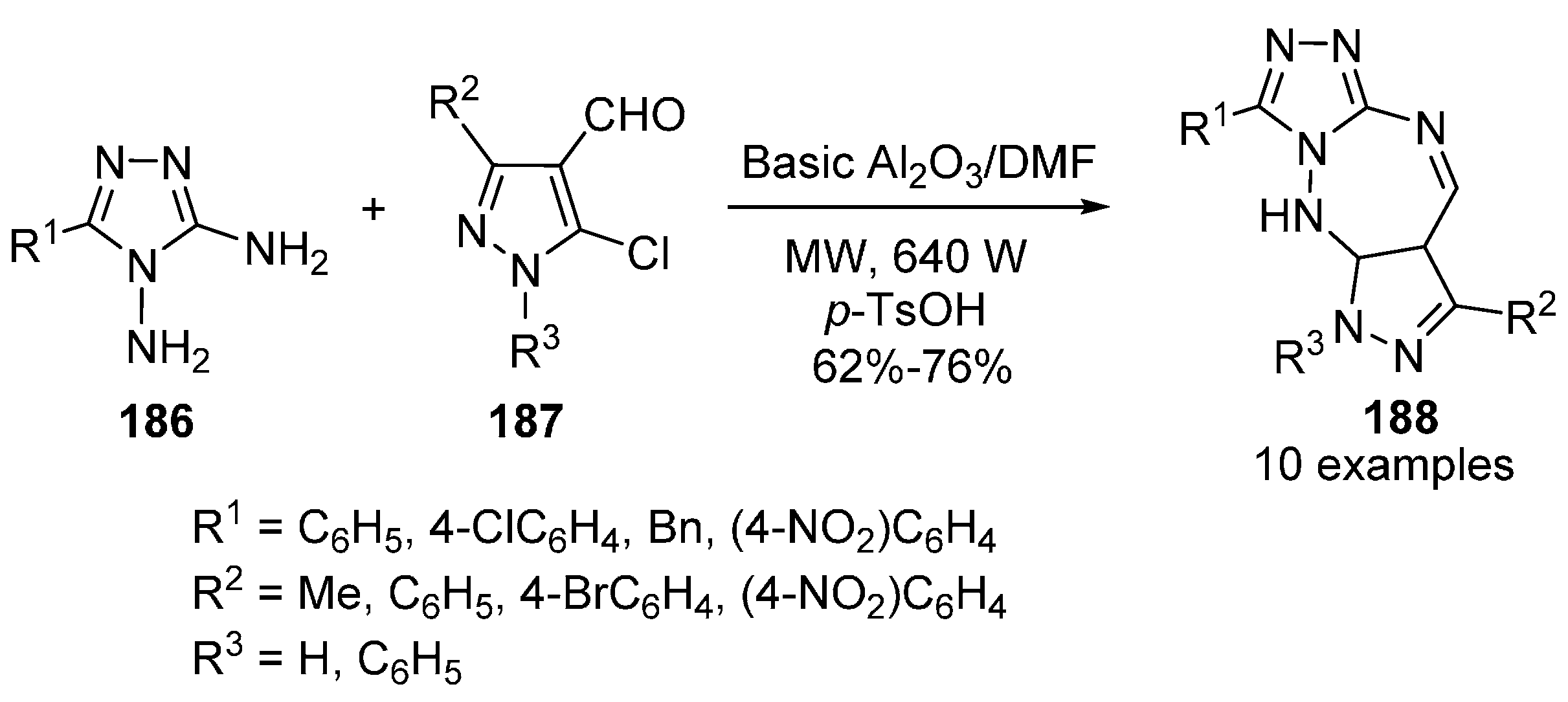
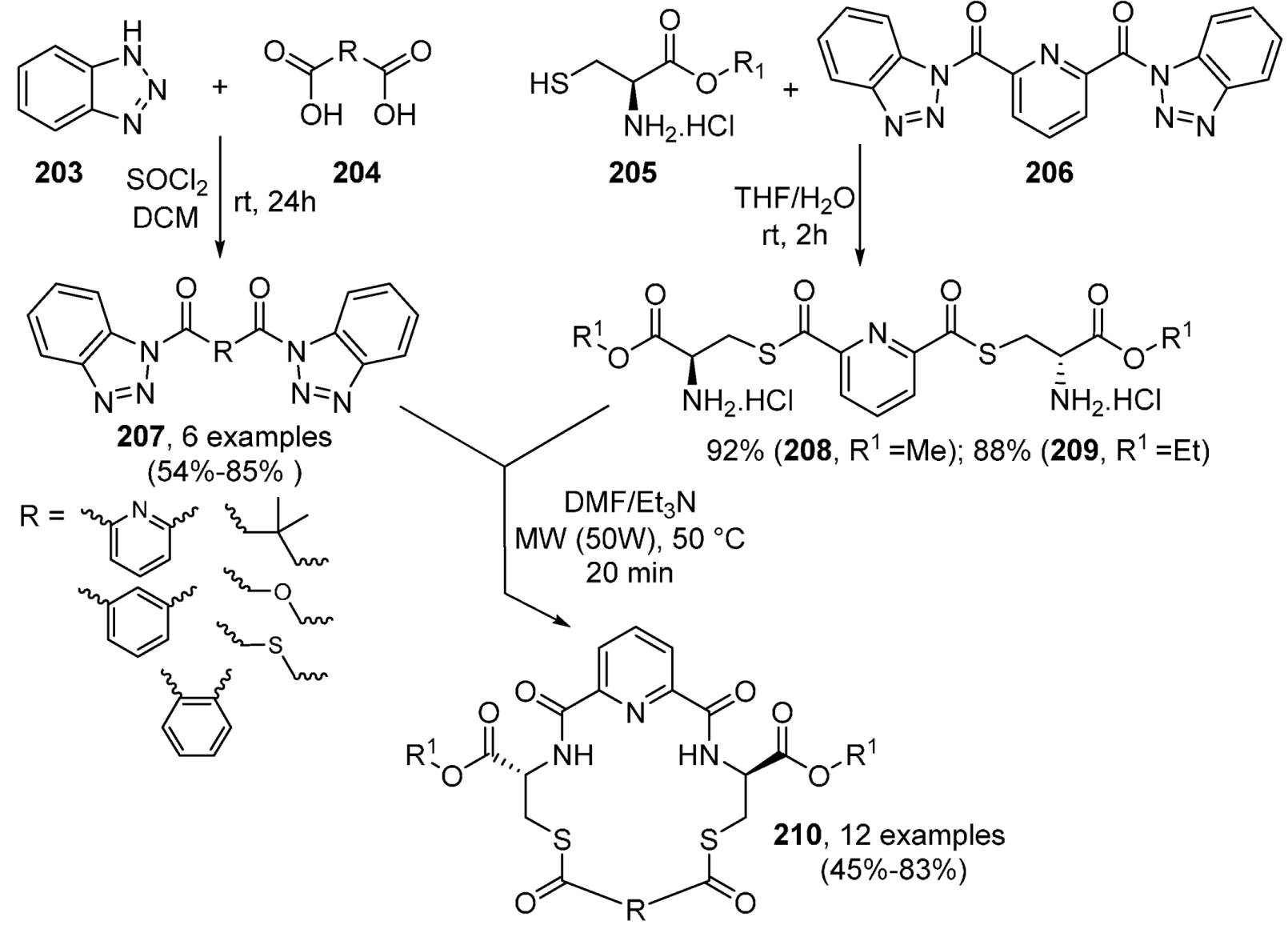
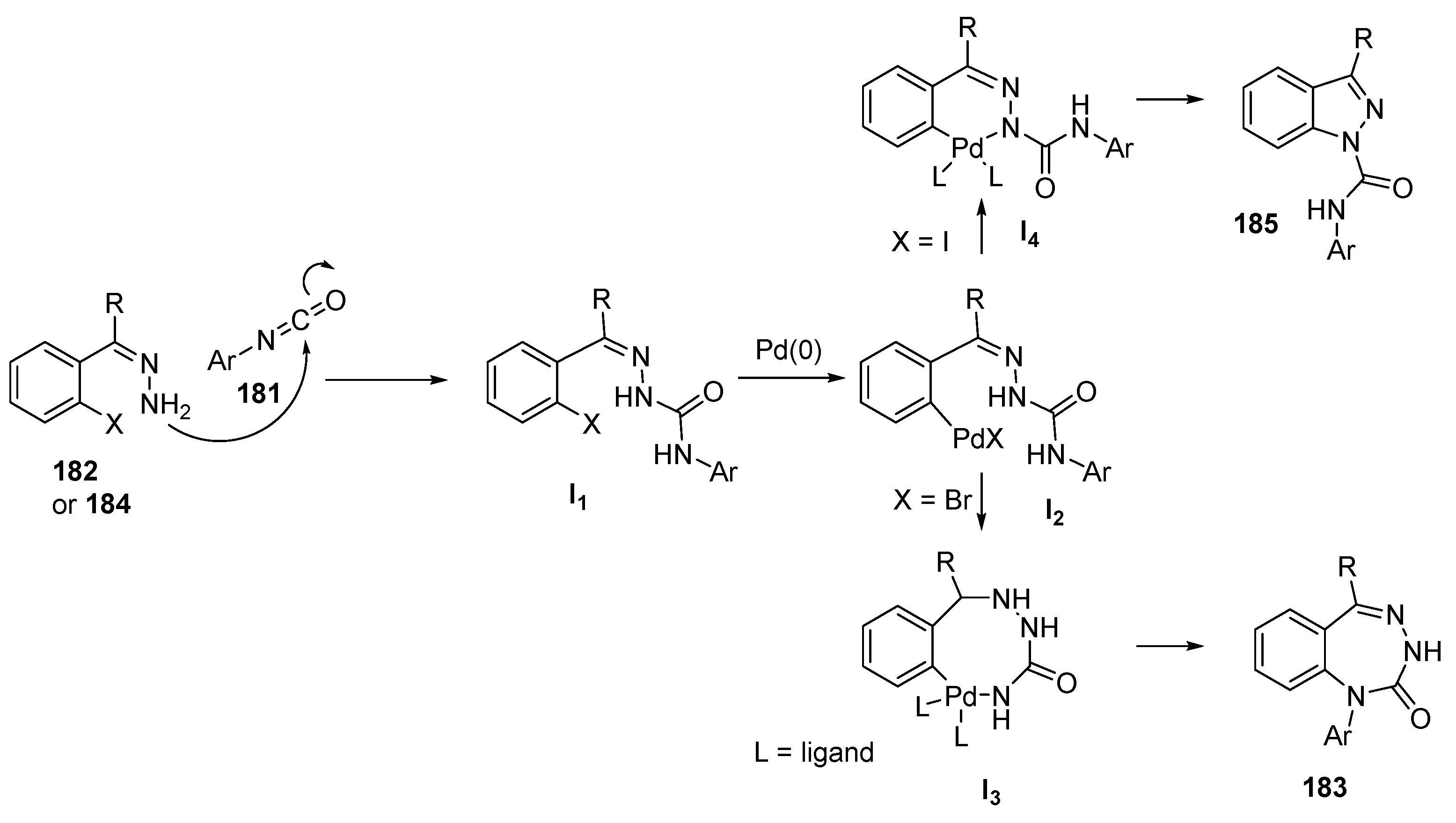
Benzotriazepines and Their Fused Analogues
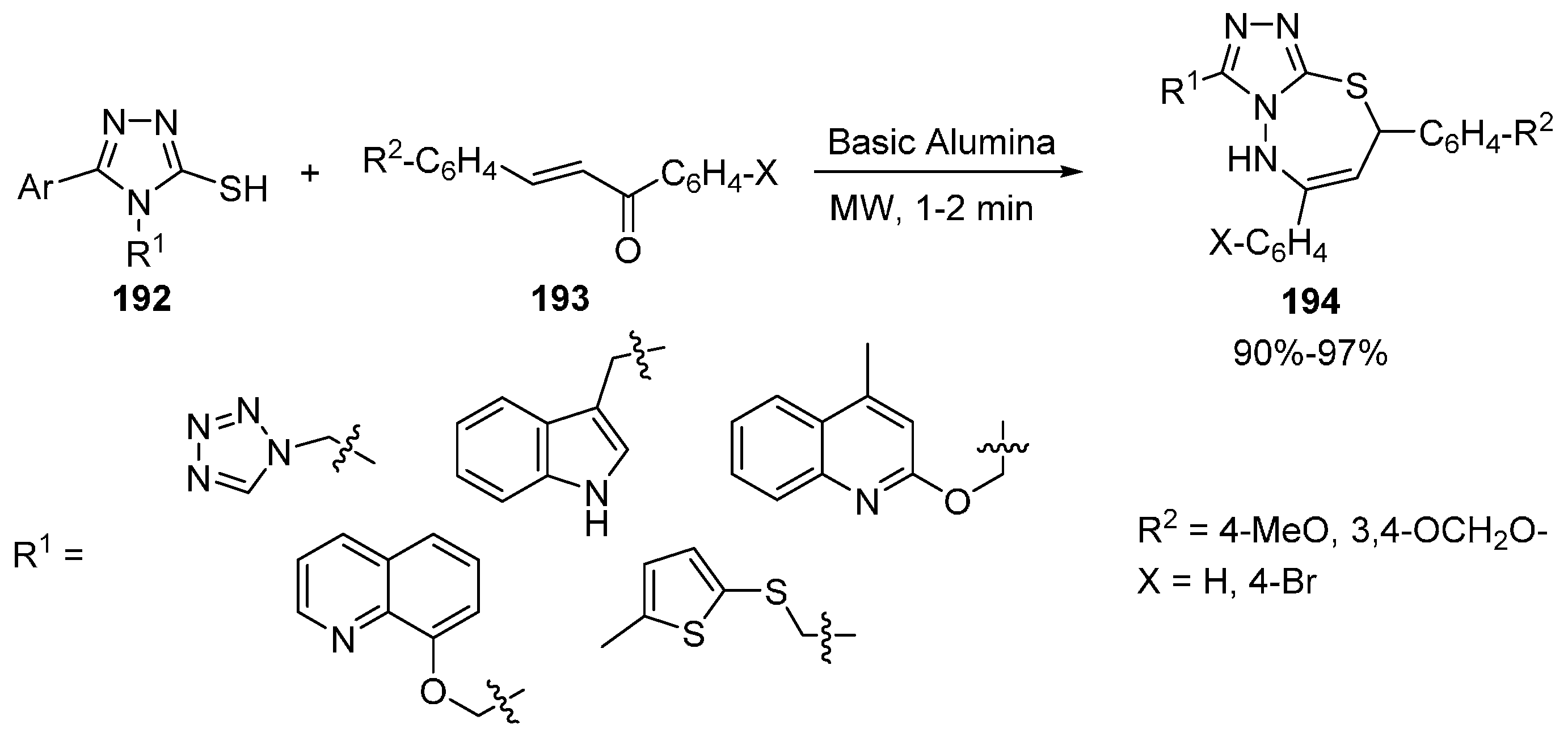
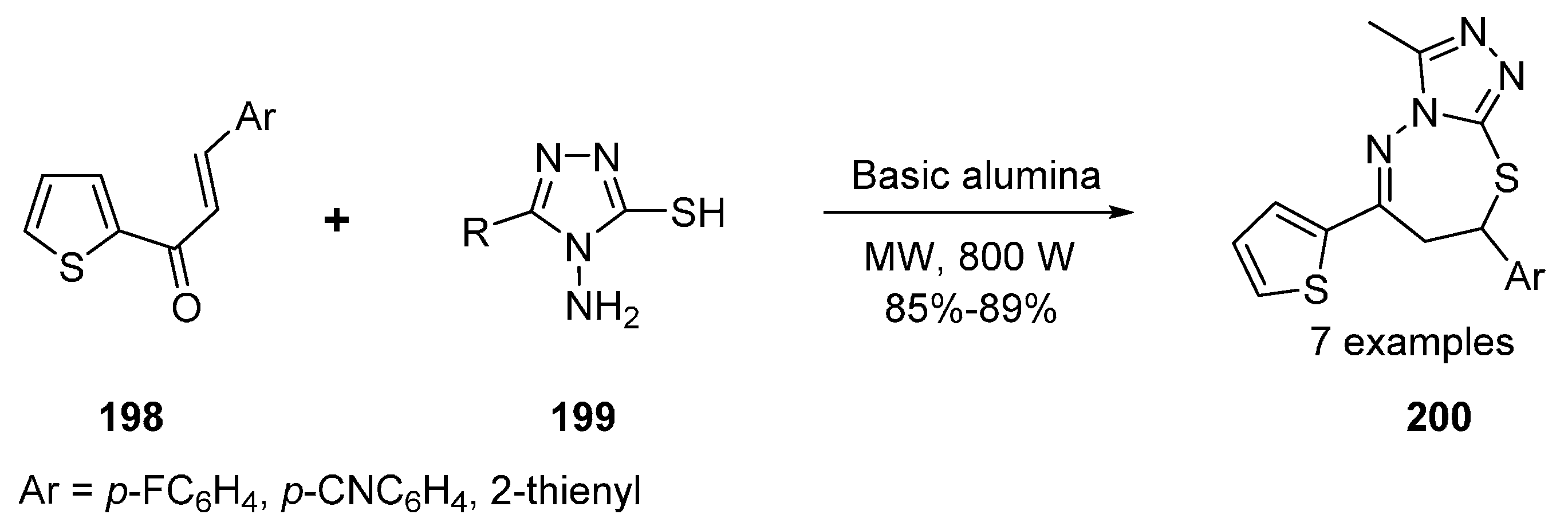
Thiadiazepines and Benzothiadiazepines
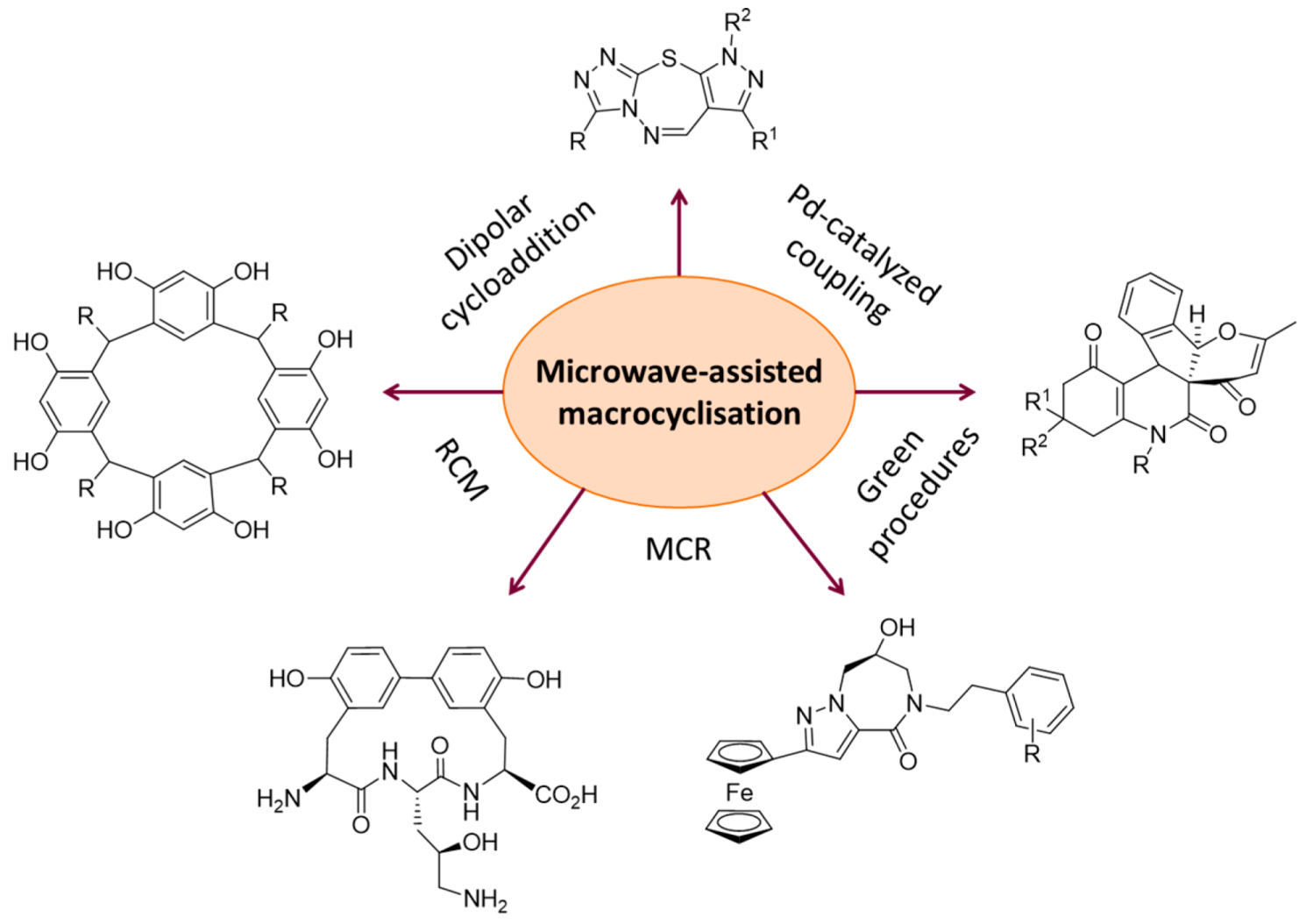
2.2. Synthesis of Other Macroheterocycles
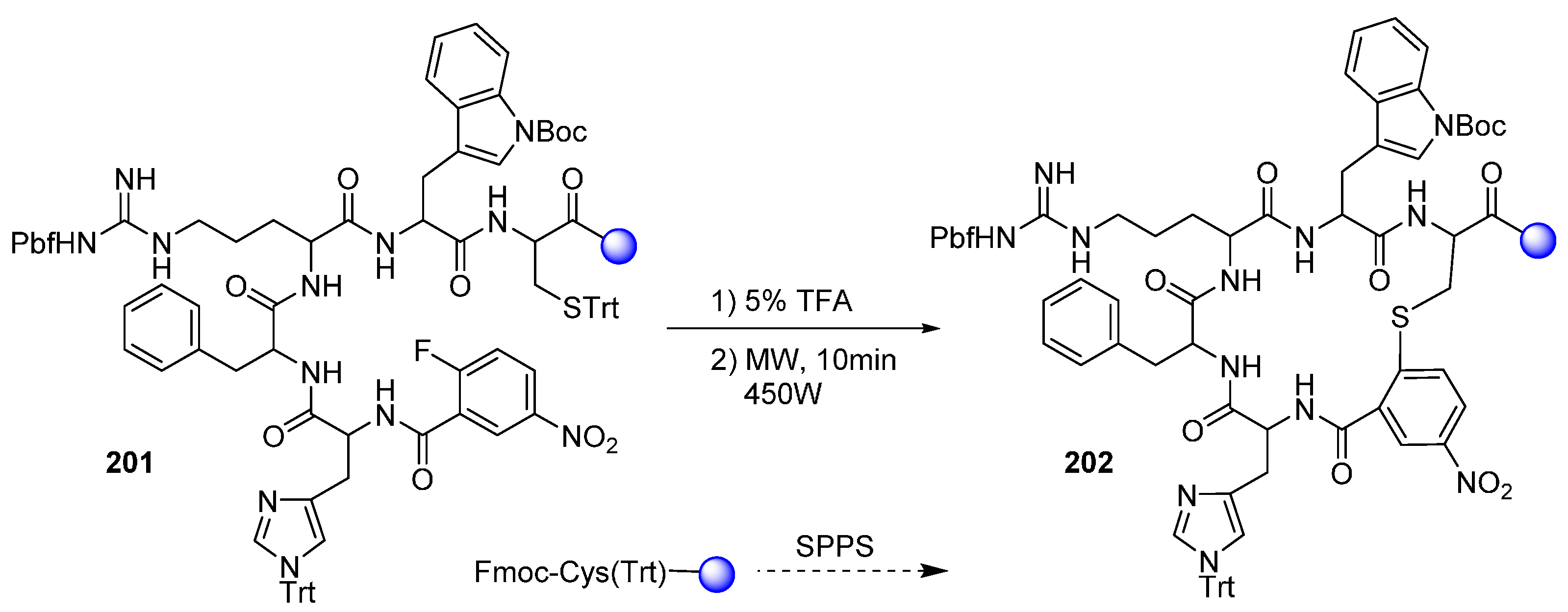
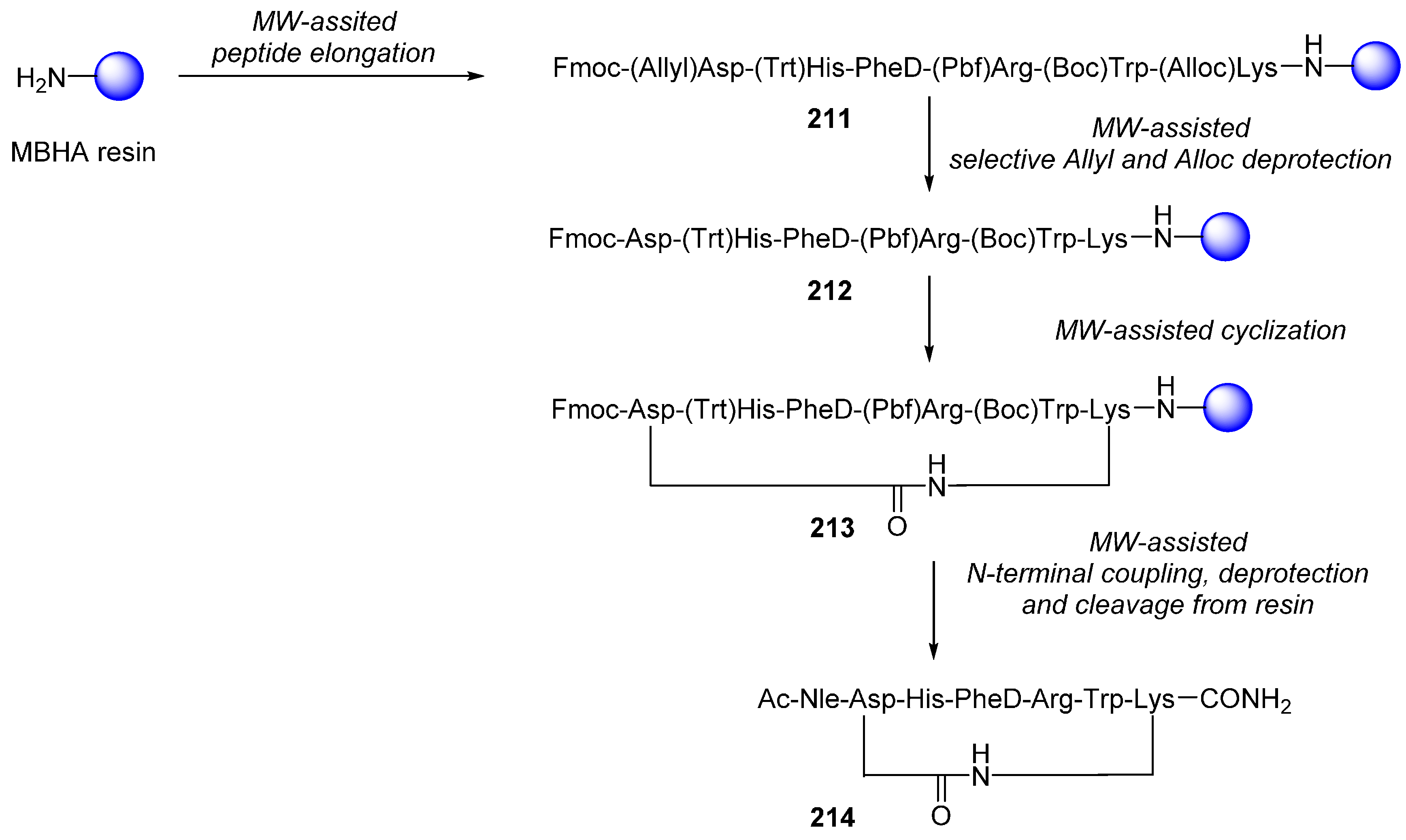
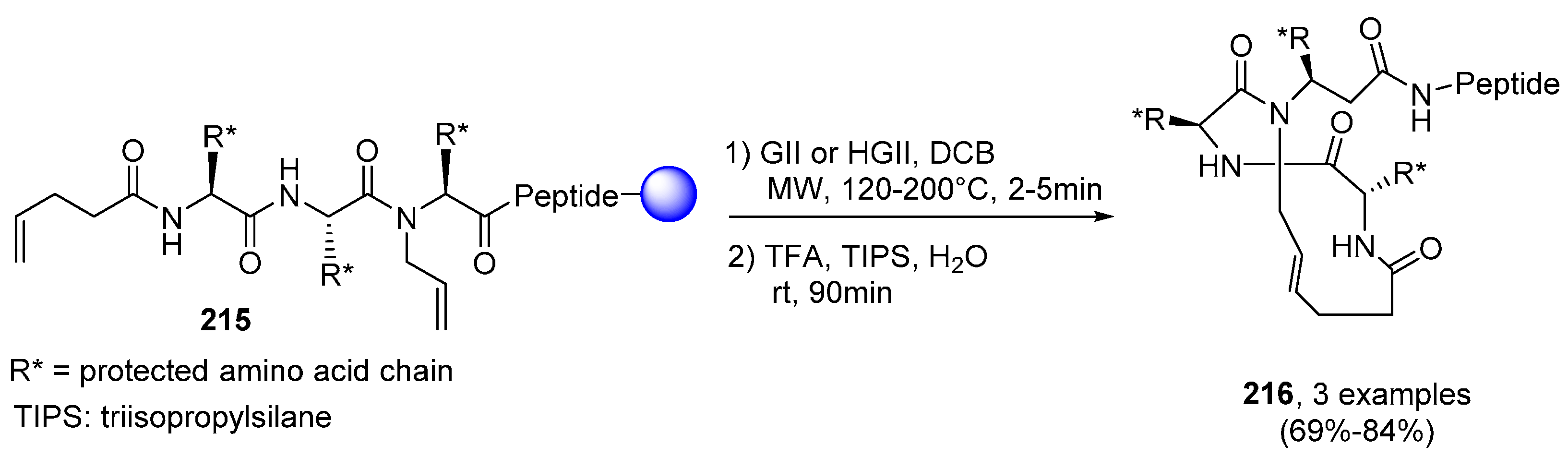
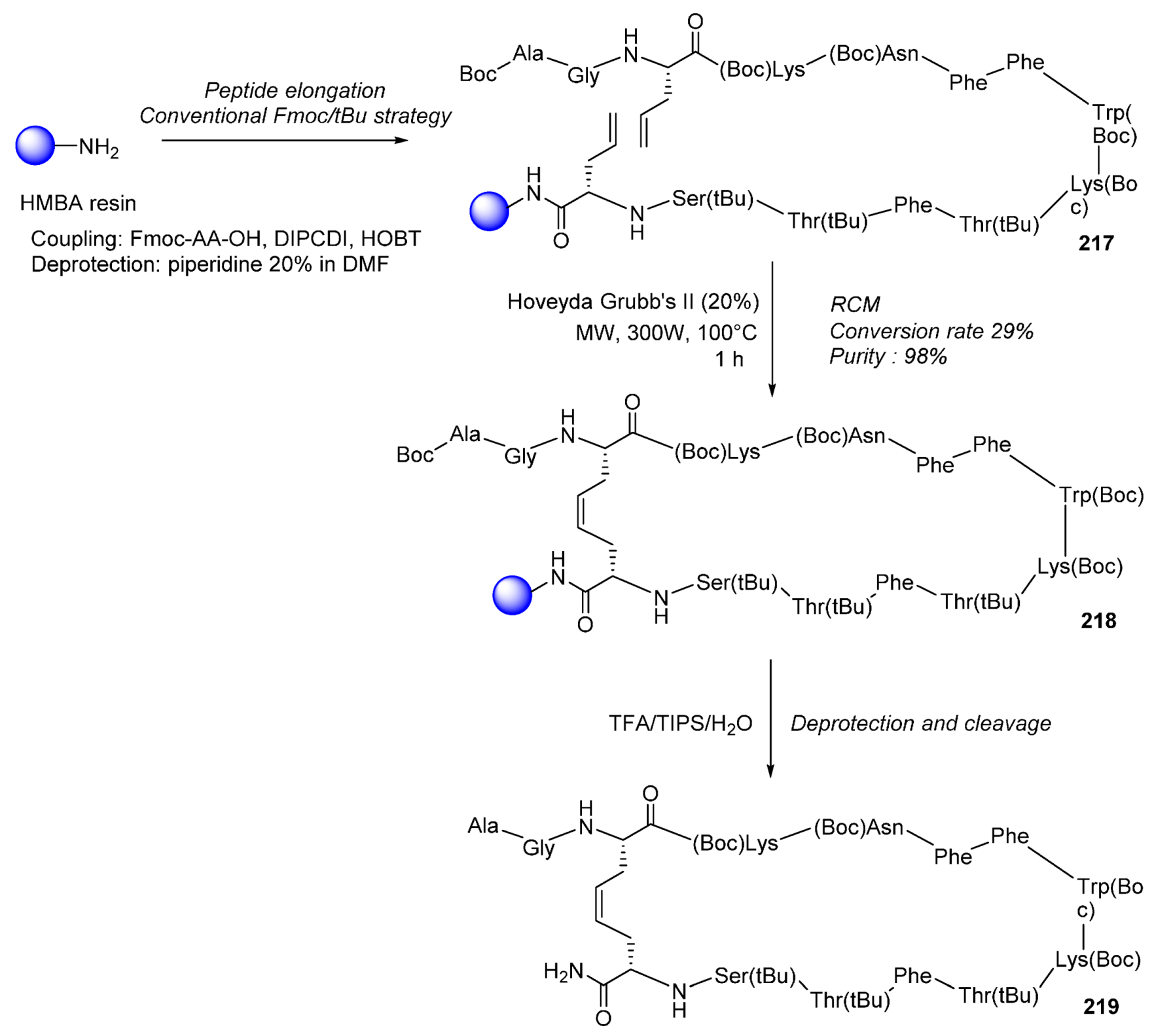
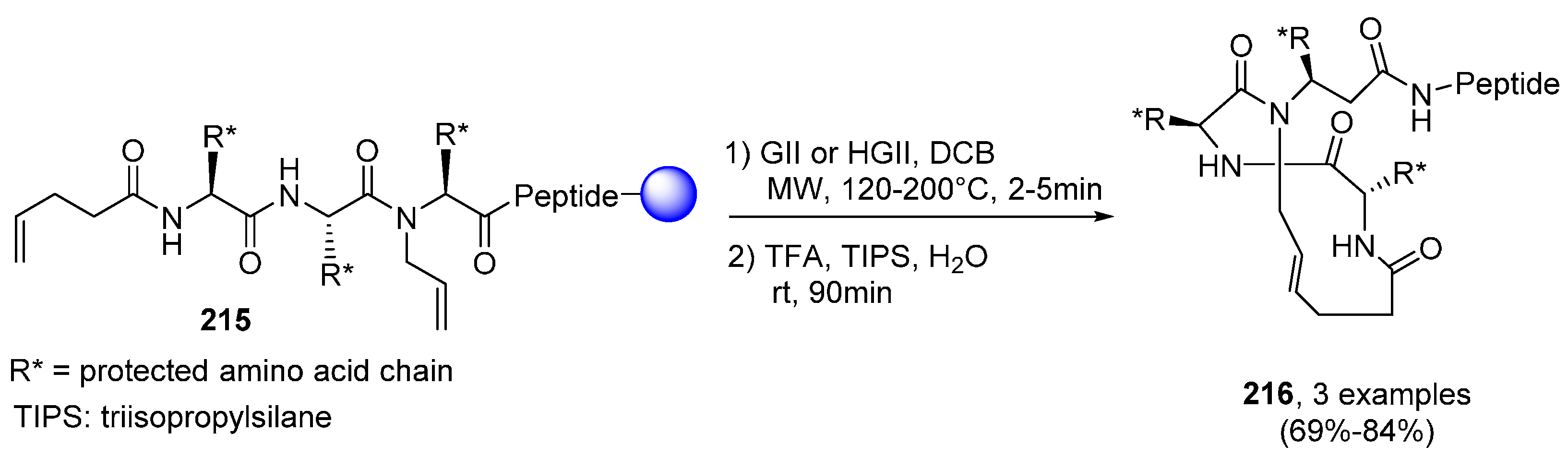
2.2.1. Cyclic Peptides and Pseudo-Peptides: MW-Irradiation Applied to SPPS
2.2.2. MW-Assisted Syntheses of Macrocyclic Natural Products and Analogues
2.2.3. MW-Assisted Syntheses of Phthalocyanines
2.2.4. MW-Assisted Syntheses of Calix-Type Derivatives
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ac | Acetyle |
| Ala | Alanine |
| Alloc | Allyloxycarbonyl |
| Arg | Arginine |
| Asn | Asparagine |
| Asp | Aspartic acid |
| BET | Bacterial endotoxin test |
| BINAP | 2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl |
| Bn | Benzyl group |
| Boc | tert-Butyloxycarbonyl |
| Bu | Butyl |
| Cbz | Carboxybenzyl |
| CCK2 | Cholecystokinin B |
| CNS | Central Nervous System |
| Cy | Cyclohexyl |
| Cys | Cysteine |
| Cu(Phen)(PPh3)Br | Bromo-(1,10-phenanthroline-N,N′)(triphenylphosphine)cuprate |
| dba | Dibenzylideneacetone |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCB | 1,4-Dichlorobenzene |
| DDA | Diketene acetone adduct |
| DCM | Dichloromethane |
| DIEA | N,N-Diisopropylethylamine |
| DIPCDI | N,N-Diisopropylcarbodiimide |
| DMAE | Dimethylethanolamine |
| DMA | Dimethylacetamide |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| Dppf | 1,1′-Bis(diphenylphosphino)ferrocene |
| dr | Diastereomeric ratio |
| ee | Enantiomeric excess |
| eq. | Equivalent |
| Et | Ethyl |
| Fmoc | Fluorenylmethyloxycarbonyl |
| F-SPE | Fluorine solid phase extraction |
| EtOH | Ethanol |
| GII | Grubbs II catalyst |
| Gly | Glycine |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid hexafluorophosphate |
| HBS | Hydrogen-bond surrogate |
| HBTU | 2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| HCT 116 | Human colon carcinoma cells |
| His | Histidine |
| HMBA | Hydroxymethylbenzoic acid |
| HGII | Hoveyda-Grubbs II catalyst |
| HOBt | Hydroxybenzotriazole |
| IC50 | Half maximal inhibitory concentration |
| IL | Ionic Liquid |
| KSAc | Potassium thioacetate |
| Lys | Lysine |
| MBHA | 4-Methylbenzhydryl amine |
| m-CPBA | meta-Chloroperoxybenzoic acid |
| MCR | Multi-component reaction |
| Me | Methyl |
| MeOH | Methanol |
| MIC | Minimum Inhibitory Concentration |
| Ms | Mesyl |
| mTOR | Mammalian Target of Rapamycin |
| min | Minute |
| MW | Microwave |
| NADH | Nicotinamide Adenine Dinucleotide Hydride |
| Nle | Norleucine |
| Pbf | 2,2,4,6,7-Pentamethyldihydrobenzofuran-5-sulfonyl |
| Pd/C | Palladium on carbon |
| PEG | Polyethylene glycol |
| Ph | Phenyl |
| Phe | Phenylalanine |
| PheD | D-Phenylalanine |
| PMB | p-Methoxybenzyl |
| PPE | Polyphosphoric ester |
| PPh3 | Triphenylphosphine |
| Pr | Propyl |
| PSSA | Polystyrene sulfonic acid |
| PTH1R | Parathyroid Hormone 1 Receptor |
| PyBOP | Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate |
| p-TsOH | para-Toluenesulfonic acid |
| RCM | Ring closure metathesis |
| r.t | Room temperature |
| Ser | Serine |
| SES | The 2-(trimethylsilyl)ethanesulfonyl group |
| SNAr | Nucleophilic aromatic substitution |
| SPPS | Solid Phase Peptide Synthesis |
| TBAB | Tetrabutylammonium bromide |
| TFA | Trifluoroacetic acid |
| Thr | Threonine |
| TIPS | Triisopropylsilyl |
| TMS | Trimethylsilyl |
| Trp | Tryptophan |
| Trt | Trityl (triphenylmethyl) |
| Ts | Tosyl |
| THF | Tetrahydrofuran |
| XO | Xanthine Oxidase |
References
- Kappe, C.O.; Dallinger, D. Controlled microwave heating in modern organic synthesis: highlights from the 2004–2008 literature. Mol. Divers. 2009, 13, 71–193. [Google Scholar] [CrossRef] [PubMed]
