
Evaluation of Biosynthetic Pathway and Engineered Biosynthesis of Alkaloids

Abstract
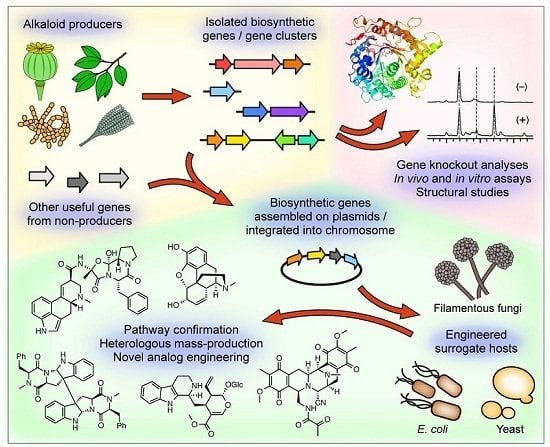
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
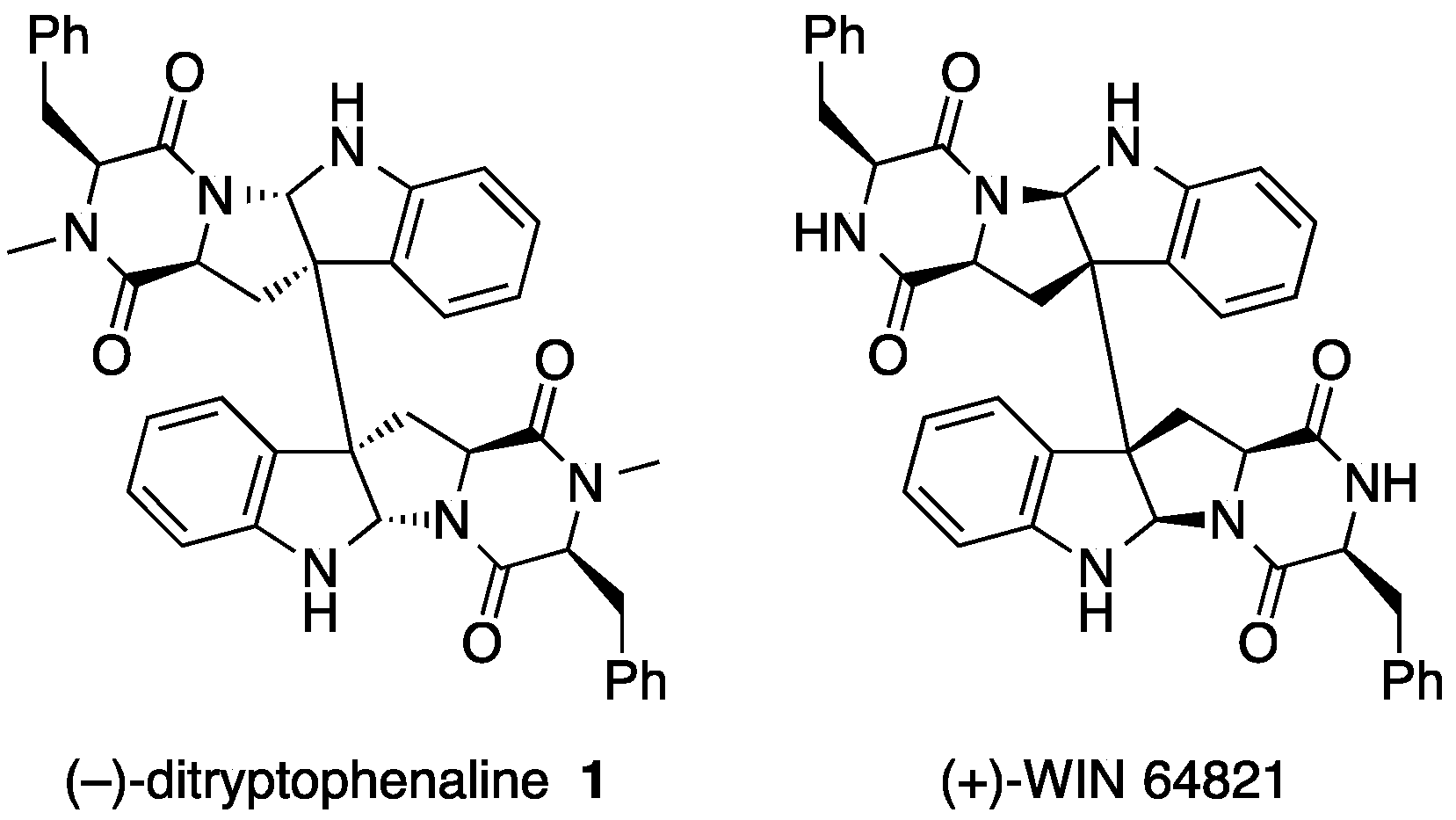
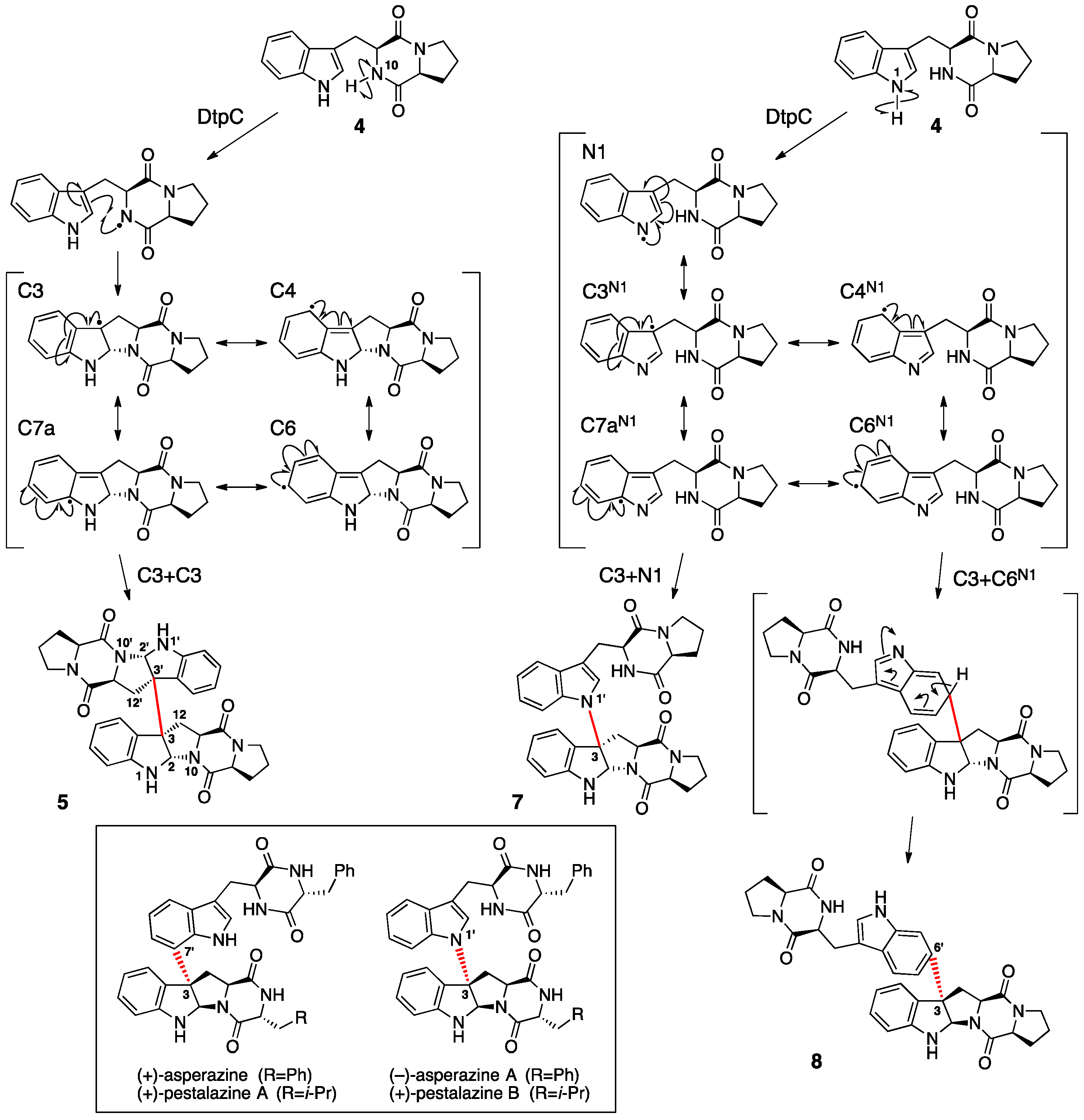
2. Ditryptophenaline
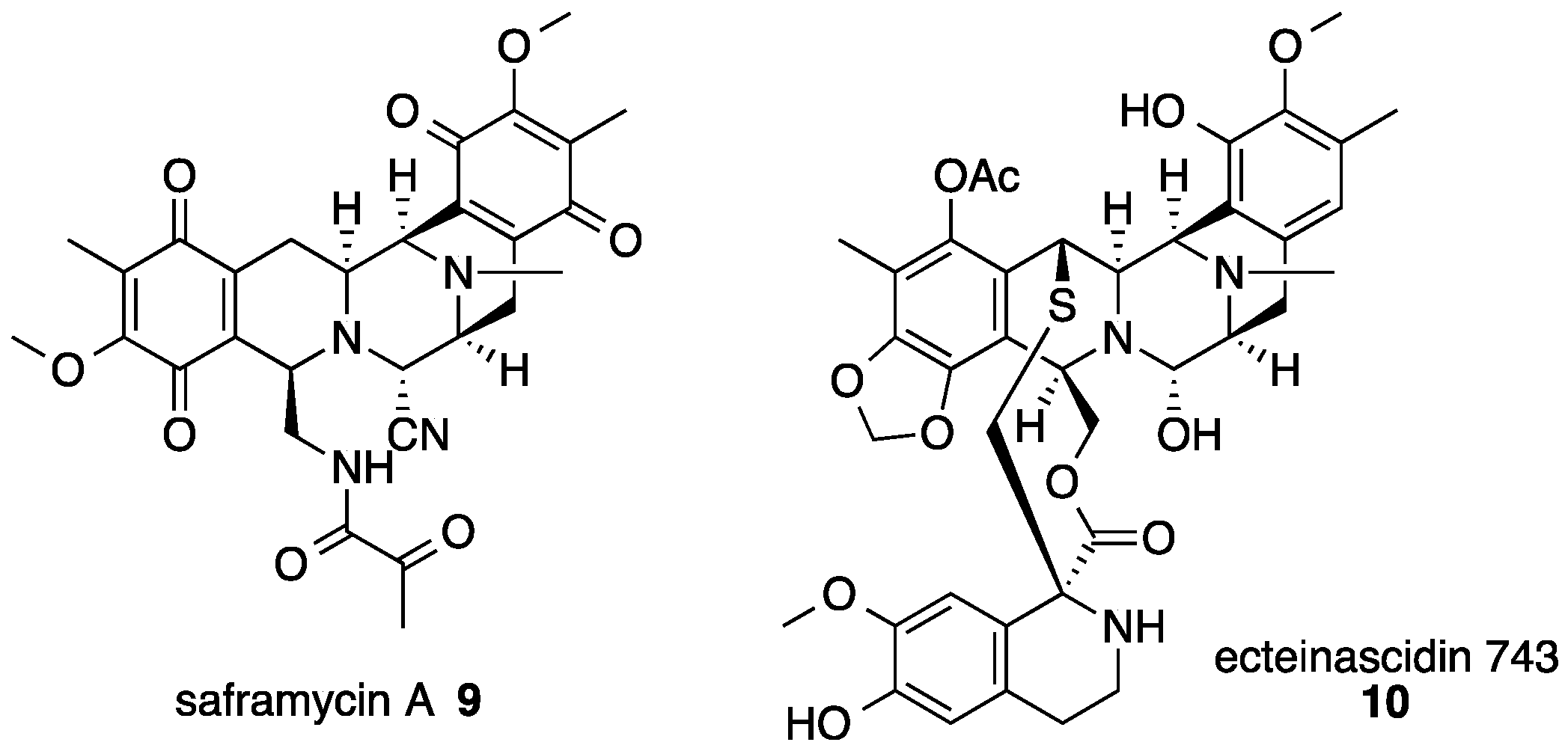
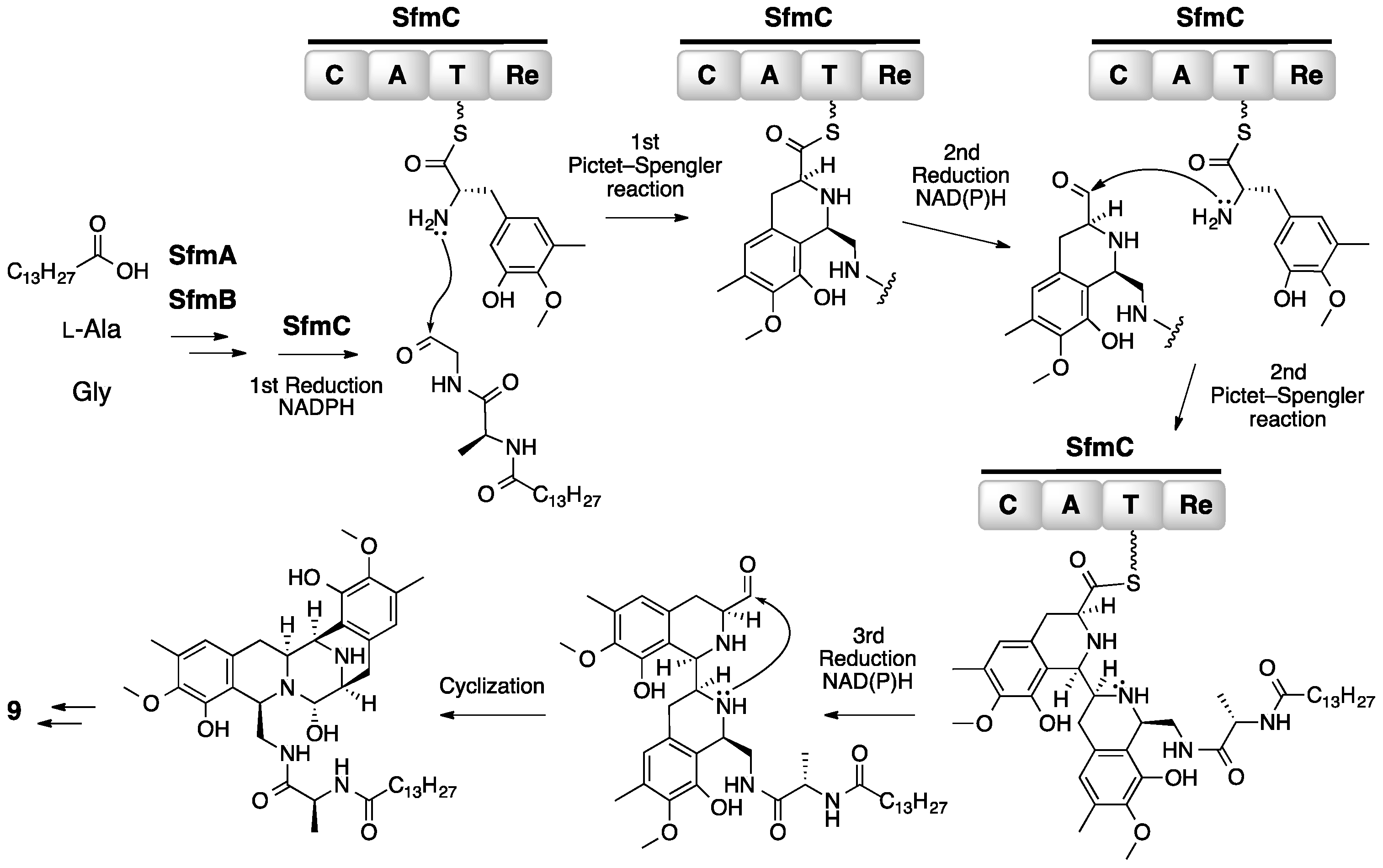
3. Saframycin
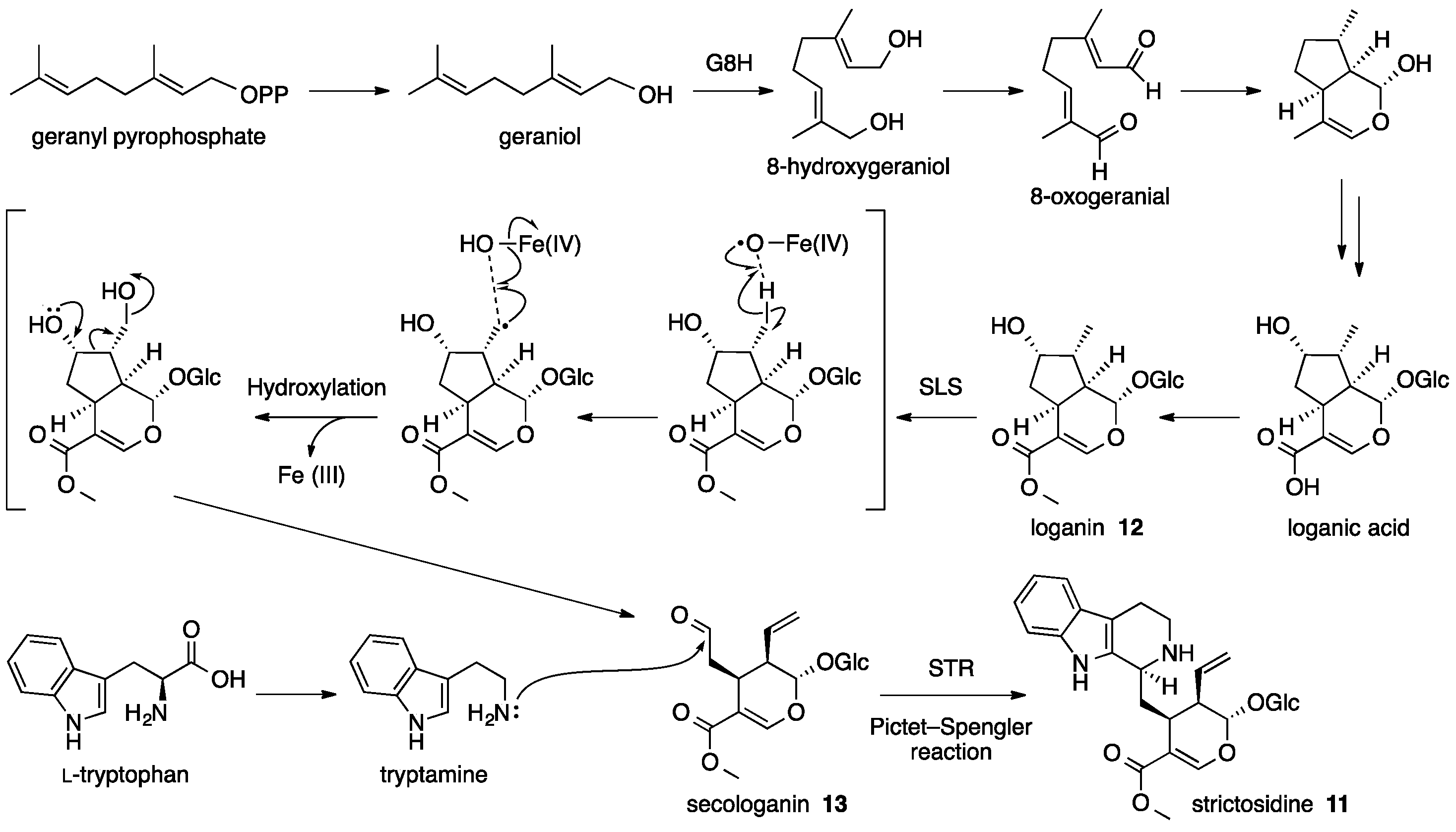
4. Strictosidine
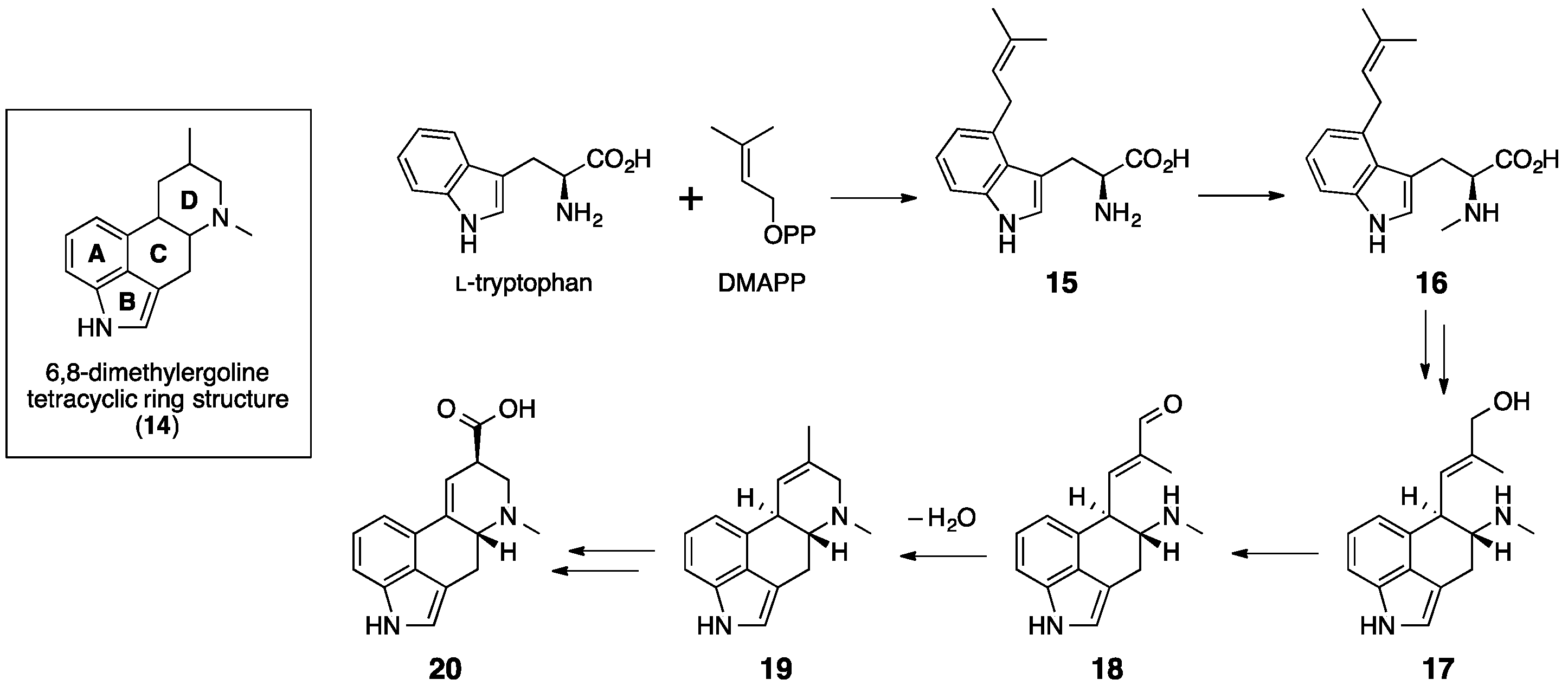
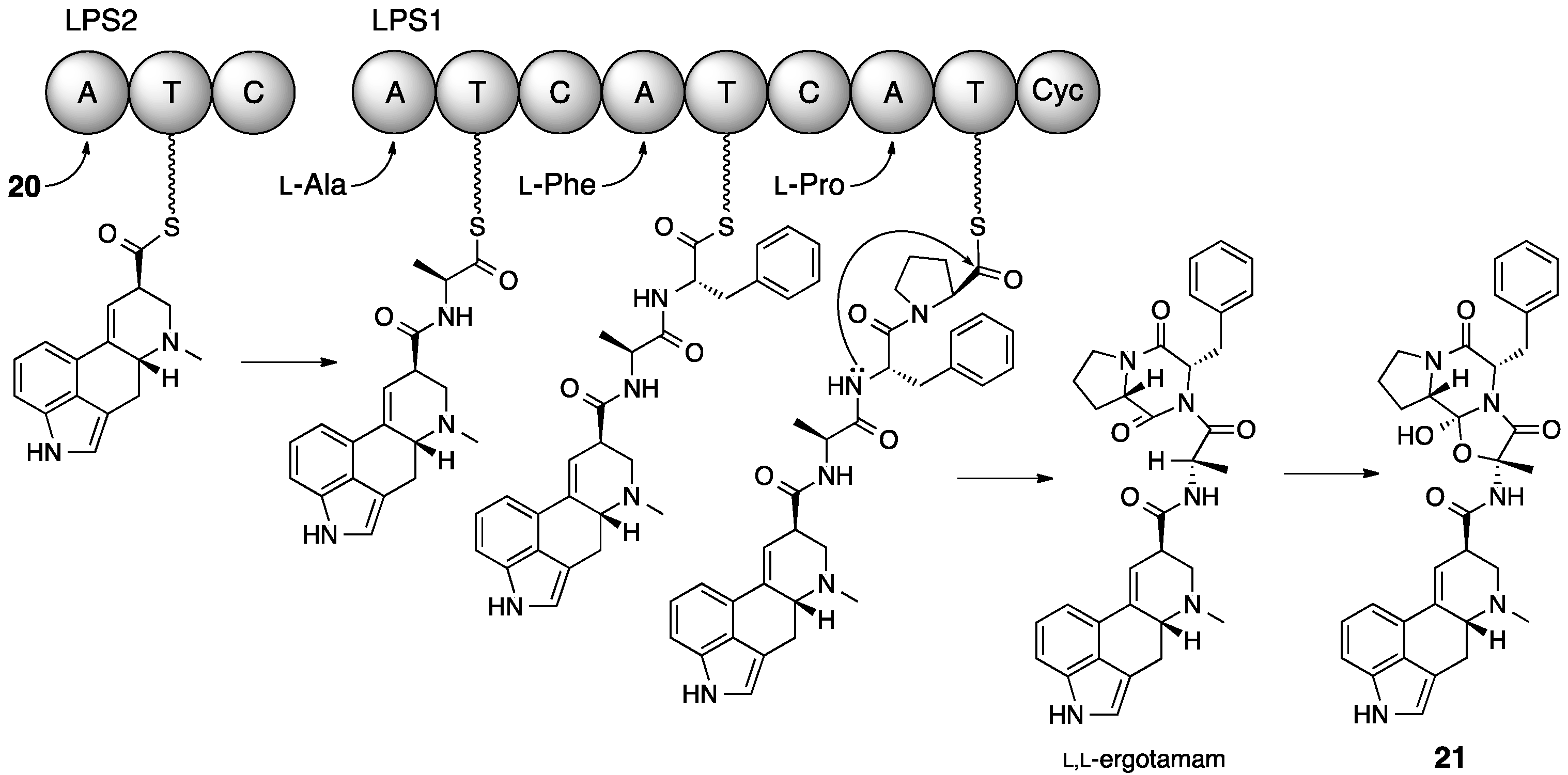
5. Ergotamine
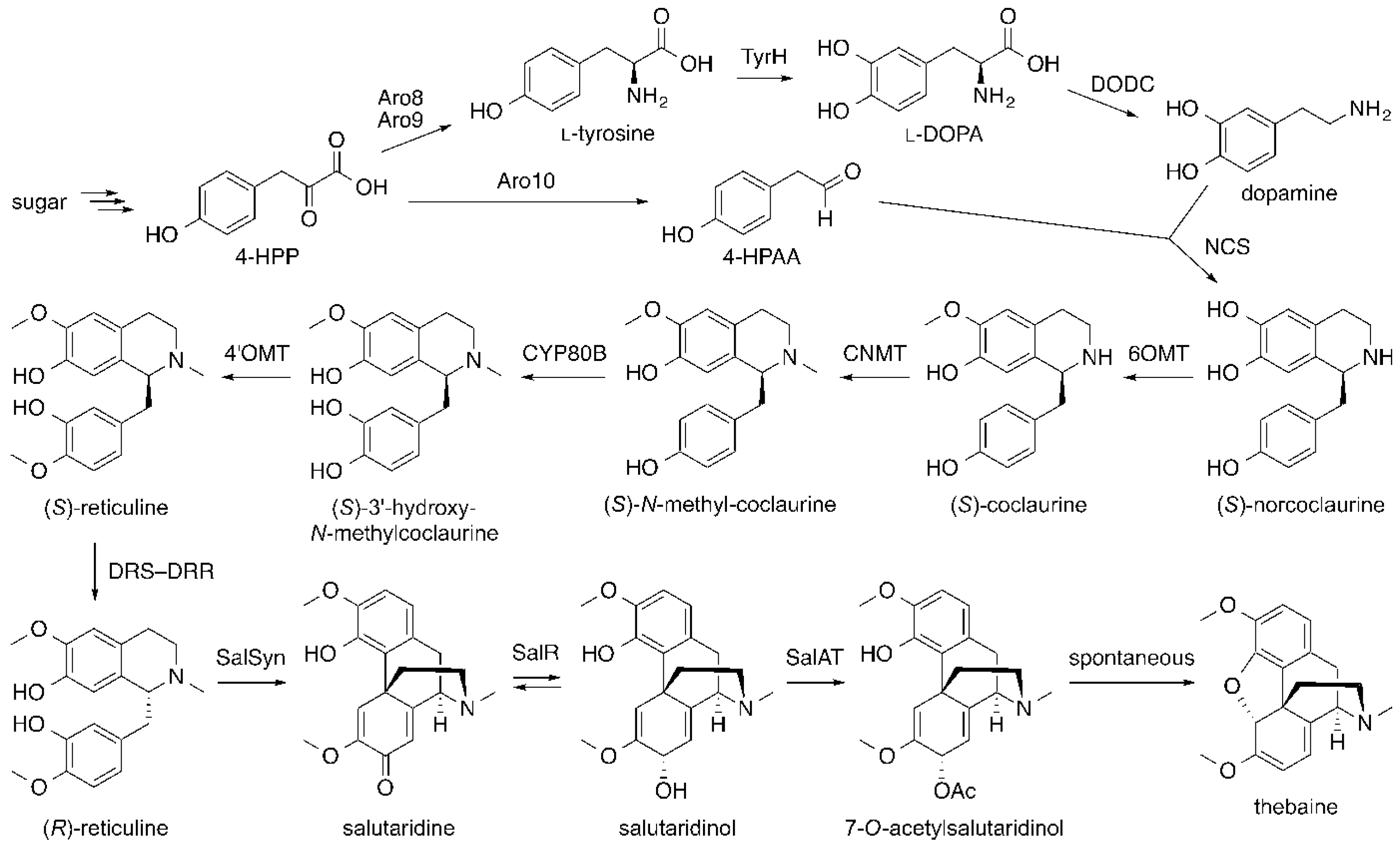
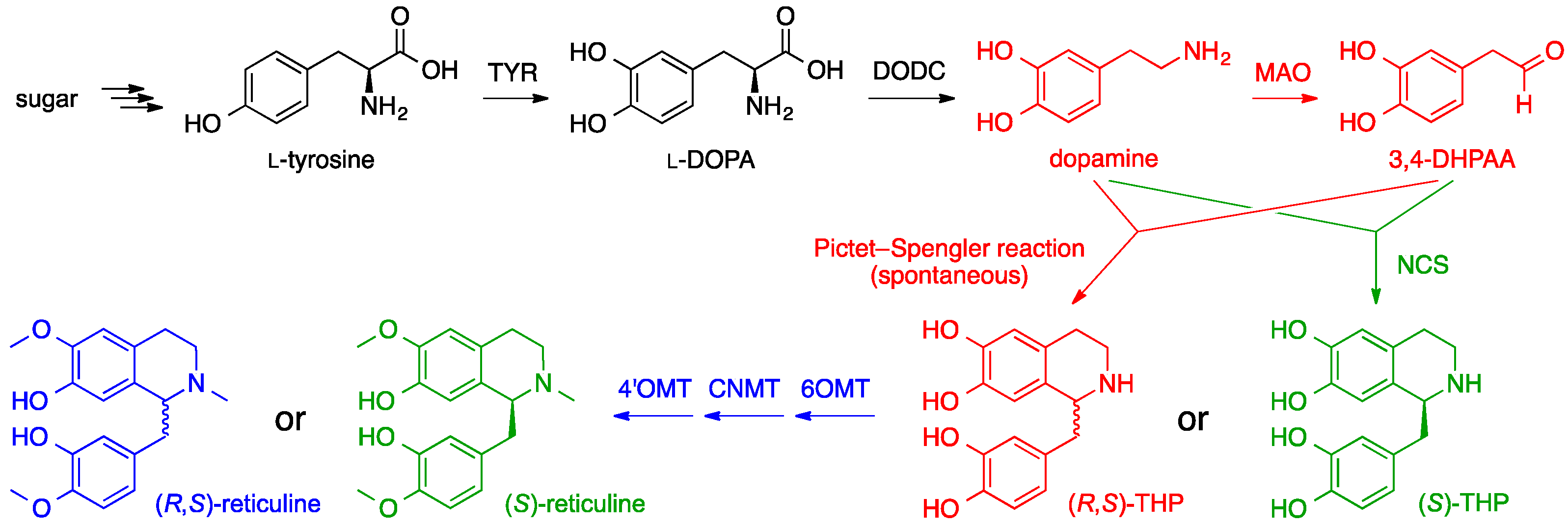
6. Opiates
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Aniszewski, T. Definition, typology, and occurrence of alkaloids. In Alkaloids, 2nd ed.; Elsevier: Boston, MA, USA, 2015; Chapter 1; pp. 1–97. [Google Scholar]
- Rathbone, D.A.; Bruce, N.C. Microbial transformation of alkaloids. Curr. Opin. Microbiol. 2002, 5, 274–281. [Google Scholar] [CrossRef]
- Diamond, A.; Desgagné-Penix, I. Metabolic engineering for the production of plant isoquinoline alkaloids. Plant Biotechnol. J. 2016, 14, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Srivastava, A.K. Biotechnology and genetic engineering for alkaloid production. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, G.K., Mérillon, J.-M., Eds.; Springer-Verlag Berlin Heidelberg: Berlin, Germany, 2013; pp. 213–250. [Google Scholar]
- Springer, J.P.; Büchi, G.; Kobbe, B.; Demain, A.L.; Clardy, J. The structure of ditryptophenaline—A new metabolite of Aspergillus flavus. Tetrahedron Lett. 1977, 18, 2403–2406. [Google Scholar] [CrossRef]
- Voloshchuk, T.; Farina, N.S.; Wauchope, O.R.; Kiprowska, M.; Haberfield, P.; Greer, A. Molecular bilateral symmetry of natural products: Prediction of selectivity of dimeric molecules by density functional theory and semiempirical calculations. J. Nat. Prod. 2004, 67, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.J.; Sedlock, D.M. 1′-(2-Phenyl-ethylene)-ditryptophenaline, a new dimeric diketopiperazine from Aspergillus flavus. J. Nat. Prod. 1994, 57, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.J.; Cai, P.; Snyder, J.K.; Sedlock, D.M.; Sun, H.H.; Cooper, R. WIN 64821, a new competitive antagonist to substance P, isolated from an Aspergillus species: Structure determination and solution conformation. J. Org. Chem. 1993, 58, 6016–6021. [Google Scholar] [CrossRef]
