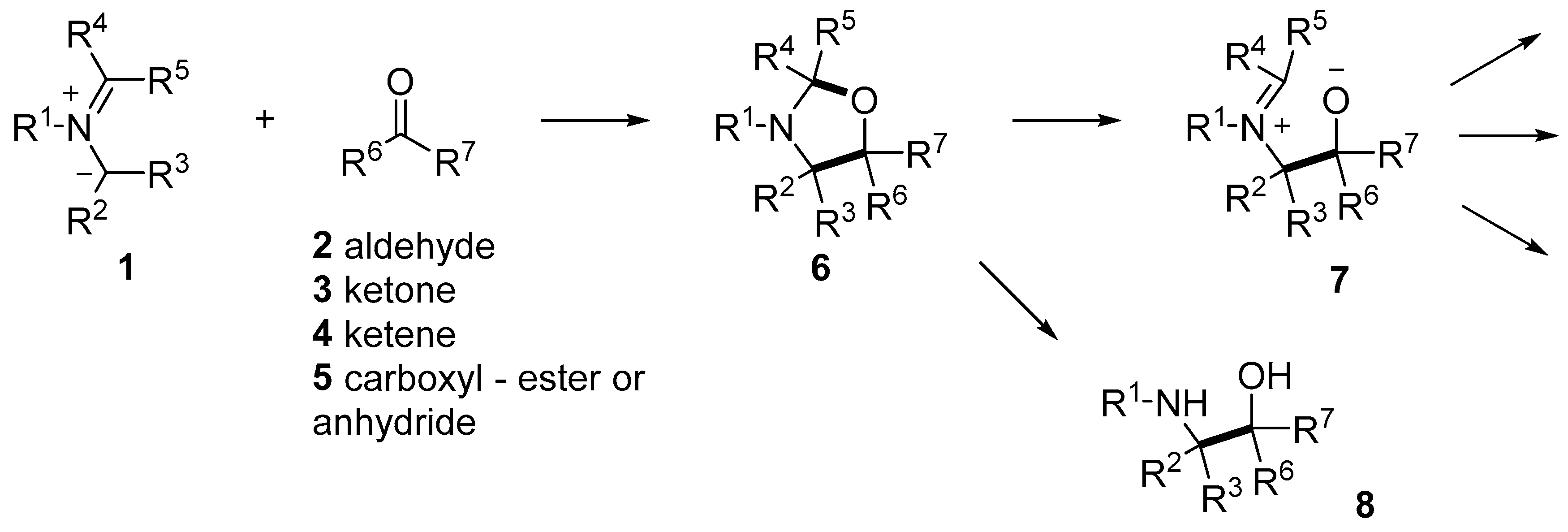
Scheme 1.
1,3-Dipolar cycloaddition reactions of azomethine ylides and carbonyl compounds to give oxazolidines and chemistry of the oxazolidines.
Scheme 1.
1,3-Dipolar cycloaddition reactions of azomethine ylides and carbonyl compounds to give oxazolidines and chemistry of the oxazolidines.
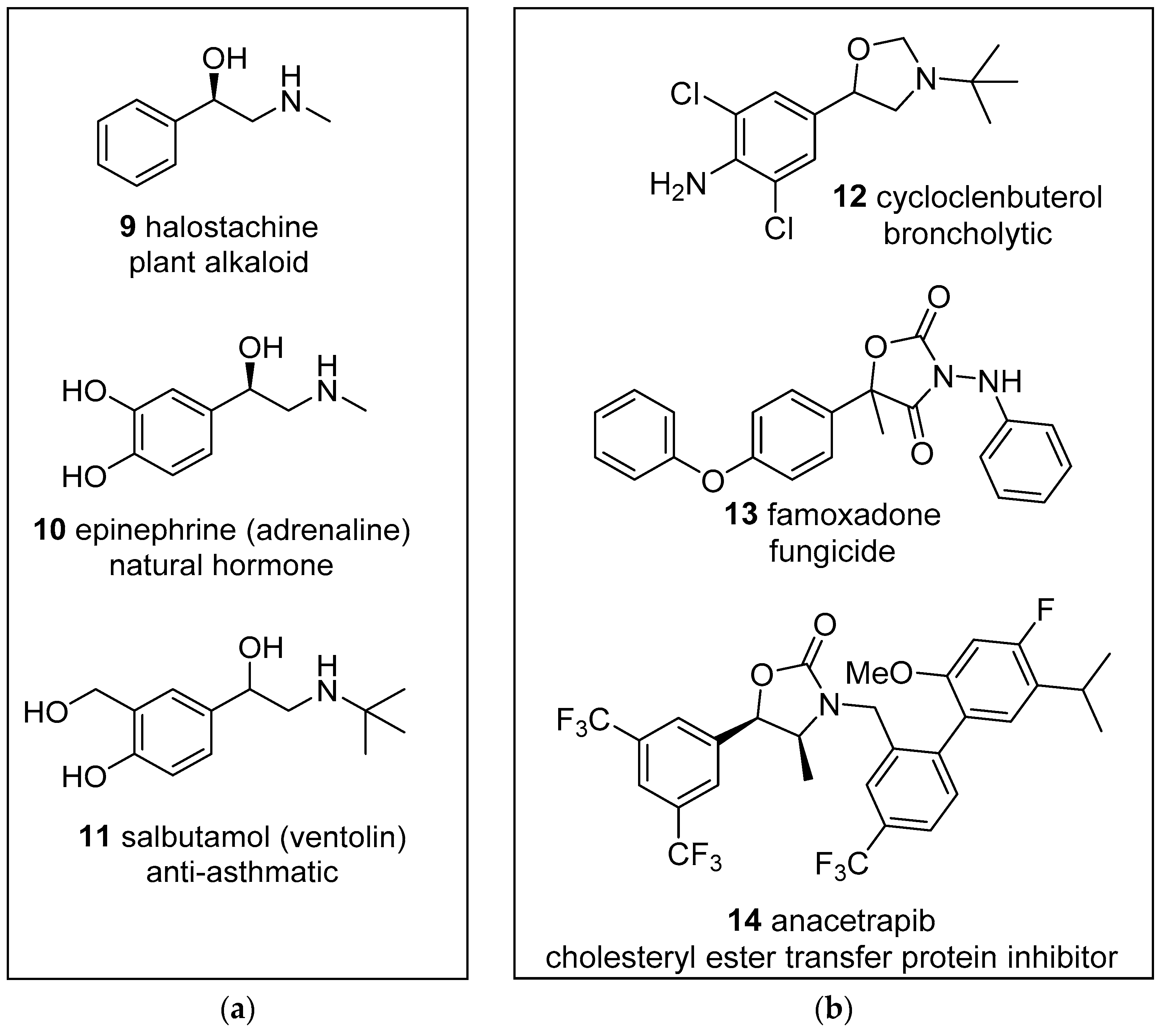
Figure 1.