- De La Hoz, A.; Loupy, A. Microwave in Organic Synthesis, 3rd ed.; Wiley-WCH: Weinheim, Germany, 2012. [Google Scholar]
- Saber, A.; Marzag, H.; Benhida, R.; Bougrin, K. Microwave-assisted cycloaddition reactions in carbo- and heterocyclic chemistry. Curr. Org. Chem. 2014, 18, 2139–2180. [Google Scholar] [CrossRef]
- Driowya, M.; Saber, A.; Marzag, H.; Demange, L.; Benhida, R.; Bougrin, K. Microwave-assisted synthesis of bioactive six-membered heterocycles and their fused analogues. Molecules 2016, 21, 492. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.H.; Hindupur, R.M.; Pati, H.N. Pharmacological and biological activities of benzazepines: An overview. Curr. Bioact. Compd. 2015, 11, 170–188. [Google Scholar] [CrossRef]
- Lin, H.-C.; Chiou, G.; Chooi, Y.-H.; MacMahon, T.C.; Xu, W.; Garg, N.K.; Tang, Y. Elucidation of the concise biosynthetic pathway of the communesin indole alkaloids. Angew. Chem. 2015, 54, 3004–3007. [Google Scholar] [CrossRef] [PubMed]
- Gozler, T.; Gozler, B.; Weiss, I.; Freyer, A.J.; Shamma, M. (+)-Turkiyenine: An unusual extension of the biogenetic sequence for the isoquinoline alkaloids. J. Am. Chem. Soc. 1984, 106, 6101–6102. [Google Scholar] [CrossRef]
- Li, H.; Wen, Y.; Wang, F.; Wu, P.; Wei, X. Cephalofortunone, a structurally unique cephalotaxus alkaloid from Cephalotaxus fortune Hook. F. Tetrahedron Lett. 2015, 56, 5735–5737. [Google Scholar] [CrossRef]
- Chung, H.-S.; Hon, P.-M.; Lin, G.; But, P.P.-H.; Dong, H. Antitussive activity of Stemona alkaloids from Stemona tuberosa. Planta Med. 2003, 69, 914–920. [Google Scholar] [PubMed]
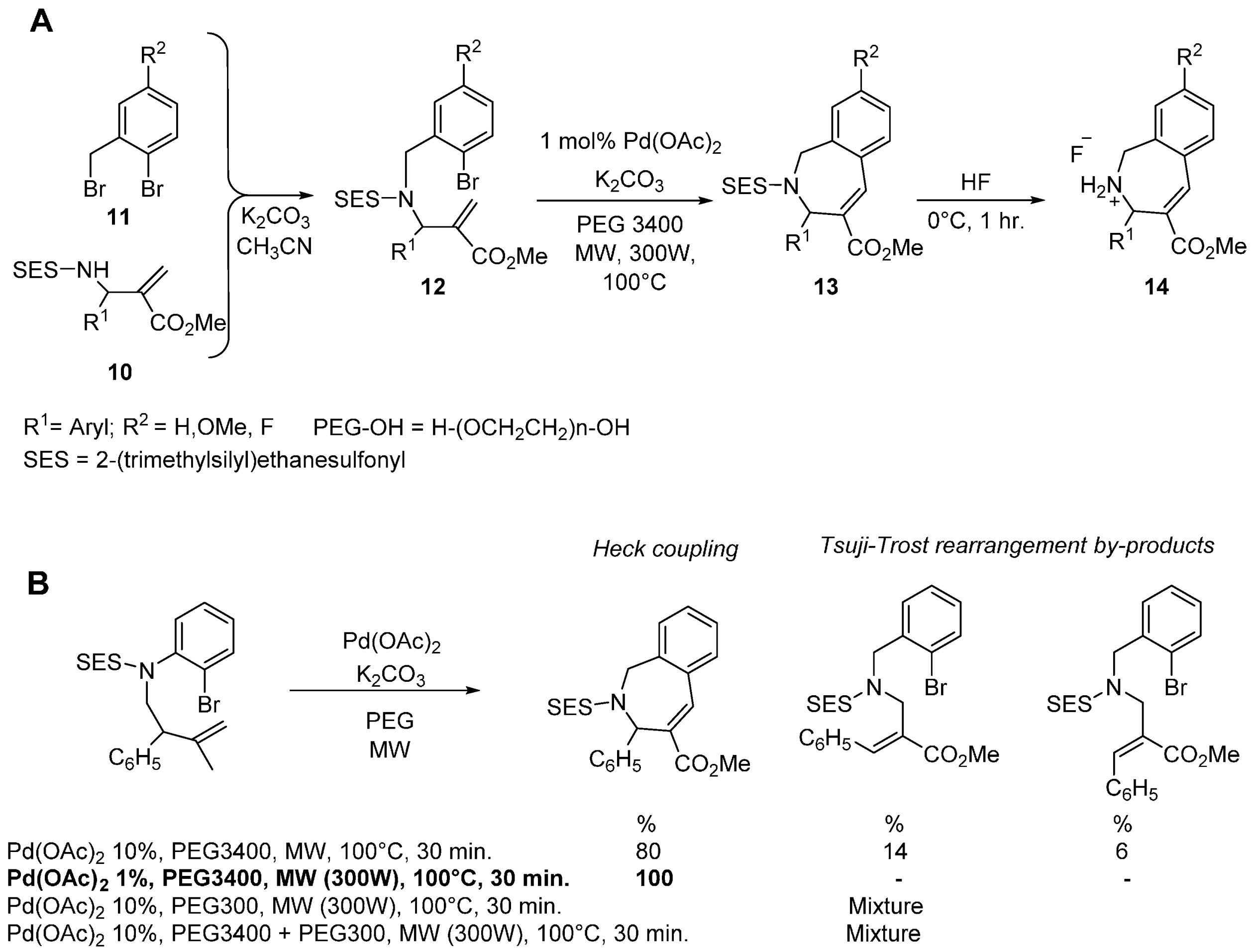
- Declerck, V.; Ribière, P.; Nédellec, Y.; Allouchi, H.; Martinez, J.; Lamaty, F. A Microwave-Assisted Heck Reaction in Poly(ethylene glycol) for the Synthesis of Benzazepines. Eur. J. Org. Chem. 2007, 38, 201–208. [Google Scholar] [CrossRef]
- Viladomat, F.; Bastida, J.; Codina, C.; Campbell, W.E.; Mathee, S. Alkaloids from Boophane flava. Phytochemistry 1995, 40, 307–311. [Google Scholar] [CrossRef]
- Ying, G.; de Andrade, J.P.; Pigni, N.B.; Torras-Claveria, L.; Tallini, L.R.; Borges, W.D.S.; Viladomat, F.; Nair, J.J.; Zuanazzi, J.A.S.; Bastida, J. New alkaloids from Hippeastrum papilio (Ravenna) van Scheepen. Helv. Chim. Acta 2016, 99, 143–147. [Google Scholar]
- Bariwal, J.B.; Ermolat’ev, D.S.; Glasnov, T.N.; Van Hecke, K.; Mehta, V.P.; Van Meervelt, L.; Kappe, C.O.; Van der Eycken, E.V. Dibenzoazocines and dibenzoazepines via a microwave-assisted intramolecular A3-coupling reaction. Org. Lett. 2010, 12, 2774–2777. [Google Scholar] [CrossRef] [PubMed]
- Donets, P.A.; van der Eycken, E.V. Synthesis of ring-expanded aza-analogues of bisbenzocyclooctadiene lignin lactones. QSAR Comb. Sci. 2007, 26, 1239–1242. [Google Scholar] [CrossRef]
- Donets, P.A.; Goeman, J.L.; Van der Eycken, J.; Robeyns, K.; van Meervelt, L.; van der Eycken, E.V. An asymmetric approach towards (−)-aphanorphine and its analogues. Eur. J. Org. Chem. 2009, 25, 793–796. [Google Scholar] [CrossRef]
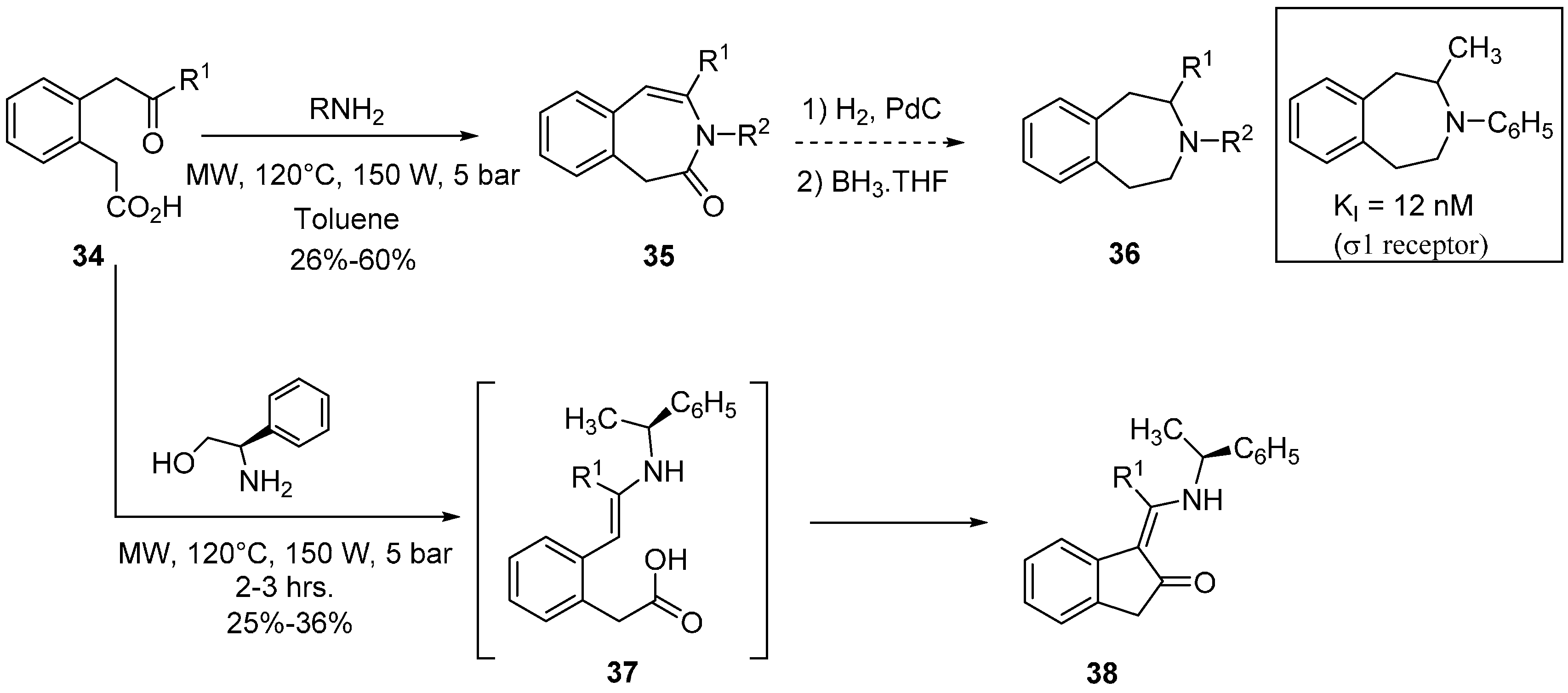
- Peshkov, V.A.; Pereshivko, O.P.; Donets, P.A.; Mehta, V.P.; van der Eycken, E.V. Diversity-oriented microwave-assisted synthesis of the 3-benzazepine framework. Eur. J. Org. Chem. 2010, 25, 4861–4867. [Google Scholar] [CrossRef]
- Sarkar, S.; Husain, S.M.; Schepmann, D.; Frölich, R.; Wünsch, B. Microwave assisted synthesis of 3-benzazepin-2-ones as building blocks for 2,3-disubstituted tetrahydro-3-benzazepines. Tetrahedron 2012, 68, 2687–2695. [Google Scholar] [CrossRef]
- Manabe, Y.; Kanematsu, M.; Yokoe, H.; Yoshida, M.; Shishido, K. Concise total synthesis of heliannuols B and D. Tetrahedron 2014, 70, 742–748. [Google Scholar] [CrossRef]
- Engler, M.; Anke, T.; Sterner, O. Pterulinic acid and pterulone, two novel inhibitors of NADH: Ubiquinone oxidoreductase (complex I) produced by a Pterula species. I. Production, isolation and biological activities. J. Antibiot. 1997, 50, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-K.; Jang, Y.-W.; Kim, Y.-S.; Yu, S.H.; Lee, K.J.; Park, S.-M.; Oh, B.-T.; Chae, J.-C.; Yun, B.-S. Xylarinols A and B, two new 2-benzoxepin derivatives from the fruiting bodies of Xylaria polymorpha. J. Antibiot. 2009, 62, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, M.; Kimber, M.C.; Moody, C.J. Synthesis of ptaeroxylin (desoxykarenin): An unusual chromone from the sneezewood tree Ptaeroxylon obliquum. Synlett 2008, 14, 2101–2102. [Google Scholar]