- Saruwatari, T.; Yagishita, F.; Mino, T.; Noguchi, H.; Hotta, K.; Watanabe, K. Cytochrome P450 as dimerization catalyst in diketopiperazine alkaloid biosynthesis. Chembiochem 2014, 15, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.A.; Nierman, W.C.; Wortman, J.R.; Pritchard, B.L.; Brown, D.; Dean, R.A.; Bhatnagar, D.; Cleveland, T.E.; Machida, M.; Yu, J. Whole genome comparison of Aspergillus flavus and A. oryzae. Med. Mycol. 2006, 44, 9–11. [Google Scholar] [CrossRef]
- Chang, P.K.; Scharfenstein, L.L.; Wei, Q.; Bhatnagar, D. Development and refinement of a high-efficiency gene-targeting system for Aspergillus flavus. J. Microbiol. Methods 2010, 81, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Tsunematsu, Y.; Ishikawa, N.; Wakana, D.; Goda, Y.; Noguchi, H.; Moriya, H.; Hotta, K.; Watanabe, K. Distinct mechanisms for spiro-carbon formation reveal biosynthetic pathway crosstalk. Nat. Chem. Biol. 2013, 9, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Maiya, S.; Grundmann, A.; Li, S.M.; Turner, G. The fumitremorgin gene cluster of Aspergillus fumigatus: Identification of a gene encoding brevianamide F synthetase. Chembiochem 2006, 7, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Saruwatari, T.; Watanabe, K. Additional unnatural diketopiperazine heterodimers produced by cytochrome P450 DtpC. 2014; unpublished results. [Google Scholar]
- Raju, R.; Piggott, A.M.; Conte, M.; Aalbersberg, W.G.; Feussner, K.; Capon, R.J. Naseseazines A and B: A new dimeric diketopiperazine framework from a marine-derived actinomycete, Streptomyces sp. Org. Lett. 2009, 11, 3862–3865. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Li, Y.L.; Zhou, J.C.; Yuan, H.Q.; Wang, X.N.; Lou, H.X. A new diketopiperazine heterodimer from an endophytic fungus Aspergillus niger. J. Asian. Nat. Prod. Res. 2015, 17, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Jiang, L.; Guo, L.; Chen, X.; Zhang, H.; Che, Y. Pestalazines and pestalamides, bioactive metabolites from the plant pathogenic fungus Pestalotiopsis theae. J. Nat. Prod. 2008, 71, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Balado, C.; de Lera, A.R. Concise total synthesis and structural revision of (+)-pestalazine B. Org. Biomol. Chem. 2010, 8, 5179–5186. [Google Scholar] [CrossRef] [PubMed]
- Varoglu, M.; Corbett, T.H.; Valeriote, F.A.; Crews, P. Asperazine, a selective cytotoxic alkaloid from a sponge-derived culture of Aspergillus niger. J. Org. Chem. 1997, 62, 7078–7079. [Google Scholar] [CrossRef] [PubMed]