Examples of biologically active: (a) 1-phenyl-1-ethanol-2-amine derivatives; (b) oxazolidine derivatives.
Figure 1.
Examples of biologically active: (a) 1-phenyl-1-ethanol-2-amine derivatives; (b) oxazolidine derivatives.
Scheme 2.
Cycloaddition of azomethine ylides 16a-cis and 16b-trans to benzaldehyde (2a).
Scheme 2.
Cycloaddition of azomethine ylides 16a-cis and 16b-trans to benzaldehyde (2a).
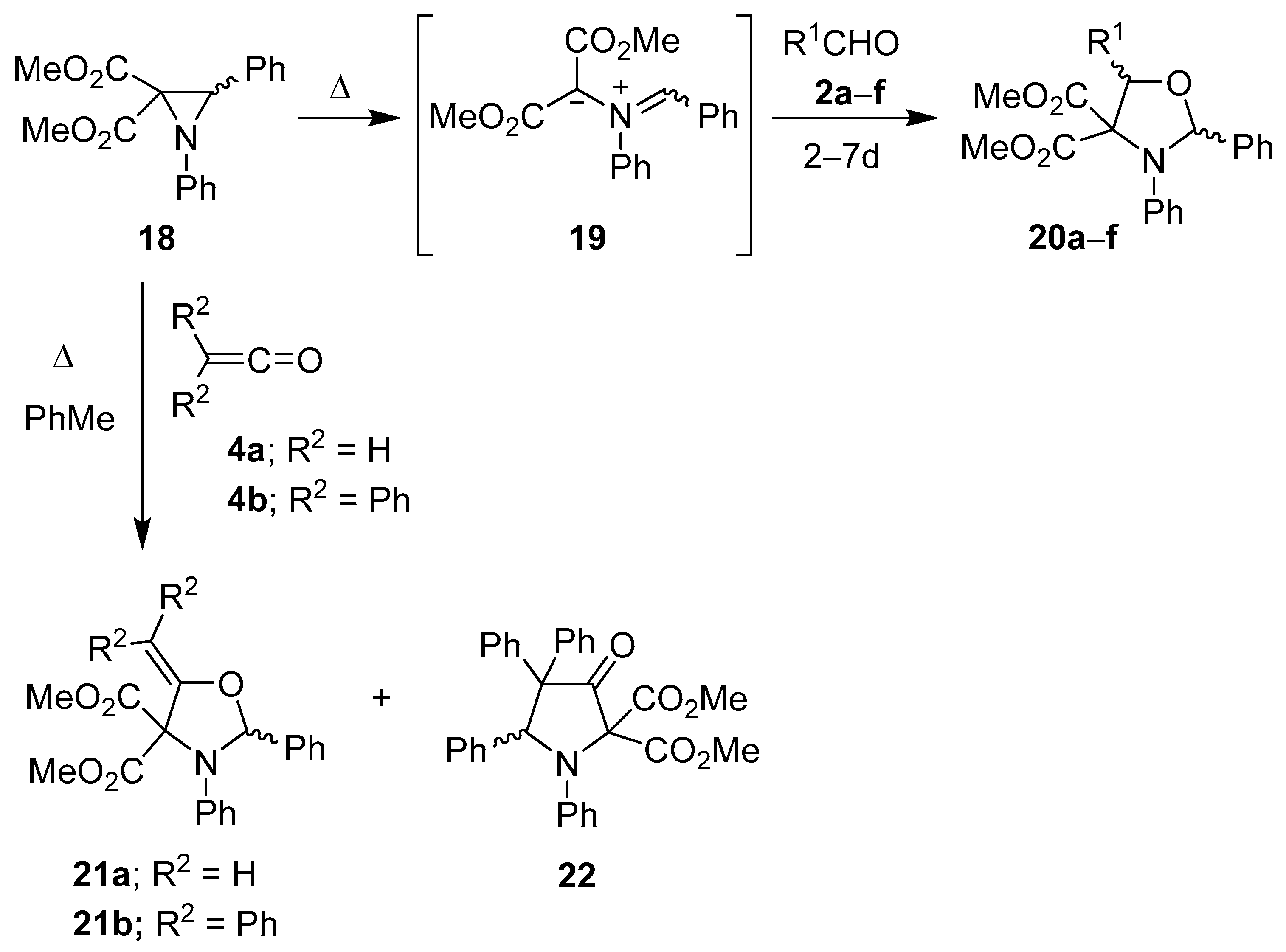
Scheme 3.
Cycloaddition of azomethine ylide 19 to aldehydes 2a–f and to ketenes 4a and 4b.
Scheme 3.
Cycloaddition of azomethine ylide 19 to aldehydes 2a–f and to ketenes 4a and 4b.
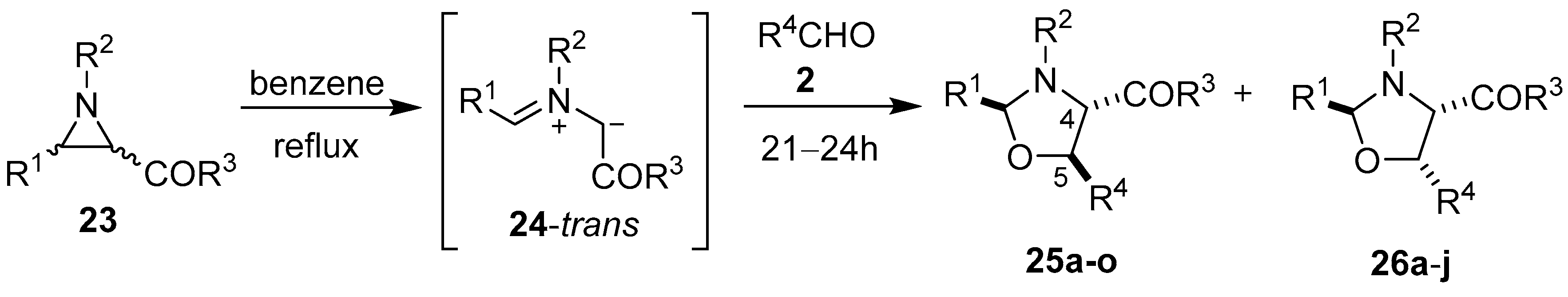
Scheme 4.
Cycloaddition of trans-azomethine ylide 24, generated from 2-benzoylaziridines 23, to aldehydes 2.
Scheme 4.
Cycloaddition of trans-azomethine ylide 24, generated from 2-benzoylaziridines 23, to aldehydes 2.