- Jiang, B.; Feng, B.-M.; Wang, S.-L.; Tu, S.-J.; Li, G. Domino constructions of pentacyclic indeno[2,1-c]quinolones and pyrano[4,3-b]oxepines by [4 + 1]/[3 + 2 + 1]/[5 + 1] and [4 + 3] multiple cyclisations. Chem. Eur. J. 2012, 18, 9823–9826. [Google Scholar] [CrossRef] [PubMed]
- Bruder, M.; Haseler, P.L.; Muscarella, M.; Lewis, W.; Moody, C.J. Synthesis of the oxepinochrome natural products ptaeroxylin (desoxykarenin), ptaeroxylinol, and eranthin. J. Org. Chem. 2010, 75, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Castedo, L.; Suau, R. Cularine alkaloids. Alkaloids 1986, 29, 287–324. [Google Scholar]
- Wu, B.; He, S.; Pan, Y.-J. New dihydrodibenzoxepins from bulbophyllum kwangtungense. Planta Med. 2006, 72, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Kittakoop, P.; Nopichai, S.; Thongon, N.; Charoenchai, P.; Thebtaranonth, Y. Bauhinoxepins A and B: New antimycobacterial dibenzo[b,f]oxepins from Bauhinia saccocalyx. Helv. Chim. Acta 2004, 87, 175–179. [Google Scholar] [CrossRef]
- Trabanco, A.A.; Alonso, J.M.; Andres, J.I.; Cid, J.M.; Fernández, J.; Iturrino, L.; Megens, A. Synthesis of 2-N,N-dimethylaminomethyl-2,3,3a, 12b-tetrahydrodibenzo[b,f]furo[2,3-d]oxepin as potential anxiolytic agents. Chem. Pharm. Bull. 2004, 52, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Irie, A.; Nakamura, H.; Hino, K.; Uno, H.; Nishimura, H. Nonsteroïdal antiinflammatory agents. 1. 10,11-Dihydro-11-oxodibenz[b,f]oxepinacetic acids and related compounds. J. Med. Chem. 1982, 25, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.R.R.; Giorgi, G.; Salas, C.O.; Tapia, R.A. New short strategy for the synthesis of the dibenzo[b,f]oxepin scaffold. Molecules 2013, 18, 14797–14806. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.I.; Hussain, M.K.; Arun, A.; Chakravarti, B.; Konwar, R.; Hajela, K. Synthesis of targeted dibenzo[b,f]thiepines and dibenzo[b,f]oxepines as potential lead molecules with promising anti-breast cancer activity. Eur. J. Med. Chem. 2015, 99, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Green, B. Zotepine: A clinical review. Expert Opin. Drig Metab. Toxicol. 2009, 5, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, J.L.; Puschl, A.; Jensen, M.; Risgaard, R.; Christiffersen, C.T.; Bang-Andersen, B.; Balle, T. Exploring the neuroleptic substituent in octoclothepin: Potential ligand for positron emission tomography with subnanomola affinity for α1-adrenocaptors. J. Med. Chem. 2010, 53, 7021–7034. [Google Scholar] [CrossRef] [PubMed]
- Bozinovic, N.; Novakovic, I.; Kostic Rajacic, S.; Opsenica, I.M.; Solaja, B.A. Synthesis and antimicrobial activity of azepine and thiepine derivatives. J. Serb. Chem. Soc. 2015, 80, 839–852. [Google Scholar] [CrossRef]
- Bozinovic, N.; Opsenica, I.; Solaja, B. Double palladium-catalyzed synthesis of azepines. Synlett 2013, 24, 49–52. [Google Scholar]
- Costantino, L.; Barlocco, D. Privileged Structures as Leads in Medicinal Chemistry. Curr. Med. Chem. 2006, 13, 65–85. [Google Scholar] [CrossRef] [PubMed]
- James, G.L.; Goldstein, J.L.; Brown, M.S.; Rawson, T.E.; Somers, T.C.; McDowell, R.S.; Crowley, C.W.; Lucas, B.K.; Levinson, A.D.; Marsters, J.C., Jr. Benzodiazepine peptidomimetics: Potent inhibitors of Ras farnesylation in animal cells. Science 1993, 260, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, C.; Borer, X. A non-peptide ligand for the somatostatin receptor having a benzodiazepinone structure. Bioorg. Med. Chem. Lett. 1996, 6, 267–272. [Google Scholar] [CrossRef]
- Manih, R.M.; Myrboh, B. A facile synthesis of 3,5,7-trisubstituted-4H-[1,2]diazepines by microwave irradiation. Indian J. Chem. Sec. B 2012, 51B, 1613–1618. [Google Scholar]