- Loach, R.P.; Fenton, O.S.; Movassaghi, M. Concise total synthesis of (+)-asperazine, (+)-pestalazine A, and (+)-iso-pestalazine A. Structure revision of (+)-pestalazine A. J. Am. Chem. Soc. 2016, 138, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Takahashi, K.; Kubo, A. New antibiotics saframycins A, B, C, D and E. J. Antibiot. 1977, 30, 1015–1018. [Google Scholar] [PubMed]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.E.; Lown, J.W. Mode of action of saframycin antitumor antibiotics: Sequence selectivities in the covalent binding of saframycins A and S to deoxyribonucleic acid. Chem. Res. Toxicol. 1990, 3, 262–267. [Google Scholar] [CrossRef]
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective total synthesis of Ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203. [Google Scholar] [CrossRef]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.; Francesch, A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009, 26, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.; Perez, M.; Martin, M.J.; Chicharro, J.L.; Fernandez-Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; de la Calle, F.; et al. Synthesis of ecteinascidin ET-743 and phthalascidin Pt-650 from cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef] [PubMed]
- Pospiech, A.; Bietenhader, J.; Schupp, T. Two multifunctional peptide synthetases and an O-methyltransferase are involved in the biosynthesis of the DNA-binding antibiotic and antitumour agent saframycin Mx1 from Myxococcus xanthus. Microbiology 1996, 142, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Velasco, A.; Acebo, P.; Gomez, A.; Schleissner, C.; Rodriguez, P.; Aparicio, T.; Conde, S.; Munoz, R.; de la Calle, F.; Garcia, J.L.; et al. Molecular characterization of the safracin biosynthetic pathway from Pseudomonas fluorescens A2-2: Designing new cytotoxic compounds. Mol. Microbiol. 2005, 56, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, W.; Song, J.; Ding, W.; Zhao, Q.F.; Peng, C.; Song, W.W.; Tang, G.L.; Liu, W. Characterization of the saframycin A gene cluster from Streptomyces lavendulae NRRL 11002 revealing a nonribosomal peptide synthetase system for assembling the unusual tetrapeptidyl skeleton in an iterative manner. J. Bacteriol. 2008, 190, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Takahashi, K.; Yazawa, K.; Arai, T.; Namikoshi, M.; Iwasaki, S.; Okuda, S. Biosynthetic studies on saframycin A, a quinone antitumor antibiotic produced by Streptomyces lavendulae. J. Biol. Chem. 1985, 260, 344–348. [Google Scholar] [PubMed]