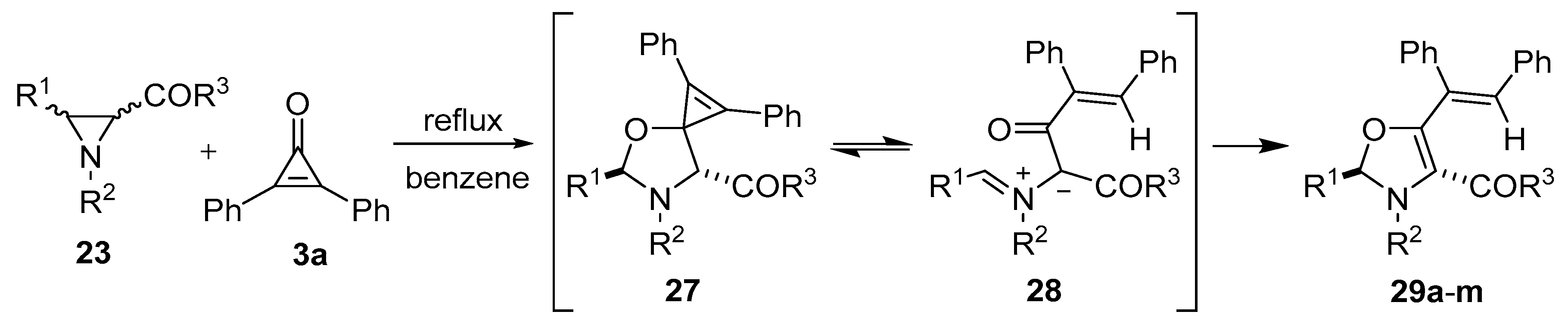
Scheme 5.
Reaction of aziridines 23 with diphenylcyclopropenone 3a to afford 4-aroyl- and 4-acyl-4-oxazolines 29.
Scheme 5.
Reaction of aziridines 23 with diphenylcyclopropenone 3a to afford 4-aroyl- and 4-acyl-4-oxazolines 29.
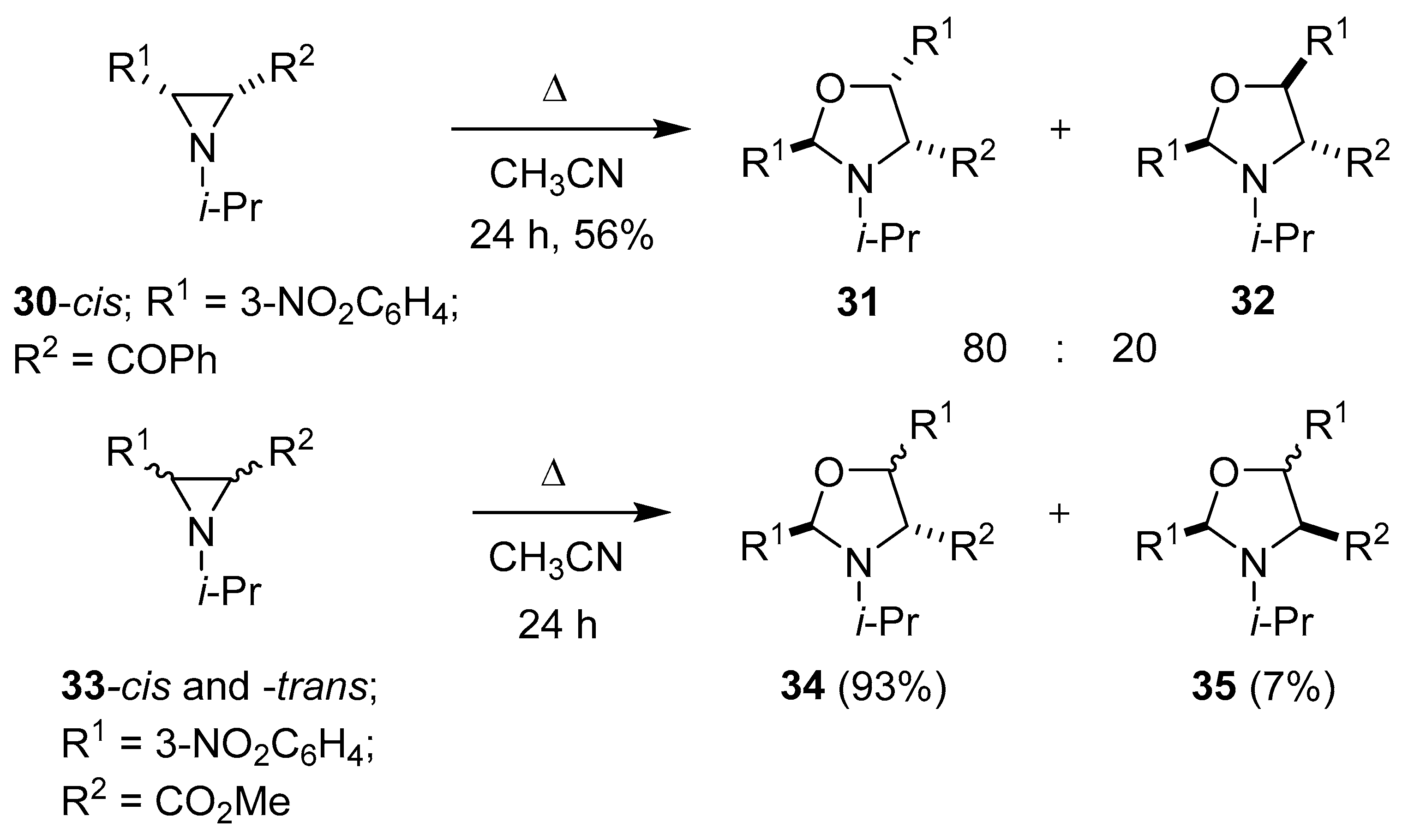
Scheme 6.
The partial fragmentation/cycloaddition reactions of aziridines 30 and 33.
Scheme 6.
The partial fragmentation/cycloaddition reactions of aziridines 30 and 33.
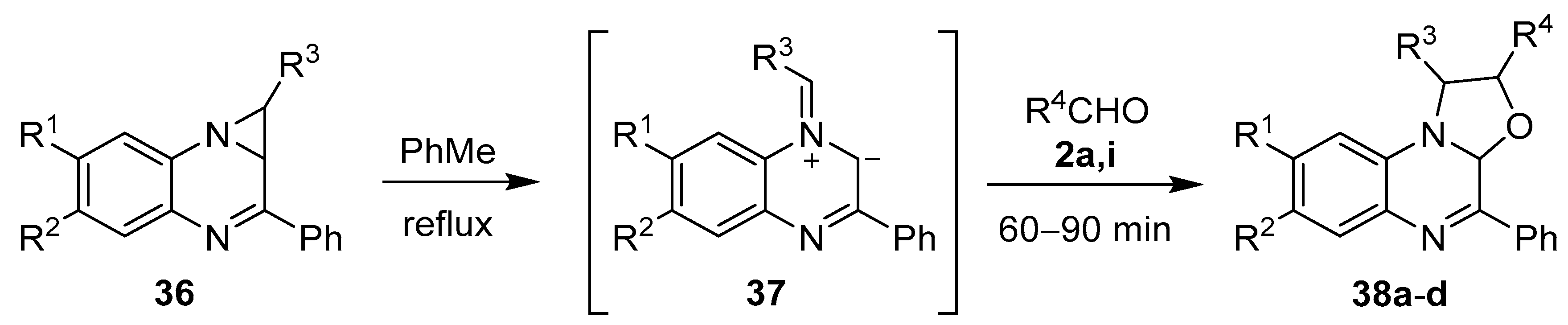
Scheme 7.