- Díaz, J.E.; Bisceglia, J.A.; Mollo, M.C.; Orelli, L.R. 1,n-Diamines. Part 2: Synthesis of acyclic and heterocyclic N-arylputrescine derivatives. Tetrahedron Lett. 2011, 52, 1895–1897. [Google Scholar] [CrossRef]
- Bisceglia, J.A.; Diaz, J.E.; Torres, R.A.; Orelli, L.R. 1,n-diamines. Part 3: Microwave-assisted synthesis of N-acyl-N’-arylhaxahydropyrimidines and hexahydo-1,3-diazepines. Tetrahedron Lett. 2011, 52, 5238–5240. [Google Scholar] [CrossRef]
- Morrison, C.S.; Lampe, J.B.; Kolodziejczyk, T.C.; Cavazos, R.J.; Petros, R.A. Rapid, quantitative, solvent-free synthesis of medium-ring diaza heterocycles from diketene-acetone adduct and diamines. Tetrahedron Lett. 2014, 55, 6547–6549. [Google Scholar] [CrossRef]
- Liang, L.; Saiz, C.; Pizzo, C.; Wipf, P. Synthesis of pyrrolo[1,3]diazepines by a dipolar cycloaddition—Retro-Mannich domino reaction. Tetrahedron Lett. 2009, 50, 6810–6813. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.L.; Shao, J.H.; Luo, J.Z.; Liu, J.T.; Miao, J.M.; Zhao, B.X. Novel chiral ferrocenylpyrazolo[1,5-a][1,4]diazepin-4-one derivatives—Synthesis, characterization and inhibition against lung cancer cells. Eur. J. Med. Chem. 2013, 63, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.L.; Zhu, J.; Li, M.; Zhao, B.X.; Miao, J.Y. Synthesis of ferrocenyl pyrazole-containing chiral aminoethanol derivatives and their inhibition against A549 and H322 lung cancer cells. Eur. J. Med. Chem. 2012, 54, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Bougrin, K.; Bennani, K.A.; Tétouani, S.F.; Soufiaoui, M. An easy route to synthesize 1,5-arylodiazepin-2-ones. Tetrahedron Lett. 1994, 35, 8373–8376. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Varma, R.S. Greener and rapid access to bio-active heterocycles: Room temperature synthesis of pyrazoles and diazepines in aqueous medium. Tetrahedron Lett. 2008, 49, 397–400. [Google Scholar] [CrossRef]
- Insuasty, B.; García, A.; Quiroga, J.; Abonia, R.; Nogueras, M.; Cobo, J. Synthesis of novel 6,6a,7,8-tetrahydro-5H-naphtho[1,2-e]pyrimido[4,5-b][1,4]diazepines under microwave irradiation as potential anti-tumor agents. Eur. J. Med Chem. 2010, 45, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Vaddula, B.R.; Varma, R.S.; Leazer, J. Mixing with microwaves: Solvent-free and catalyst-free synthesis of pyrazoles and diazepines. Tetrahedron Lett. 2013, 54, 1538–1541. [Google Scholar] [CrossRef]
- Willy, B.; Dallos, T.; Rominger, F.; Schönhaber, J.; Müller, T.J.J. Three-Component Synthesis of Cryofluorescent 2,4-Disubstituted 3H-1,5-Benzodiazepines—Conformational Control of Emission Properties. Eur. J. Org. Chem. 2008, 4796–4805. [Google Scholar] [CrossRef]
- Zhang, W. Fluorous-Enhanced Multicomponent Reactions for Making Drug-Like Library Scaffolds. Comb. Chem. High Throughput Screen. 2007, 10, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Cheng, C.; Wu, F.Y.; Jiang, B.; Shi, F.; Tu, S.J.; Rajale, T.; Li, G. Microwave-assisted multi-component reaction in water leading to highly regioselective formation of benzo[f]azulen-1-ones. Tetrahedron 2011, 67, 4485–4493. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Chiba, H.; Oishi, S.; Fujii, N.; Ohno, H. Concise Synthesis of Indole-Fused 1,4-Diazepines through Copper(I)-Catalyzed Domino Three-Component Coupling-Cyclization-N-Arylation under Microwave Irradiation. Org. Lett. 2008, 10, 3535–3538. [Google Scholar] [CrossRef] [PubMed]
- Acosta, P.; Becerra, D.; Goudedranche, S.; Quiroge, J.; Constantieux, T.; Bonne, D.; Rodriguez, J. Exploiting the reactivity of 1,2-ketoamides: Enantioselective synthesis of functionalized pyrrolidines and pyrrolo-1,4-benzodiazepine-2,5-diones. Synlett 2015, 26, 1591–1595. [Google Scholar] [CrossRef]