- Arai, T.; Yazawa, K.; Takahashi, K.; Maeda, A.; Mikami, Y. Directed biosynthesis of new saframycin derivatives with resting cells of Streptomyces lavendulae. Antimicrob. Agents Chemother. 1985, 28, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.T.; Lee, J.; Sims, J.W.; Schmidt, E.W. Characterization of SafC, a catechol 4-O-methyltransferase involved in saframycin biosynthesis. Appl. Environ. Microbiol. 2007, 73, 3575–3580. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Nelson, J.T.; Fillmore, J.P. Synthesis of tyrosine derivatives for saframycin MX1 biosynthetic studies. Tetrahedron Lett. 2004, 45, 3921–3924. [Google Scholar] [CrossRef]
- Koketsu, K.; Watanabe, K.; Suda, H.; Oguri, H.; Oikawa, H. Reconstruction of the saframycin core scaffold defines dual Pictet-Spengler mechanisms. Nat. Chem. Biol. 2010, 6, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.B.; Bumpus, S.B.; Aron, Z.D.; Kelleher, N.L.; Walsh, C.T. The loading module of mycosubtilin: An adenylation domain with fatty acid selectivity. J. Am. Chem. Soc. 2007, 129, 6366–6367. [Google Scholar] [CrossRef] [PubMed]
- Baltz, R.H.; Miao, V.; Wrigley, S.K. Natural products to drugs: Daptomycin and related lipopeptide antibiotics. Nat. Prod. Rep. 2005, 22, 717–741. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, S.E.; Maresh, J.J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 2006, 23, 532–547. [Google Scholar] [CrossRef] [PubMed]
- De Luca, V.; Salim, V.; Thamm, A.; Masada, S.A.; Yu, F. Making iridoids/secoiridoids and monoterpenoid indole alkaloids: Progress on pathway elucidation. Curr. Opin. Plant Biol. 2014, 19, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Slim, H.B.; Black, H.R.; Thompson, P.D. Older blood pressure medications—Do they still have a place? Am. J. Cardiol. 2011, 108, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.E.; Senol, F.S. Alkaloids and inhibitory effects against enzymes linked to neurodegenerative diseases (physostigmine, galanthamine, huperzine, etc.). In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, G.K., Mérillon, J.-M., Eds.; Springer-Verlag: Berlin, Germany, 2013; pp. 1525–1539. [Google Scholar]