Cycloaddition of azomethine ylide 37, generated from 1,1a-dihydro-1,2-diarylazirino[1,2-a]quinoxalines 36, to aromatic aldehydes 2a and 2i.
Scheme 7.
Cycloaddition of azomethine ylide 37, generated from 1,1a-dihydro-1,2-diarylazirino[1,2-a]quinoxalines 36, to aromatic aldehydes 2a and 2i.
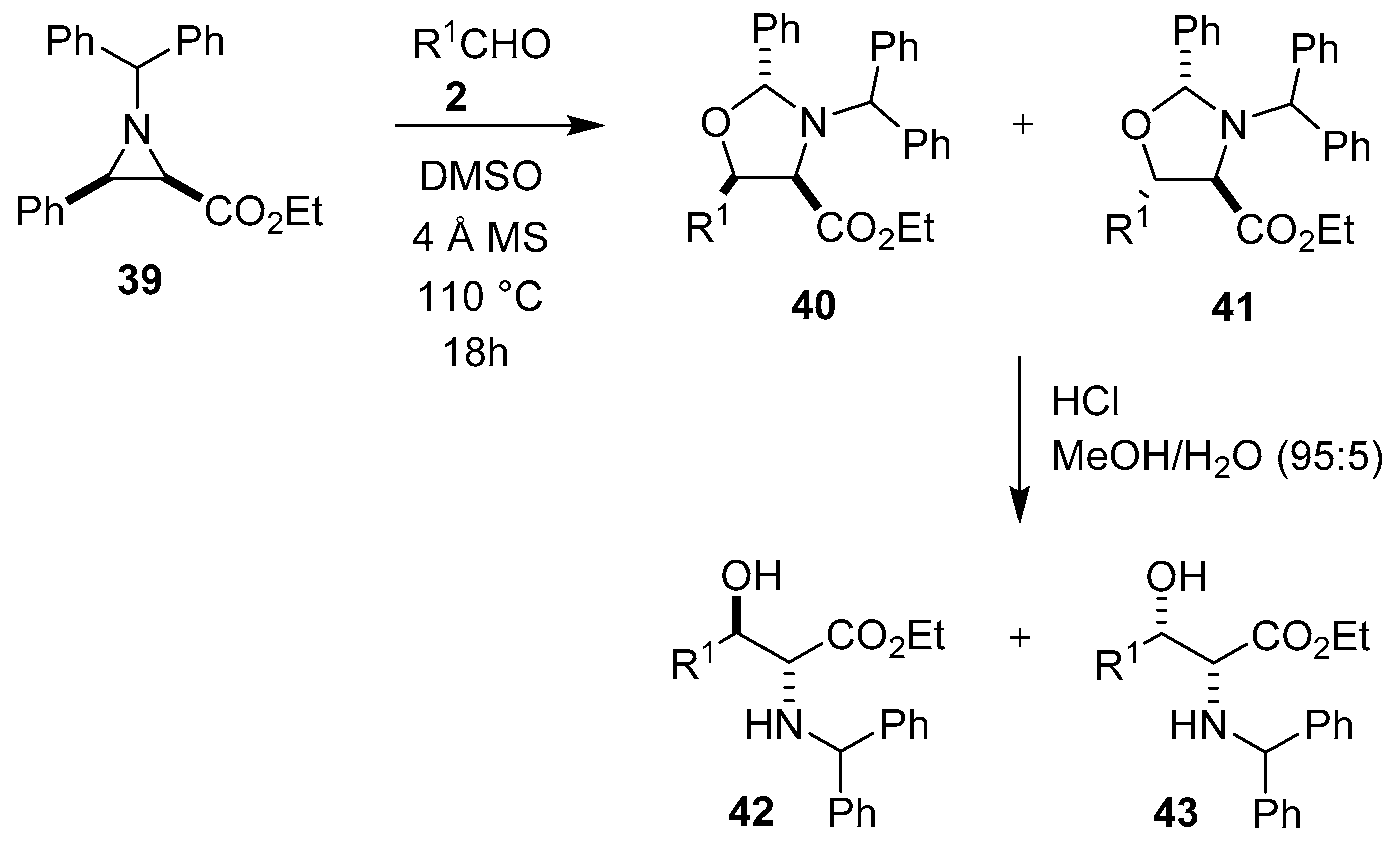
Scheme 8.
Reaction of aziridine 39 with aldehydes 2 to afford oxazolidines 40 and 41.
Scheme 8.
Reaction of aziridine 39 with aldehydes 2 to afford oxazolidines 40 and 41.