- Hopenwasser, J.; Mozayani, A.; Danielson, T.J.; Harbin, A.; Narula, H.S.; Posey, D.H.; Shrode, P.W.; Wilson, S.K.; Li, R.; Sanchez, L. Postmortem Distribution of the Novel Antipsychotic Drug Quetiapine. J. Anal. Toxicol. 2004, 28, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Bocquet, A.; Sablayrolles, S.; Vacher, B.; Le Grand, B. F15845, a new blocker of the persistent sodium current prevents consequences of hypoxia in rat femoral artery. Br. J. Pharmacol. 2010, 161, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.B.; Morgans, G.L.; Aderibigbe, B.A.; Madeley, L.G.; Fernandes, M.A.; Michael, J.P.; de Koning, C.B.; Van Otterlo, W.A.L. Application of an isomerization-ring-closing metathesis strategy to the synthesis of unsaturated seven-membered, benzo-fused heterocycles containing two heteroatoms. Tetrahedron 2011, 67, 2991–2997. [Google Scholar] [CrossRef]
- Saha, D.; Wadhwa, P.; Sharma, A. A sequential synthetic strategy towards unexplored dibenzo[b,f][1,4]thiazepine carboxamides: Copper catalysed C-S cyclisation followed by Ugi type 3CC cascade. RSC Adv. 2015, 5, 33067–33076. [Google Scholar] [CrossRef]
- Tu, S.J.; Cao, X.D.; Hao, W.G.; Zhang, X.H.; Yan, S.; Wu, S.S.; Han, Z.G.; Shi, F. An efficient and chemoselective synthesis of benzo[e][1,4]thiazepin-2(1H,3H,5H)-ones via a microwave-assisted multi-component reaction in water. Org. Biomol. Chem. 2009, 7, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Zeng, X.N.; Cao, X.D.; Zhang, S.; Jiang, B.; Zheng, W.F.; Tu, S.G. Design and diversity-oriented synthesis of novel 1,4-thiazepan-3-ones fused with bioactive heterocyclic skeletons and evaluation of their antioxidant and cytotoxic activities. Bioorg. Med. Chem. Lett. 2012, 22, 743–746. [Google Scholar] [CrossRef] [PubMed]
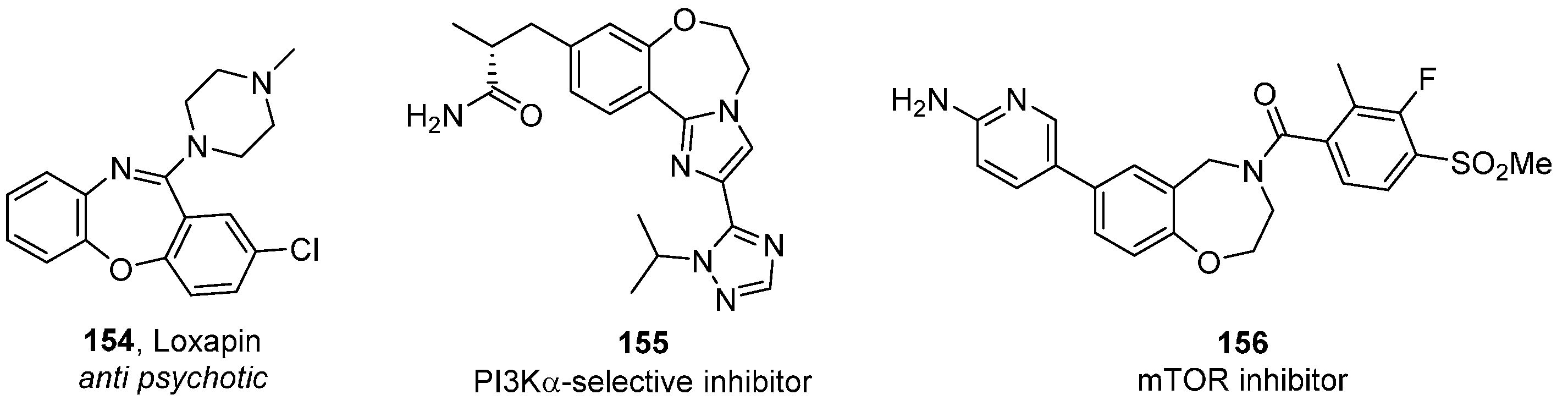
- Keating, G.M. Loxapine inhalation powder: A review of its use in the acute treatment of agitation in patients with bipolar disorders of schizophrenia. CNS Drugs 2013, 27, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Blaquiere, N.; Do, S.; Dudley, D.; Folkes, A.J.; Heald, R.; Heffron, T.; Jones, M.; Kolesnikov, A.; Ndubaku, C.; Olivero, A.G.; et al. Benzoxazepine as PI3K Inhibitor and Their Preparation and Use in the Treatment of Cancer. PCT Patent WO 2011036280 A1 20110331, 31 March 2011. [Google Scholar]
- Takeuchi, C.S.; Kim, B.G.; Blazey, C.M.; Ma, S.; Johnson, W.B.; Anand, N.K.; Arcalas, A.; Baik, T.G.; Buhr, C.A.; Cannoy, J.; et al. Discovery of a novel class of highly potent, selective, ATP-competitive, and orally bioavailable inhibitors of the mammalian target of rapamycin (mTOR). J. Med. Chem. 2013, 56, 2218–2234. [Google Scholar] [CrossRef] [PubMed]
- De Moliner, F.; Bigatti, M.; De Rosas, C.; Banfi, L.; Riva, R.; Basso, A. Synthesis of triazolo-fused benzoxazepines and benzoxazepinones via Paserini reactions followed by 1,3-dipolar cycloadditions. Mol. Divers. 2014, 18, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Sarkar, S.; Pal, R.; Sen, A.K. An approach toward the syntheses of triazolo benzoxazines, triazolo quinoxaline, triazolo benzodiazepines, triazolo benzoxazepines, and triazolo benzothiazines via a simple and convenient protocol using basic alumina as solid support. Tetrahedron Lett. 2014, 55, 2261–2265. [Google Scholar] [CrossRef]
- Xing, X.; Wu, J.; Luo, J.; Wei-Min, D. C-N bond-linked conjugates of dibenz[b,f][1,4]oxazepines with 2-oxindole. Synlett 2006, 13, 2099–2103. [Google Scholar] [CrossRef]
- Dabholkar, V.V.; Moris, G.D. Synthesis of 4,10-dihydro-5/7substituted-9-oxo-quinolino[2,3-e]-2-amino-1,3,4- thiadiazine and Schiff base by microwave irradiation. Indian J. Chem. 2004, 43B, 682–684. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Fedorov, O.; Keller, M.; Wrobel, M.; Morgenstern, O.; Bracher, F.; Knapp, S. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorg. Med. Chem. 2012, 20, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Mc Donald, I.M.; Black, J.W.; Buck, I.M.; Dunstone, D.J.; Griffin, E.P.; Harper, E.A.; Hull, R.A.D.; Kalindjian, S.B.; Lilley, E.J.; Linney, I.D.; et al. Optimization of 1,3,4-Benzotriazepine-Based CCK2 Antagonists to Obtain Potent, Orally Active Inhibitors of Gastrin-Mediated Gastric Acid Secretion. J. Med. Chem. 2007, 50, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, G.K.; Kumaraswamy, M.N.; Vaidya, V.P.; Mahadevan, K.M. Microwave assisted synthesis of naphtho[2,1-b]furan-1, 3, 4- benzotriazepines: a potent antimicrobial agent. ARKIVOC 2006, 10, 211–219. [Google Scholar]