- Li, J.; Wang, T.; Yu, P.; Peterson, A.; Weber, R.; Soerens, D.; Grubisha, D.; Bennett, D.; Cook, J.M. General approach for the synthesis of ajmaline/sarpagine indole alkaloids: Enantiospecific total synthesis of (+)-ajmaline, alkaloid G, and norsuaveoline via the asymmetric Pictet-Spengler reaction. J. Am. Chem. Soc. 1999, 121, 6998–7010. [Google Scholar] [CrossRef]
- Ishikawa, H.; Colby, D.A.; Boger, D.L. Direct coupling of catharanthine and vindoline to provide vinblastine: Total synthesis of (+)- and ent-(−)-vinblastine. J. Am. Chem. Soc. 2008, 130, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Irmler, S.; Schroder, G.; St-Pierre, B.; Crouch, N.P.; Hotze, M.; Schmidt, J.; Strack, D.; Matern, U.; Schroder, J. Indole alkaloid biosynthesis in Catharanthus roseus: New enzyme activities and identification of cytochrome P450 CYP72A1 as secologanin synthase. Plant J. 2000, 24, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Katano, N.; Ooi, A.; Inoue, K. Secologanin synthase which catalyzes the oxidative cleavage of loganin into secologanin is a cytochrome P450. Phytochemistry 2000, 53, 7–12. [Google Scholar] [CrossRef]
- Maresh, J.J.; Giddings, L.A.; Friedrich, A.; Loris, E.A.; Panjikar, S.; Trout, B.L.; Stöckigt, J.; Peters, B.; O′Connor, S.E. Strictosidine synthase: Mechanism of a Pictet-Spengler catalyzing enzyme. J. Am. Chem. Soc. 2008, 130, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Clastre, M.; Courdavault, V.; O’Connor, S.E. De novo production of the plant-derived alkaloid strictosidine in yeast. Proc. Natl. Acad. Sci. USA 2015, 112, 3205–3210. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, K.; Dong, L.; Navrot, N.; Schneider, T.; Burlat, V.; Pollier, J.; Woittiez, L.; van der Krol, S.; Lugan, R.; Ilc, T.; et al. The seco-iridoid pathway from Catharanthus roseus. Nat. Commun. 2014, 5, 3606. [Google Scholar] [CrossRef] [PubMed]
- Nevoigt, E. Progress in metabolic engineering of Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2008, 72, 379–412. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.; Croteau, R. Geranyl diphosphate synthase from Abies grandis: cDNA isolation, functional expression, and characterization. Arch. Biochem. Biophys. 2002, 405, 130–136. [Google Scholar] [CrossRef]
- Stanley Fernandez, S.M.; Kellogg, B.A.; Poulter, C.D. Farnesyl diphosphate synthase. Altering the catalytic site to select for geranyl diphosphate activity. Biochemistry 2000, 39, 15316–15321. [Google Scholar] [CrossRef] [PubMed]
- Schardl, C.L.; Panaccione, D.G.; Tudzynski, P. Ergot alkaloids—Biology and molecular biology. Alkaloids Chem. Biol. 2006, 63, 45–86. [Google Scholar] [PubMed]