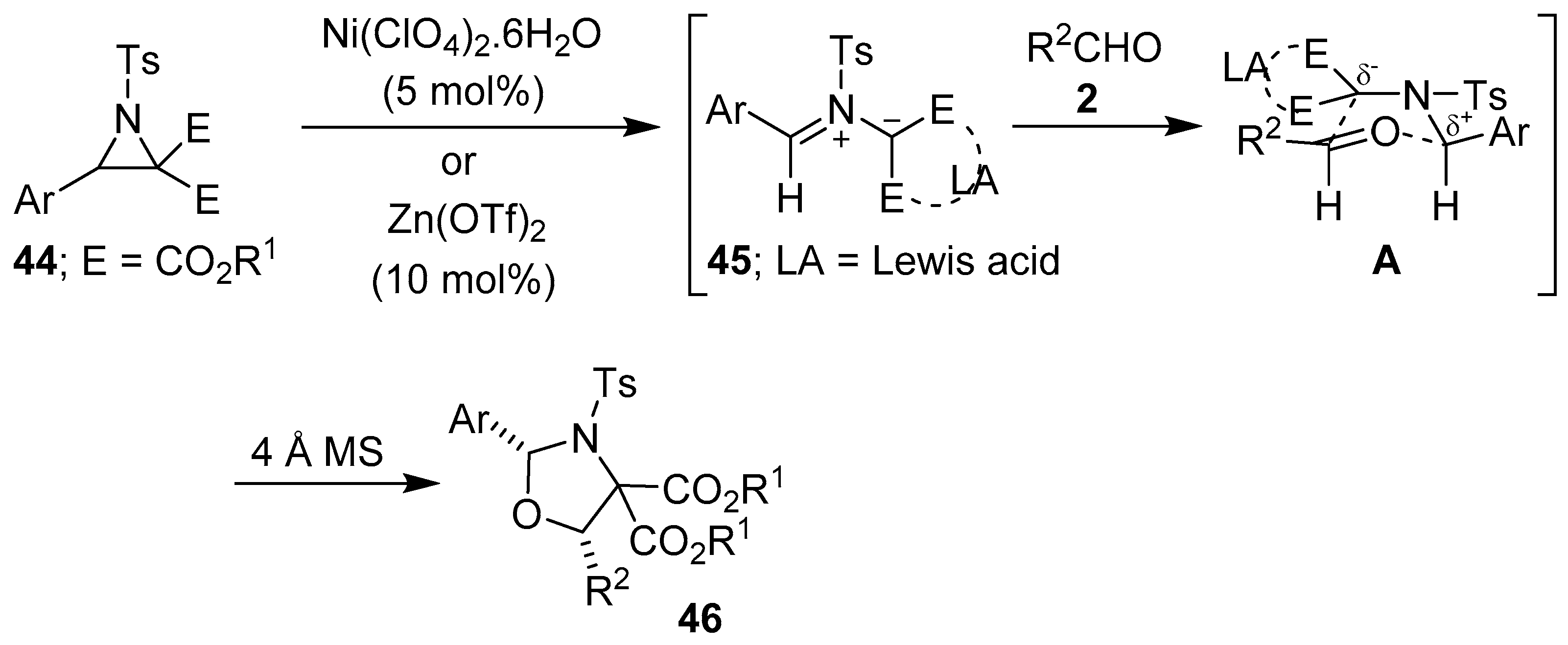
Scheme 9.
Lewis acid-promoted cycloaddition of azomethine ylides 45, generated from aziridines 44, to aldehydes 2.
Scheme 9.
Lewis acid-promoted cycloaddition of azomethine ylides 45, generated from aziridines 44, to aldehydes 2.
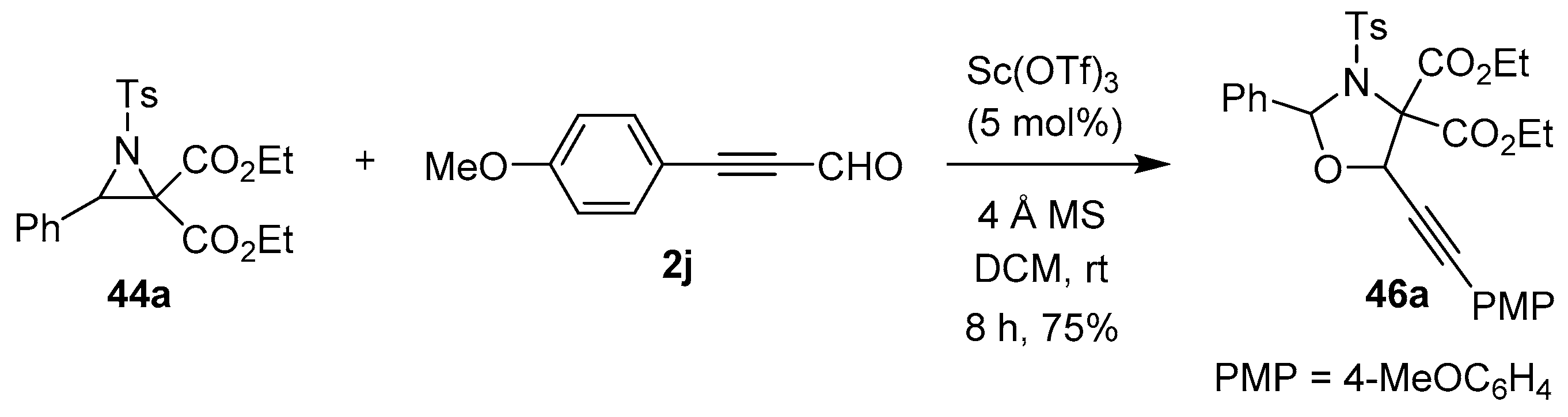
Scheme 10.
Reaction of N-tosylaziridine 44a with (4-methoxyphenyl)propiolaldehyde (2j) to form 1,3-oxazolidine 46a.
Scheme 10.
Reaction of N-tosylaziridine 44a with (4-methoxyphenyl)propiolaldehyde (2j) to form 1,3-oxazolidine 46a.
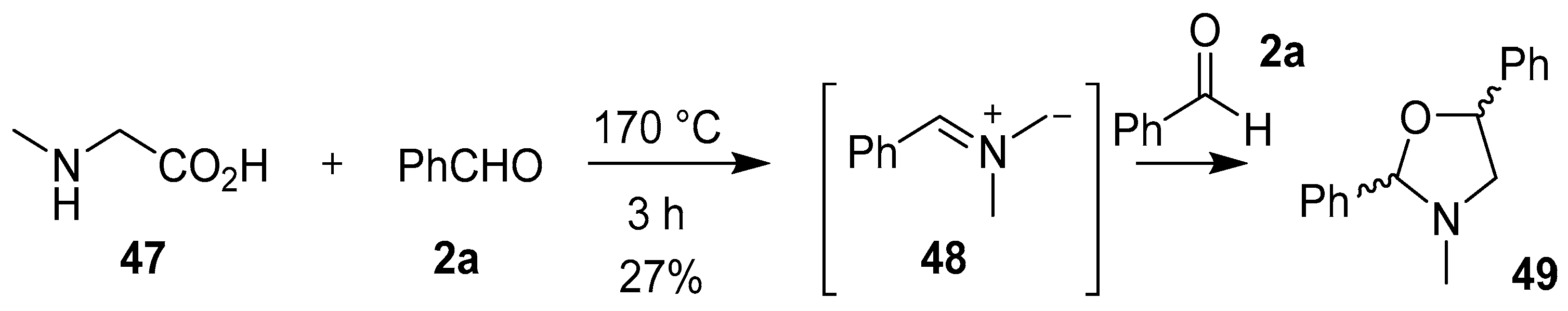
Scheme 11.
Cycloaddition of azomethine ylide 48 to benzaldehyde (2a).
Scheme 11.
Cycloaddition of azomethine ylide 48 to benzaldehyde (2a).