- Dong, C.; Xie, L.; Mou, X.; Zhong, Y.; Su, W. Facile synthesis of 1,3,4-benzotriazepines and 1-arylamide-1H-indazoles via palladium-catalyzed cyclization of aryl isocyanates and aryl hydrazones under microwave irradiation. Org. Biomol. Chem. 2010, 8, 4827–4830. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Paul, S.; Gupta, R. Efficient and novel one-pot synthesis of antifungal active 1-substituted-8-aryl-3-alkyl/aryl-4H-pyrazolo[4,5-f][1,2,4]triazolo[4,3-b][1,2,4]triazepines using solid support. Eur. J. Med. Chem. 2011, 46, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M. Efficient synthesis of antifungal active 9-substituted-3-aryl-5H,13aH-quinolino[3,2-f][1,2,4]triazolo[4,3-b][1,2,4]triazepines in ionic liquids. Bioorg. Med. Chem. Lett. 2011, 21, 4919–4923. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, M.; Sapra, P.; Misra, P.; Saxena, R.K.; Singh, M. Microwave assisted solid support synthesis of novel 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazepines as potent antimicrobial agents. Bioorg. Med. Chem. 2001, 9, 217–220. [Google Scholar] [CrossRef]
- Raghavendra, M.; Naik, H.S.B.; Naik, T.; Sherigara, B.S. p-TsOH Catalysed a Facile One-Pot Synthesis of Some New Substituted 1,2,4 Triazolo[3,4-b]-1,3,4-thiadiazepines under Microwave Irradiation in Solvent Free Conditions. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 1823–1831. [Google Scholar] [CrossRef]
- Gupta, M.; Paul, S.; Gupta, R. Microwave-assisted one-pot synthesis of antifungal active 1-substituted-3,7-dialkyl/aryl-4H-pyrazolo[4,5-f][1,3,4]thiadiazepines using solid support. Indian J. Chem. Sect. B 2009, 48B, 460–466. [Google Scholar]
- Saleh, T.S.; Abd El-Rahman, N.; Assaker, R.S.A. Microwave promoted a green protocol for solvent free synthesis of 1,5-benzothiazepine and [1,3,4]-thiadiazepine derivatives incorporating thiophene moiety. Green Chem. Lett. Rev. 2012, 5, 315–312. [Google Scholar] [CrossRef]
- Pedersen, S.L.; Tofteng, A.P.; Malik, L.; Jensen, J. Microwave heating in solid-phase synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. [Google Scholar] [CrossRef] [PubMed]
- Rizzolo, F.; Sabatino, G.; Chelli, M.; Rovero, P.; Papini, A. A convenient microwave-enhanced solid-phase synthesis of difficult peptide sequences: Case study of gramicidin A and CSF114(Glc). Int. J. Pept. Res. Ther. 2007, 13, 203–208. [Google Scholar] [CrossRef]
- Ieronymaki, M.; Androutsou, M.E.; Pantelia, A.; Friligou, I.; Crisp, M.; High, K.; Penkman, K.; Gaton, D.; Tselios, T. Use of 2-chlorotrityl chloride resin for microwave-assisted solid phase peptide synthesis. Pept. Sci. 2015, 104, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Bacsa, B.; Horvati, K.; Bosze, S.; Andreae, F.; Kappe, C.O. Solid-phase synthesis of difficult peptides sequences at elevated temperatures: A critical comparison of microwave and conventional heating technologies. J. Org. Chem. 2008, 73, 7532–7542. [Google Scholar] [CrossRef] [PubMed]
- Grieco, P.; Cai, M.; Liu, L.; Mayorov, A.; Chandler, K.; Trivedi, D.; Lin, G.; Campiglia, P.; Novellino, E.; Hruby, V.J. Design and Microwave-Assisted Synthesis of Novel Macrocyclic Peptides Active at Melanocortin Receptors: Discovery of Potent and Selective hMC5R Receptor Antagonists. J. Med. Chem. 2008, 51, 2701–2707. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Panda, S.S.; Oliferenko, A.A.; Oliferenko, P.V.; Girgis, A.S.; Elagawany, M.; Küçükbay, Z.; Panda, C.S.; Pillai, G.G.; Samir, A.; et al. Macrocyclic peptidomimetics with antimicrobial activity: Synthesis, bioassay, and molecular modeling studies. Org. Biomol. Chem. 2015, 13, 9492–9503. [Google Scholar] [CrossRef] [PubMed]