- Gröcer, D.; Floss, H.G. Biochemistry of ergot alkaloids—Achievements and challenges. Alkaloids Chem. Biol. 1998, 50, 171–218. [Google Scholar]
- Mrusek, M.; Seo, E.J.; Greten, H.J.; Simon, M.; Efferth, T. Identification of cellular and molecular factors determining the response of cancer cells to six ergot alkaloids. Investig. New Drugs 2015, 33, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.L. Ergot and its alkaloids. Am. J. Pharm. Educ. 2006, 70, Z1. [Google Scholar] [CrossRef]
- Coyle, C.M.; Panaccione, D.G. An ergot alkaloid biosynthesis gene and clustered hypothetical genes from Aspergillus fumigatus. Appl. Environ. Microbiol. 2005, 71, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- Gebler, J.C.; Poulter, C.D. Purification and characterization of dimethylallyl tryptophan synthase from Claviceps purpurea. Arch. Biochem. Biophys. 1992, 296, 308–313. [Google Scholar] [CrossRef]
- Gebler, J.C.; Woodside, A.B.; Poulter, C.D. Dimethylallyltryptophan synthase. An enzyme-catalyzed electrophilic aromatic substitution. J. Am. Chem. Soc. 1992, 114, 7354–7360. [Google Scholar] [CrossRef]
- Otsuka, H.; Quigley, F.R.; Groeger, D.; Anderson, J.A.; Floss, H.G. d-Lysergyl peptide synthetase from the ergot fungus Claviceps purpurea. Planta Med. 1980, 40, 109–119. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Wu, J.P.; Shibuya, M.; Floss, H.G. Probing ergot alkaloid biosynthesis: Identification of advanced intermediates along the biosynthetic pathway. J. Am. Chem. Soc. 1988, 110, 1970–1971. [Google Scholar] [CrossRef]
- Riederer, B.; Han, M.; Keller, U. d-Lysergyl peptide synthetase from the ergot fungus Claviceps purpurea. J. Biol. Chem. 1996, 271, 27524–27530. [Google Scholar] [CrossRef] [PubMed]
- Walzel, B.; Riederer, B.; Keller, U. Mechanism of alkaloid cyclopeptide synthesis in the ergot fungus Claviceps purpurea. Chem. Biol. 1997, 4, 223–230. [Google Scholar] [CrossRef]
- Ortel, I.; Keller, U. Combinatorial assembly of simple and complex d-lysergic acid alkaloid peptide classes in the ergot fungus Claviceps purpurea. J. Biol. Chem. 2009, 284, 6650–6660. [Google Scholar] [CrossRef] [PubMed]