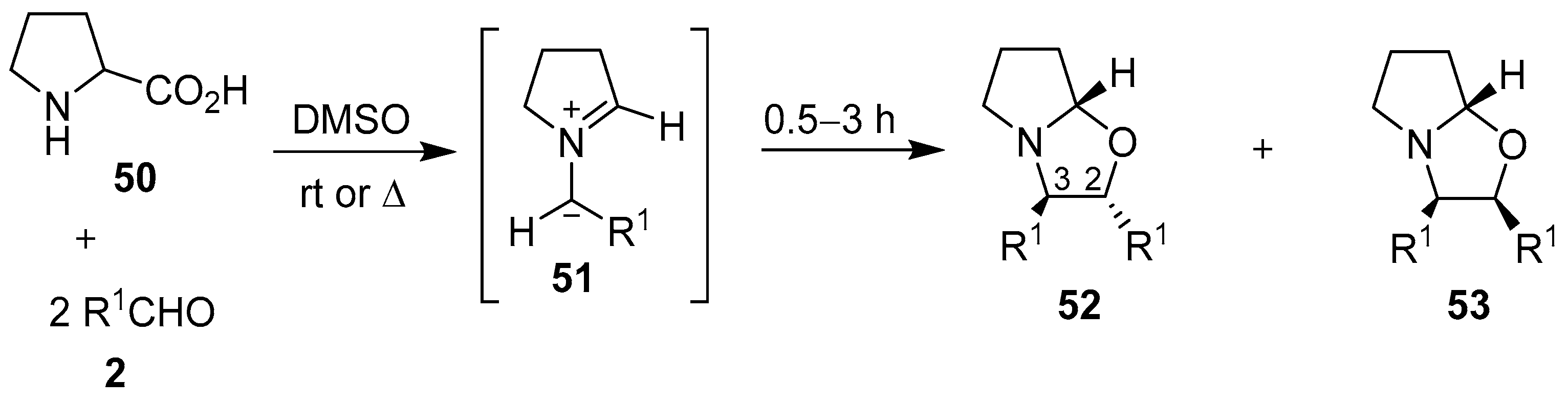
Scheme 12.
Cycloaddition of azomethine ylides 51, generated from proline (50) and aromatic aldehydes 2, to a second mole of aldehyde.
Scheme 12.
Cycloaddition of azomethine ylides 51, generated from proline (50) and aromatic aldehydes 2, to a second mole of aldehyde.
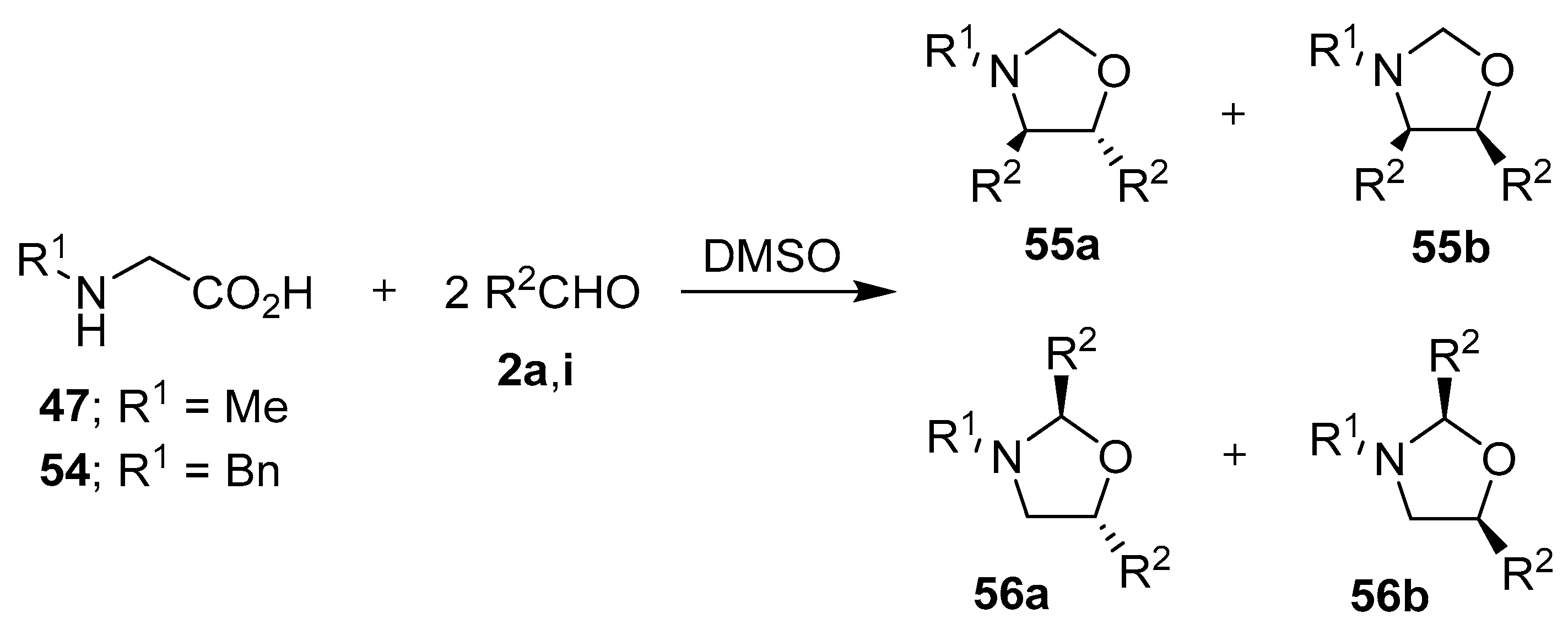
Scheme 13.
Reactions of N-substituted glycine derivatives 47 or 54 with aldehydes 2a and 2i to afford the respective 4,5- and 2,5-disubstituted-1,3-oxazolidines 55 and 56.
Scheme 13.
Reactions of N-substituted glycine derivatives 47 or 54 with aldehydes 2a and 2i to afford the respective 4,5- and 2,5-disubstituted-1,3-oxazolidines 55 and 56.