- Tala, S.R.; Schnell, S.M.; Haskell-Luevano, C. Microwave-assisted solid-phase synthesis of side-chain to side-chain lactam bridge cyclic peptides. Bioorg. Med. Chem. Lett. 2015, 25, 5708–5711. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.N.; Arora, PS. Optimized Synthesis of Hydrogen-Bond Surrogate Helices: Surprising Effects of Microwave Heating on the Activity of Grubbs Catalysts. Org. Lett. 2006, 8, 5825–5828. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gago, P.; Ramon, R.; Aragon, E.; Fernandez-Carneado, J.; Martin-Malpartida, P.; Verdaguer, X.; Lopez-Ruiz, P.; Colas, B.; Cortes, M.A.; Ponsati, B.; et al. A tetradecapeptide somatostatin dicarba-analog: Synthesis, structural impact and biological activity. Bioorg. Med. Chem. Lett. 2014, 24, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Lépine, R.; Zhu, J. Microwave-Assisted Intramolecular Suzuki-Miyaura Reaction to Macrocycle, a Concise Asymmetric Total Synthesis of Biphenomycin B. Org. Lett. 2005, 7, 2981–2984. [Google Scholar] [CrossRef] [PubMed]
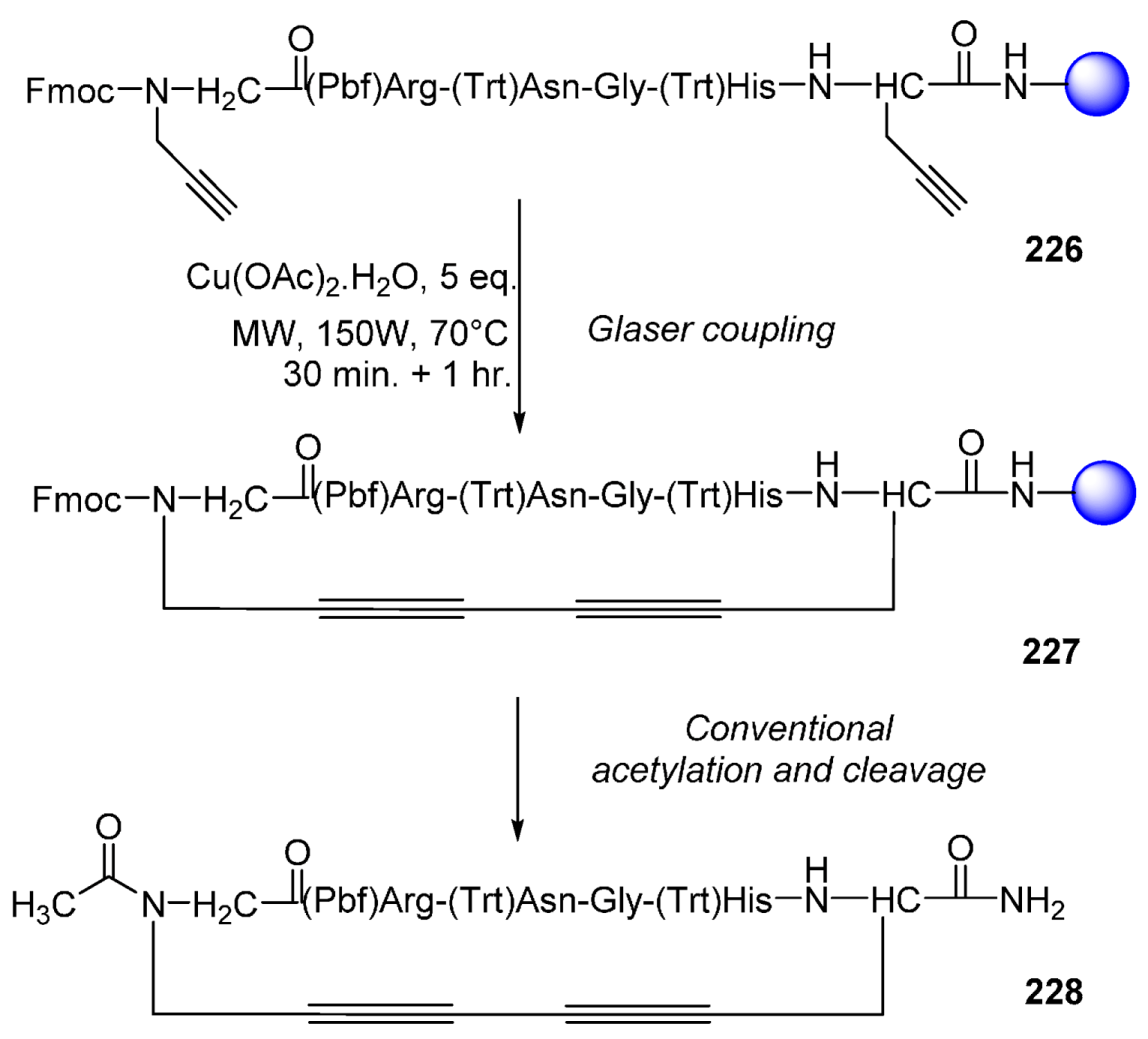
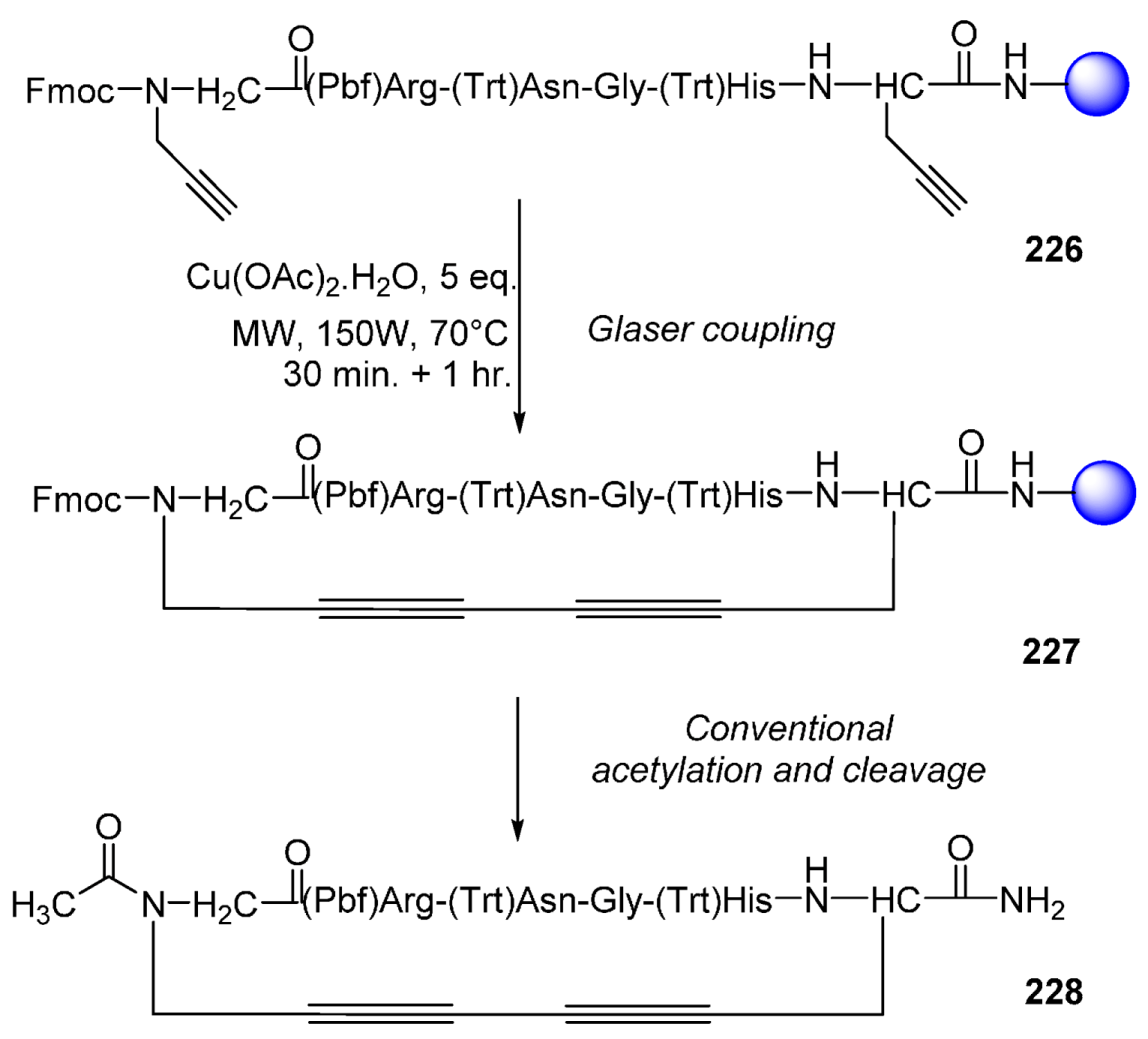
- Auberger, N.; Di Pisa, M.; Larregola, M.; Chassaing, G.; Peroni, E.; Lavielle, S.; Papini, A.-M.; Lequin, O.; Mallet, J.-M. Glaser oxidative coupling on peptides: Stabilization of β-turn structure via a 1,3-butadiyne constraint. Bioorg. Med. Chem. 2014, 22, 6924–6932. [Google Scholar] [CrossRef] [PubMed]
- Kaniraj, P.J.; Maayan, G. A facile strategy for the construction of cyclic peptoids under microwave irradiation through a simple substitution reaction. Org. Lett. 2015, 17, 2110–2113. [Google Scholar] [CrossRef] [PubMed]
- Décor, A.; Monse, B.; Martin, M.T.; Chiaroni, A.; Thoret, S.; Guénard, D.; Guéritte, F.; Baudoin, O. Synthesis and biological evaluation of B-ring analogues of (−)-rhazinilam. Bioorg. Med. Chem. 2006, 14, 2314–2332. [Google Scholar] [CrossRef] [PubMed]
- Beryozkina, T.; Appukkuttan, P.; Mont, N.; Van der Eycken, E. Microwave-Enhanced Synthesis of New (−)-Steganacin and (−)-Steganone Aza Analogues. Org. Lett. 2006, 8, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Appukkuttan, P.; Dehaen, W.; Van der Eycken, E. Microwave-Enhanced synthesis of N-Shifted Buflavine Analogues via a Suzuki-Ring-Closing Metathesis Protocol. Org. Lett. 2005, 7, 2723–2726. [Google Scholar] [CrossRef] [PubMed]
- Appukkuttan, P.; Dehaen, W.; van der Eycken, E. Microwave-Assisted Transition-Metal-Catalyzed Synthesis of N-Shifted and Ring-Expanded Buflavine Analogues. Chem Eur. J. 2007, 13, 6452–6460. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Charles, J.S.; Sun, D. Microwave-assisted synthesis of macrocycles via intramolecular and/or bimolecular Ullmann coupling. Tetrahedron Lett. 2012, 53, 4173–4178. [Google Scholar] [CrossRef] [PubMed]
- Pitsinos, E.N.; Vidali, V.P.; Couladouros, E.A. Diaryl Ether Formation in the Synthesis of Natural Products. Eur. J. Org. Chem. 2011, 1207–1222. [Google Scholar] [CrossRef]
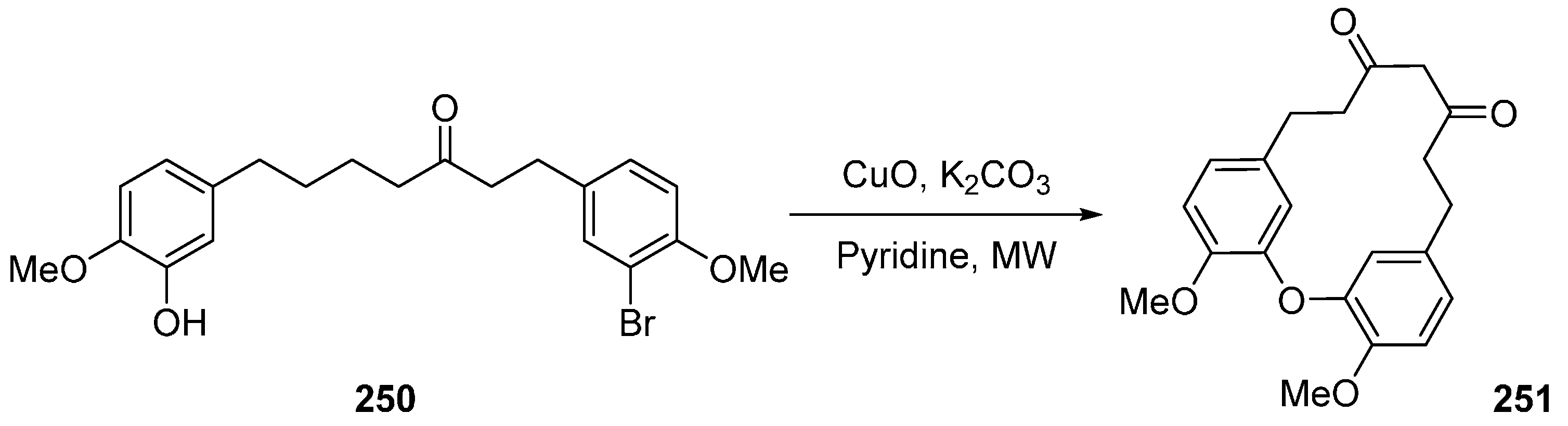
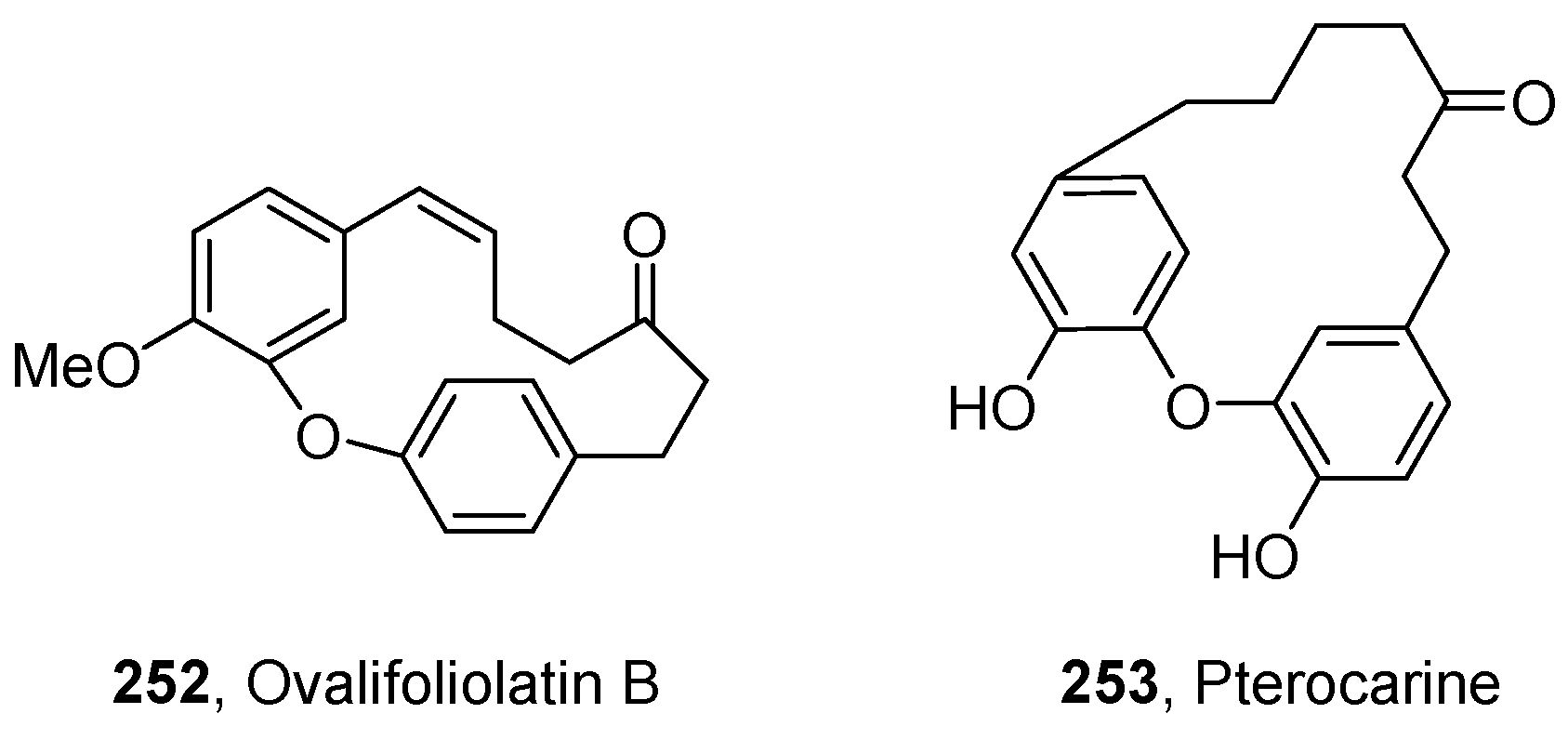
- Reddy, L.N.V.; Ravinder, K.; Srinivasulu, M.; Goud, V.T.; Reddy, M.S.; Srujankumar, D.; Rao, P.T.; Murty, S.U.; Venkateswarlu, Y. Two New Macrocyclic Diaryl Ether Heptanoids from Boswellia ovalifoliolata. Chem. Pharm. Bull. 2003, 51, 1081–1084. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, W.; Jiang, N.; Niu, Z.; Bao, K.; Zhang, L.; Liu, D.; Pan, C.; Yao, X. Total Synthesis of Bulbophylol-B. J. Nat. Prod. 2008, 71, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
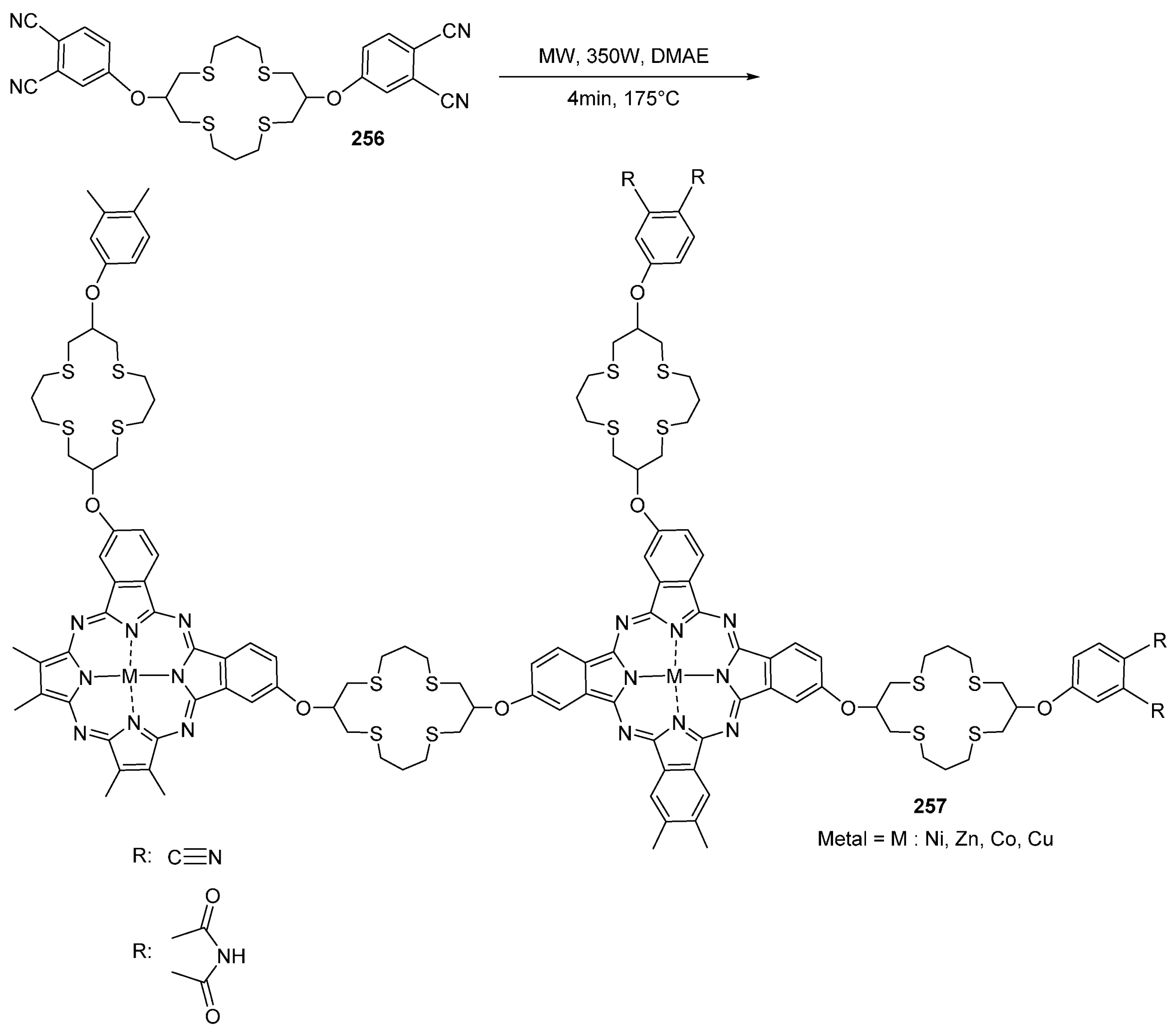
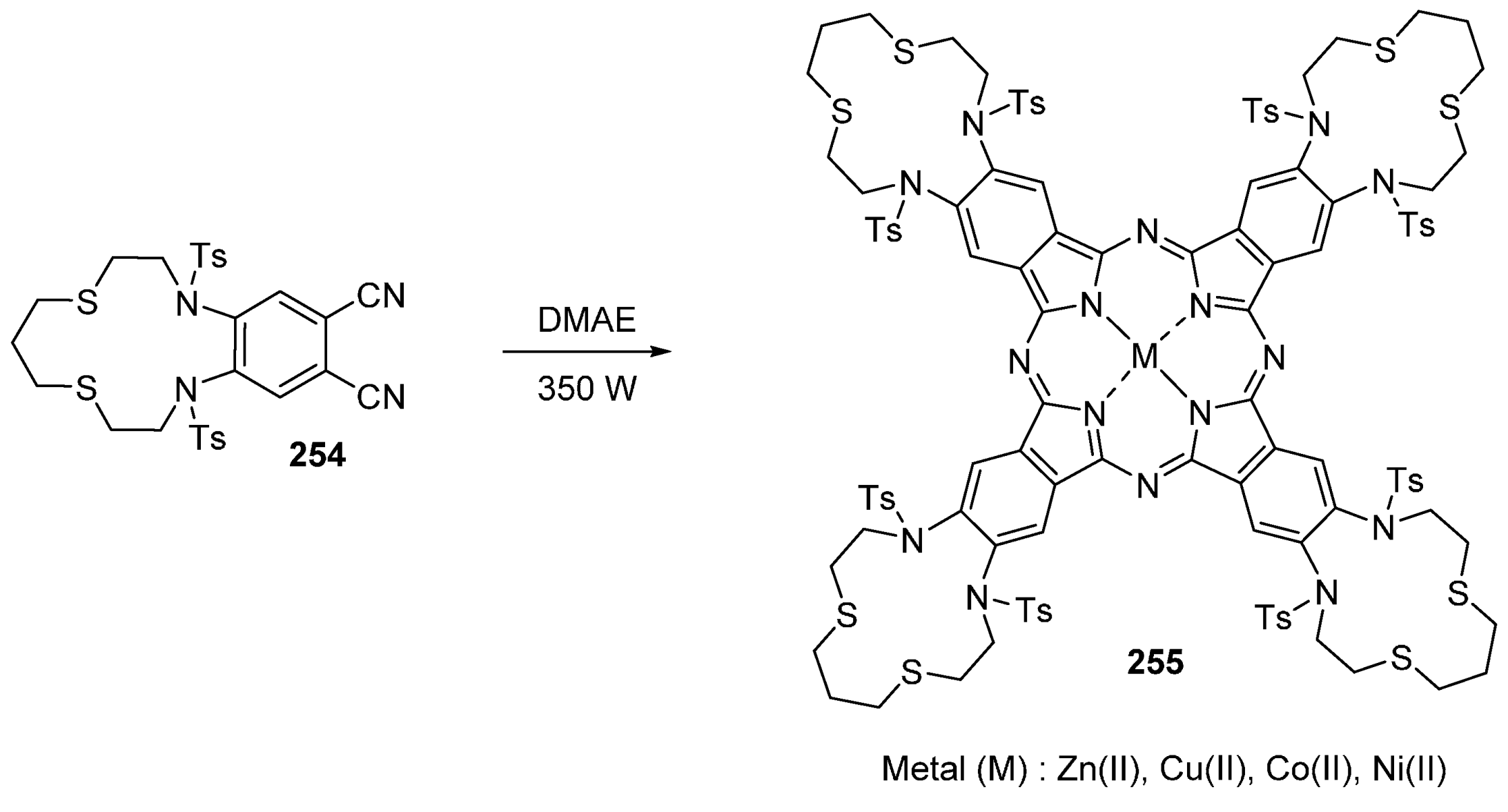
- Biyiklioglu, Z.; Kantekin, H.; Özil, M. Microwave-assisted synthesis and characterization of novel metal-free and metallophthalocyanines containing four 14-membered tetraaza macrocycles. J. Organomet. Chem. 2007, 692, 2436–2440. [Google Scholar] [CrossRef]
- Kantekin, H.; Biyiklioglu, Z. Microwave-assisted synthesis and characterization of novel metal-free and metallophthalocyanines containing four 13-membered dithiadiaza macrocycles. Dyes Pigment. 2008, 77, 98–102. [Google Scholar] [CrossRef]
- Biyiklioglu, Z.; Acar, I.; Kantekin, H. Microwave-assisted synthesis and characterization of new soluble metal-free and metallophthalocyanines substituted with four tetrathiamacrocycles through oxy bridges. Inorg. Chem. Commun. 2008, 11, 630–632. [Google Scholar] [CrossRef]