- Havemann, J.; Vogel, D.; Loll, B.; Keller, U. Cyclolization of d-lysergic acid alkaloid peptides. Chem. Biol. 2014, 21, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.L.; Moore, C.T.; Panaccione, D.G. Partial reconstruction of the ergot alkaloid pathway by heterologous gene expression in Aspergillus nidulans. Toxins 2013, 5, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Wink, M. A Short History of Alkaloids. In Alkaloids: Biochemistry, Ecology, and Medicinal Applications; Roberts, M.F., Wink, M., Eds.; Springer US: Boston, MA, USA, 1998; pp. 11–44. [Google Scholar]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Inui, T.; Takemura, T. Metabolic engineering in isoquinoline alkaloid biosynthesis. Curr. Pharm. Biotechnol. 2007, 8, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Keasling, J.D. Production of isoprenoid pharmaceuticals by engineered microbes. Nat. Chem. Biol. 2006, 2, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Minami, H.; Kim, J.S.; Ikezawa, N.; Takemura, T.; Katayama, T.; Kumagai, H.; Sato, F. Microbial production of plant benzylisoquinoline alkaloids. Proc. Natl. Acad. Sci. USA 2008, 105, 7393–7398. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, K.M.; Smolke, C.D. Production of benzylisoquinoline alkaloids in Saccharomyces cerevisiae. Nat. Chem. Biol. 2008, 4, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Galanie, S.; Smolke, C.D. Optimization of yeast-based production of medicinal protoberberine alkaloids. Microb. Cell Fact. 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Thodey, K.; Galanie, S.; Smolke, C.D. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol. 2014, 10, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Tsujita, T.; Katagiri, Y.; Yoshida, S.; Yamada, Y. Purification and characterization of S-adenosyl-l-methionine: Norcoclaurine 6-O-methyltransferase from cultured Coptis japonica cells. Eur. J. Biochem. 1994, 225, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Ounaroon, A.; Decker, G.; Schmidt, J.; Lottspeich, F.; Kutchan, T.M. (R,S)-Reticuline 7-O-methyltransferase and (R,S)-norcoclaurine 6-O-methyltransferase of Papaver somniferum—cDNA cloning and characterization of methyl transfer enzymes of alkaloid biosynthesis in opium poppy. Plant J. 2003, 36, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Gesell, A.; Chavez, M.L.; Kramell, R.; Piotrowski, M.; Macheroux, P.; Kutchan, T.M. Heterologous expression of two FAD-dependent oxidases with (S)-tetrahydroprotoberberine oxidase activity from Argemone mexicana and Berberis wilsoniae in insect cells. Planta 2011, 233, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Fossati, E.; Ekins, A.; Narcross, L.; Zhu, Y.; Falgueyret, J.P.; Beaudoin, G.A.; Facchini, P.J.; Martin, V.J. Reconstitution of a 10-gene pathway for synthesis of the plant alkaloid dihydrosanguinarine in Saccharomyces cerevisiae. Nat. Commun. 2014, 5, 3283. [Google Scholar] [CrossRef] [PubMed]