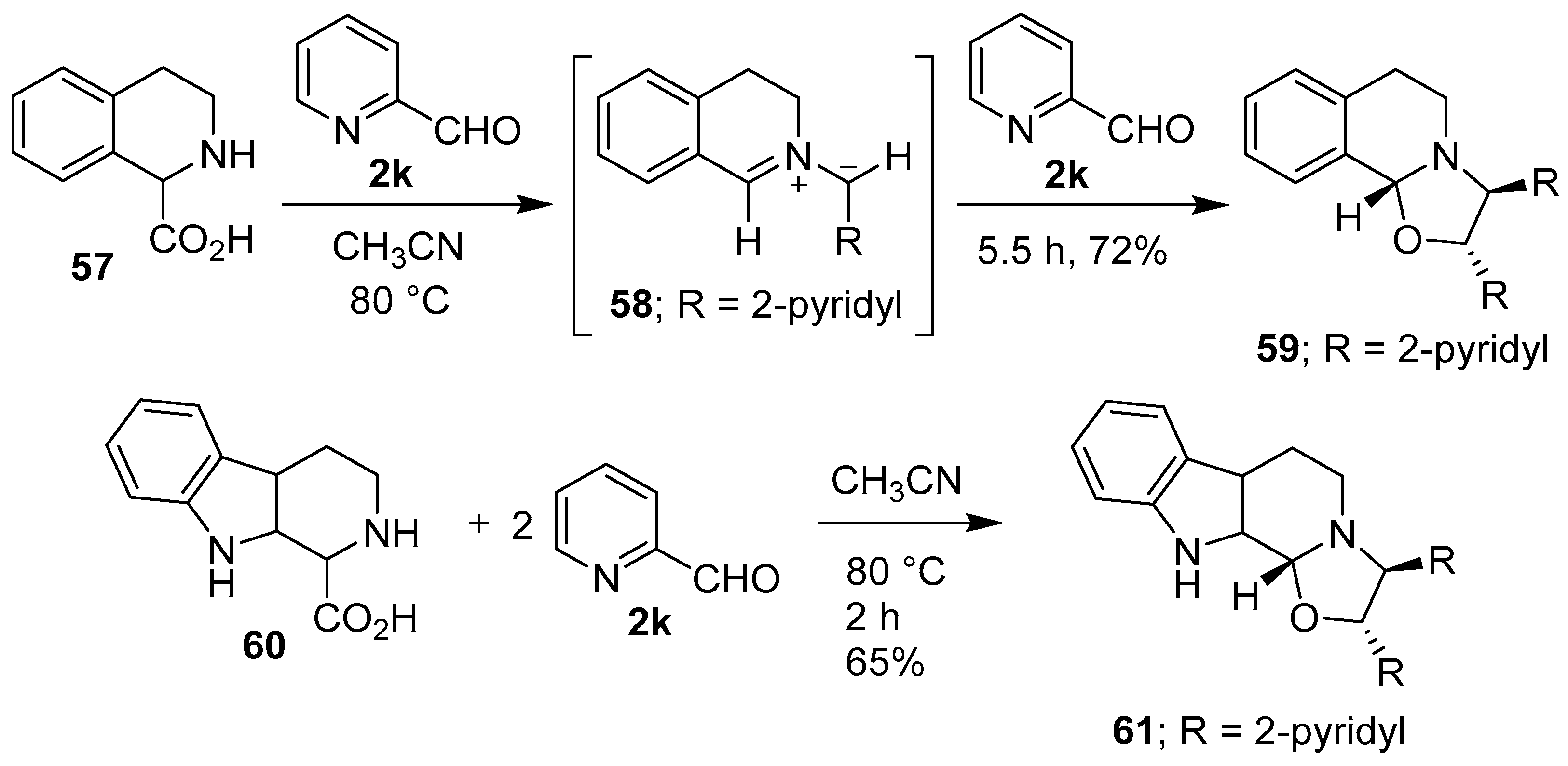
Scheme 14.
Reactions of cyclic secondary amino acids 57 and 60 with 2-pyridylcarboxaldehyde (2k).
Scheme 14.
Reactions of cyclic secondary amino acids 57 and 60 with 2-pyridylcarboxaldehyde (2k).
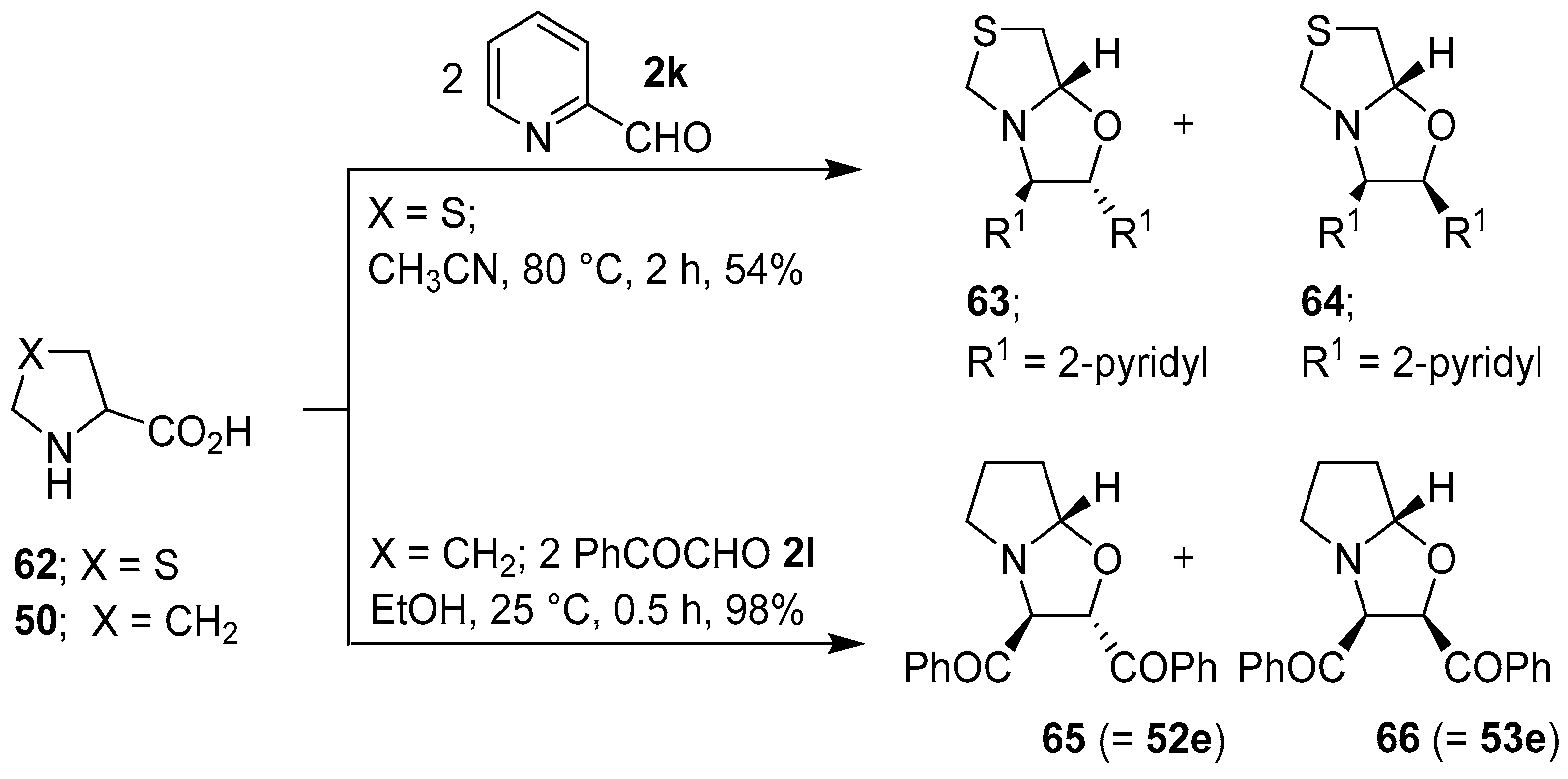
Scheme 15.