- Kantekin, H.; Biyiklioglu, Z.; Çelenk, E. Synthesis and characterization of new metal-free and metallophthalocyanines peripherally fused to four 15-membered tetraoxamonoazamacrocycles by microwave irradiation. Inorg. Chem. Commun. 2008, 11, 633–635. [Google Scholar] [CrossRef]
- Çelenk, E.; Kantekin, H. The microwave-assisted synthesis and characterization of novel metal-free and metallophthalocyanines peripherally fused to four 13-membered diazadithia macrocycles. Dyes Pigment. 2009, 80, 93–97. [Google Scholar] [CrossRef]
- Kantekin, H.; Dilber, G.; Biyiklioglu, Z.J. A new polymeric phthalocyanine containing 16-membered tetrathia macrocyclic moieties by microwave irradiation: Synthesis and characterization. Organometal. Chem. 2008, 693, 1038–1042. [Google Scholar] [CrossRef]
- Kantar, G.K.; Baltas, N.; Mentese, E.; Sasmaz, S. Microwave-assisted synthesis and investigation of xanthine oxidase inhibition of new phthalonitrile and phthalocyanines containing morpholino substituted 1,2,4-triazole-3-one. J. Organomet. Chem. 2015, 787, 8–13. [Google Scholar] [CrossRef]
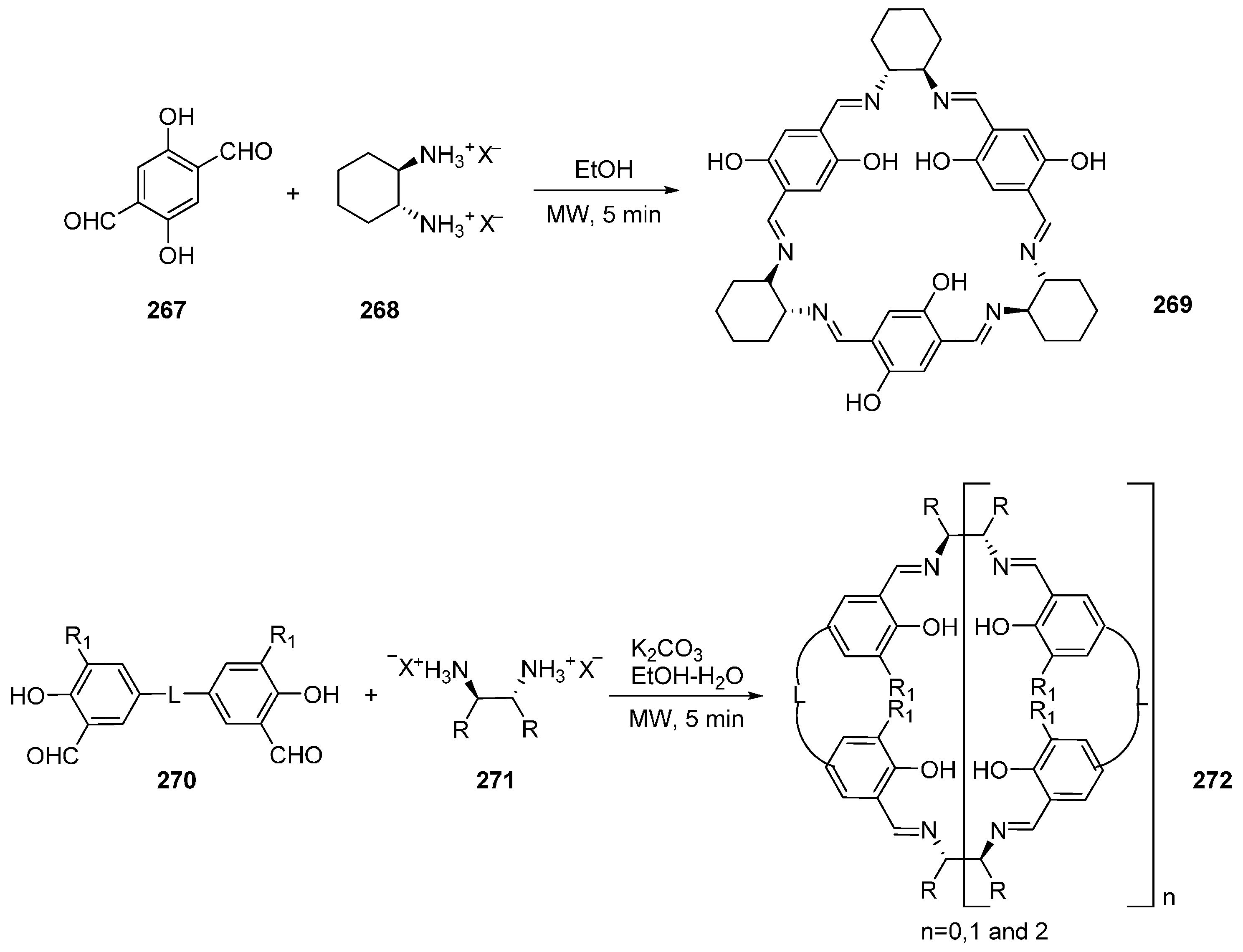
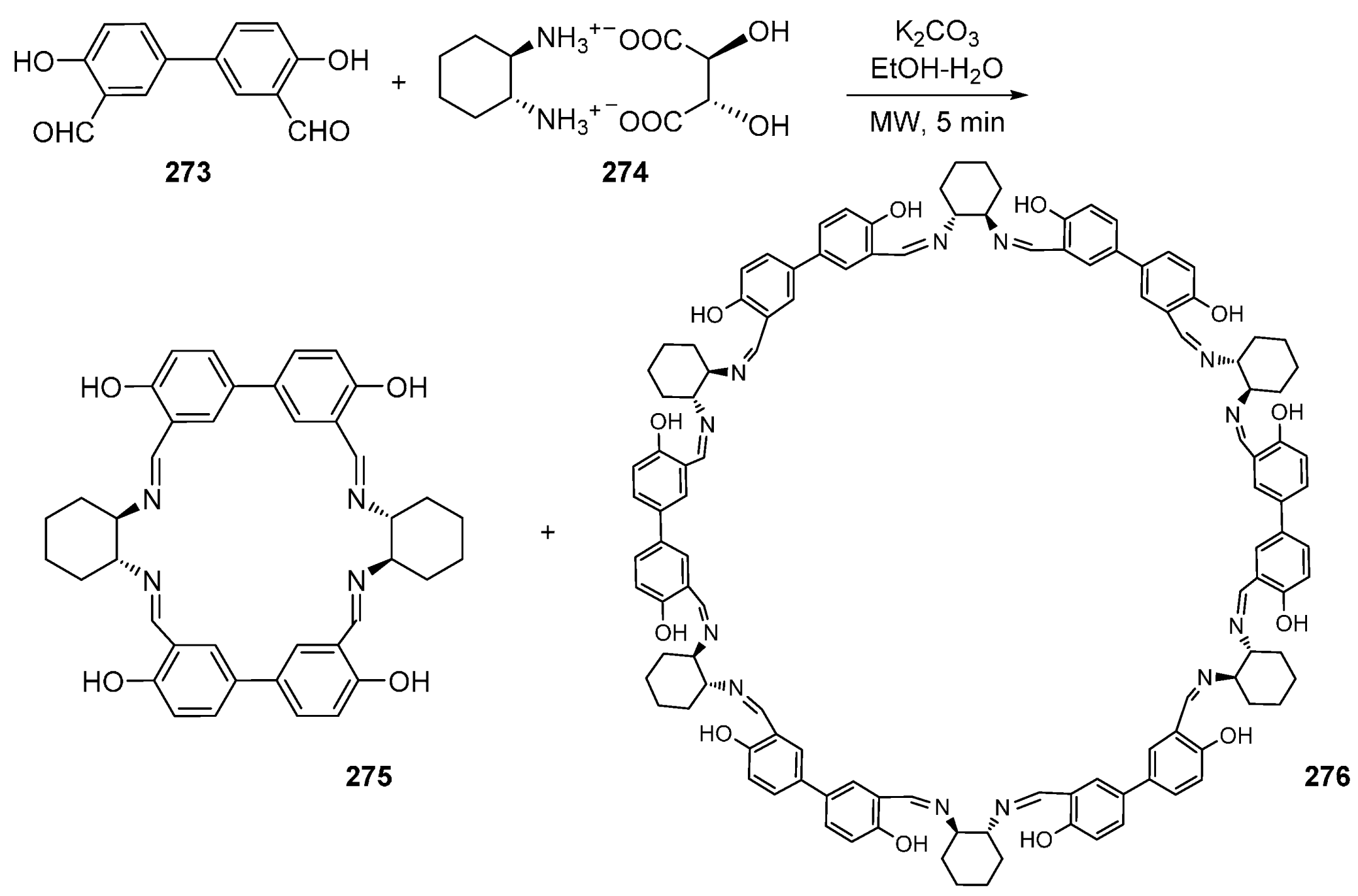
- Srimurugan, S.; Viswanathan, B.; Varadarajan, T.K.; Varghese, B. Microwave assisted cyclocondensation of dialdehydes with chiral diamines forming calixsalen type macrocycles. Tetrahedron Lett. 2005, 46, 3151–3155. [Google Scholar] [CrossRef]
- Srimurugan, S.; Suresh, P.; Pati, H.N. Microwave assisted synthesis of 72-membered chiral hexanuclear [6 + 6] macrocyclic Schiff base. J. Incl. Phenom. Macrocycl. Chem. 2007, 59, 383–388. [Google Scholar] [CrossRef]
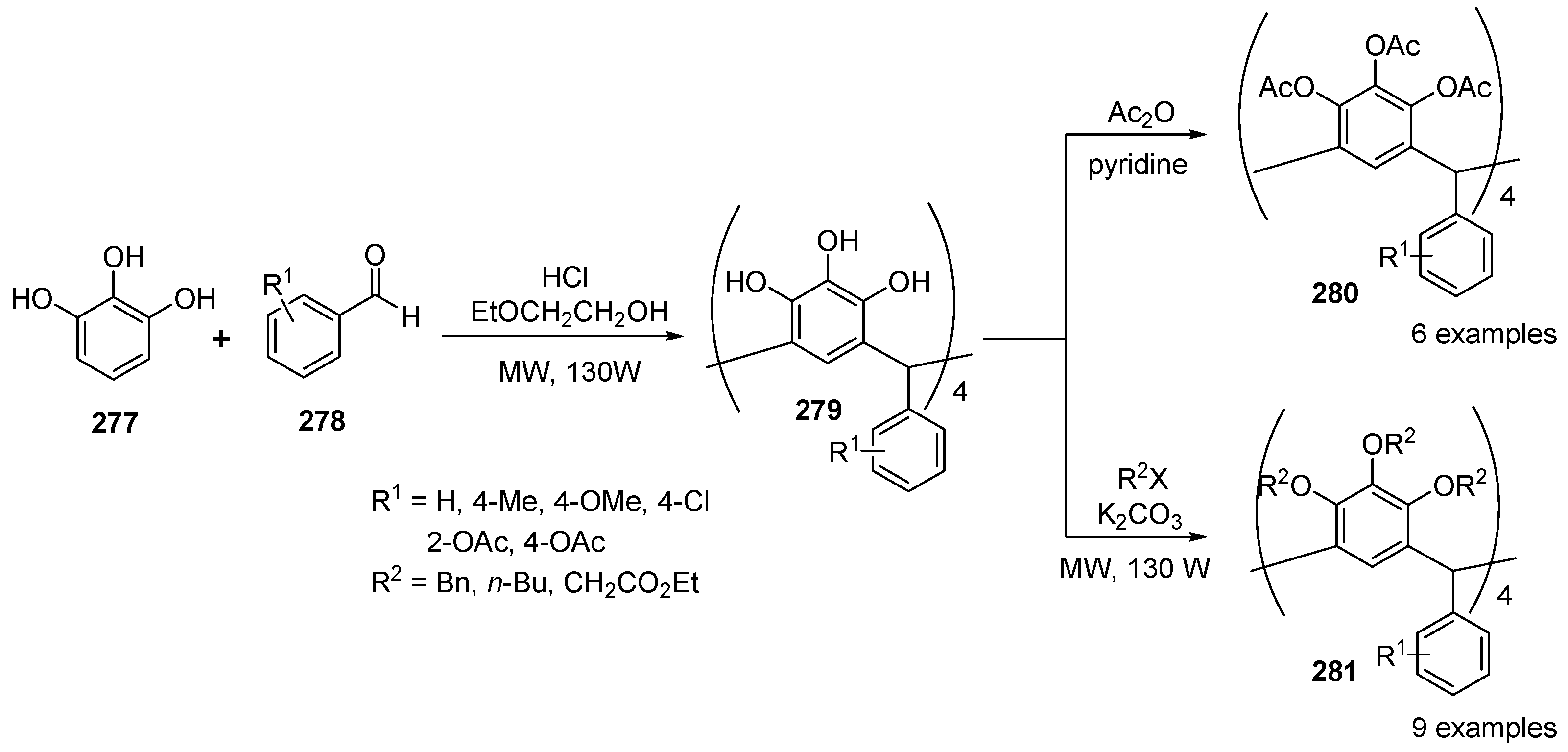
- Yan, C.; Xhen, W.; Chen, J.; Jiang, T.; Yao, Y. Microwave irradiation assisted synthesis, alkylation reaction, and configuration analysis of aryl pyrogallo[4]arenes. Tetrahedron 2007, 63, 9614–9620. [Google Scholar] [CrossRef]
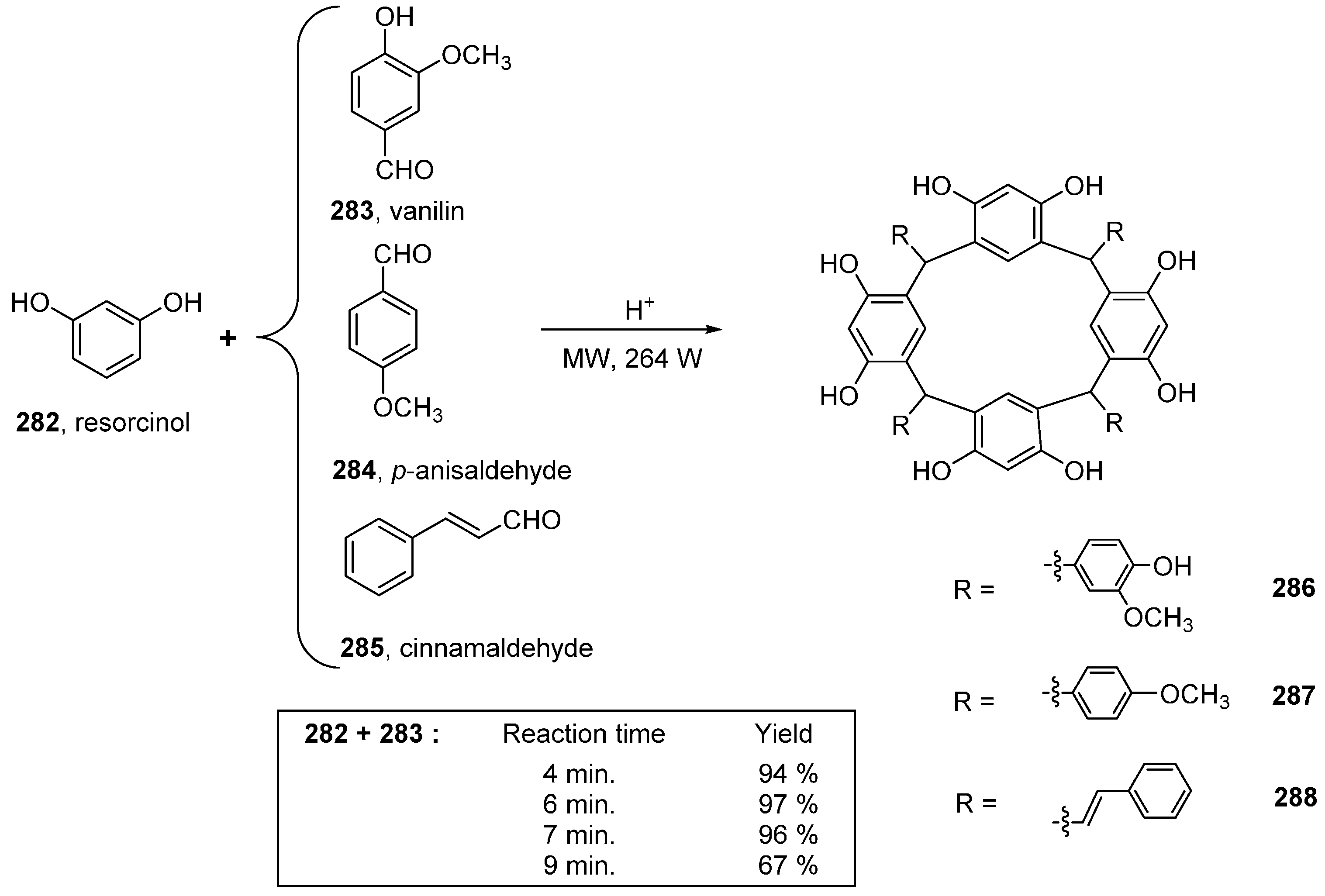
- Sardjono, R.E.; Kadarohman, A.; Mardhiyah, A. Green synthesis of some calix[4]resorcinarene under microwave irradiation. Procedia Chem. 2012, 4, 224–231. [Google Scholar] [CrossRef]
- Nayak, S.K.; Choudhary, M.K. Microwave assisted synthesis of 1,3-dialkyl ethers of calix[4]arenes: Application to the synthesis of cesium selective calix[4]crown-6 ionophores. Tetrehedron Lett. 2012, 53, 141–144. [Google Scholar] [CrossRef]
- Burilov, V.A.; Nugmanov, R.I.; Ibragimova, R.R.; Solovieva, S.E.; Antipin, I.S.; Konovalov, A.I. Microwave-assisted alkylation of p-tert-butylcalix[4]arene lower rim: The effect of alkyl halides. Mendeleev Commun. 2013, 23, 113–115. [Google Scholar] [CrossRef]
- Galan, H.; de Mendoza, J.; Prados, P. Microwave-assisted synthesis of a nitro-m-xylylenedioxycalix[6]arene building block functionalized at the upper rim. Eur. J. Org. Chem. 2010, 36, 7005–7011. [Google Scholar] [CrossRef]
- Garska, B.; Tabatabai, M.; Ritter, H. Calix[4]arene-click-cyclodextrin and supramolecular structures with watersoluble NIPAAM-copolymers bearing adamantly units: “Rings on ring on chain”. Beilstein J. Org. Chem. 2010, 6, 784–788. [Google Scholar] [CrossRef] [PubMed]

































































© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Driowya, M.; Saber, A.; Marzag, H.; Demange, L.; Bougrin, K.; Benhida, R. Microwave-Assisted Syntheses of Bioactive Seven-Membered, Macro-Sized Heterocycles and Their Fused Derivatives. Molecules 2016, 21, 1032. https://doi.org/10.3390/molecules21081032
Driowya M, Saber A, Marzag H, Demange L, Bougrin K, Benhida R. Microwave-Assisted Syntheses of Bioactive Seven-Membered, Macro-Sized Heterocycles and Their Fused Derivatives. Molecules. 2016; 21(8):1032. https://doi.org/10.3390/molecules21081032
Chicago/Turabian StyleDriowya, Mohsine, Aziza Saber, Hamid Marzag, Luc Demange, Khalid Bougrin, and Rachid Benhida. 2016. "Microwave-Assisted Syntheses of Bioactive Seven-Membered, Macro-Sized Heterocycles and Their Fused Derivatives" Molecules 21, no. 8: 1032. https://doi.org/10.3390/molecules21081032