- Galanie, S.; Thodey, K.; Trenchard, I.J.; Filsinger Interrante, M.; Smolke, C.D. Complete biosynthesis of opioids in yeast. Science 2015, 349, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Michener, J.K.; Nielsen, J.; Smolke, C.D. Identification and treatment of heme depletion attributed to overexpression of a lineage of evolved P450 monooxygenases. Proc. Natl. Acad. Sci. USA 2012, 109, 19504–19509. [Google Scholar] [CrossRef] [PubMed]
- Hazelwood, L.A.; Daran, J.M.; van Maris, A.J.; Pronk, J.T.; Dickinson, J.R. The Ehrlich pathway for fusel alcohol production: A century of research on Saccharomyces cerevisiae metabolism. Appl. Environ. Microbiol. 2008, 74, 2259–2266. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, A.; Minami, H.; Kim, J.S.; Koyanagi, T.; Katayama, T.; Sato, F.; Kumagai, H. A bacterial platform for fermentative production of plant alkaloids. Nat. Commun. 2011, 2, 326. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, A.; Matsuzaki, C.; Matsumura, E.; Koyanagi, T.; Katayama, T.; Yamamoto, K.; Sato, F.; Kumagai, H.; Minami, H. (R,S)-tetrahydropapaveroline production by stepwise fermentation using engineered Escherichia coli. Sci. Rep. 2014, 4, 6695. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, A.; Matsumura, E.; Koyanagi, T.; Katayama, T.; Kawano, N.; Yoshimatsu, K.; Yamamoto, K.; Kumagai, H.; Sato, F.; Minami, H. Total biosynthesis of opiates by stepwise fermentation using engineered Escherichia coli. Nat. Commun. 2016, 7, 10390. [Google Scholar] [CrossRef] [PubMed]
















© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishimoto, S.; Sato, M.; Tsunematsu, Y.; Watanabe, K. Evaluation of Biosynthetic Pathway and Engineered Biosynthesis of Alkaloids. Molecules 2016, 21, 1078. https://doi.org/10.3390/molecules21081078
Kishimoto S, Sato M, Tsunematsu Y, Watanabe K. Evaluation of Biosynthetic Pathway and Engineered Biosynthesis of Alkaloids. Molecules. 2016; 21(8):1078. https://doi.org/10.3390/molecules21081078
Chicago/Turabian StyleKishimoto, Shinji, Michio Sato, Yuta Tsunematsu, and Kenji Watanabe. 2016. "Evaluation of Biosynthetic Pathway and Engineered Biosynthesis of Alkaloids" Molecules 21, no. 8: 1078. https://doi.org/10.3390/molecules21081078