Reaction of thiazolidine-4-carboxylic acid (62) with 2-pyridylcarboxaldehyde (2k), and of proline (50) with phenylglyoxal (2l).
Scheme 15.
Reaction of thiazolidine-4-carboxylic acid (62) with 2-pyridylcarboxaldehyde (2k), and of proline (50) with phenylglyoxal (2l).
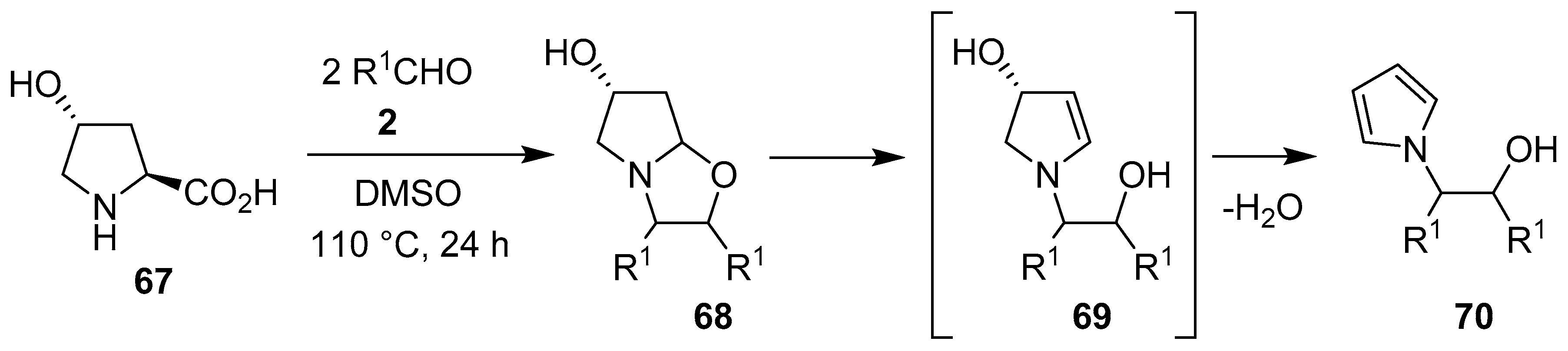
Scheme 16.
Reaction of trans-4-hydroxyproline (67) with aromatic aldehydes 2 to afford N-β-hydroxyethyl pyrroles 70.
Scheme 16.
Reaction of trans-4-hydroxyproline (67) with aromatic aldehydes 2 to afford N-β-hydroxyethyl pyrroles 70.
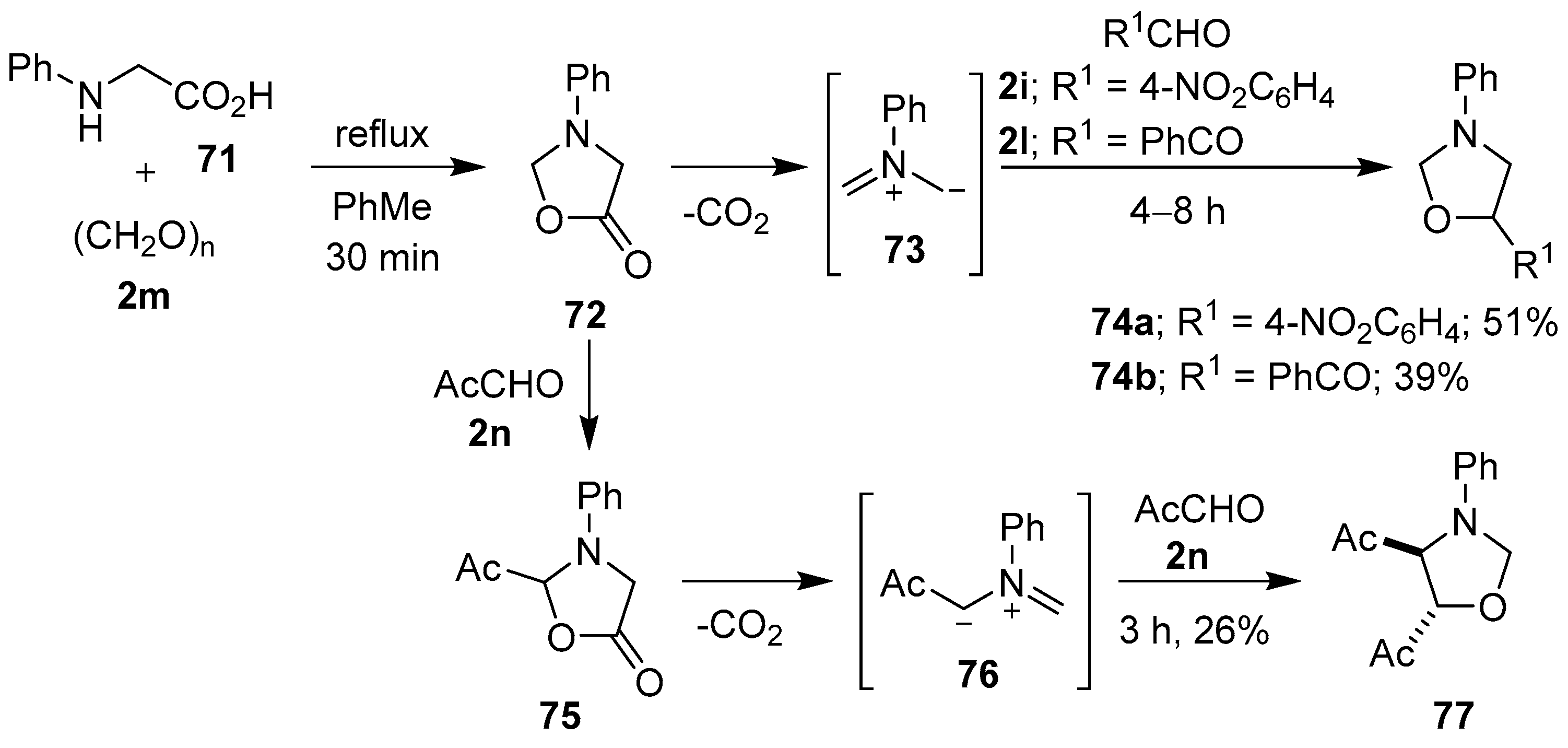
Scheme 17.
Cycloadditions of non-stabilized azomethine ylide 73 to aldehydes 2i and 2l, and of stabilized azomethine ylide 76 to methyl glyoxal (2n).
Scheme 17.
Cycloadditions of non-stabilized azomethine ylide 73 to aldehydes 2i and 2l, and of stabilized azomethine ylide 76 to methyl glyoxal (2n).
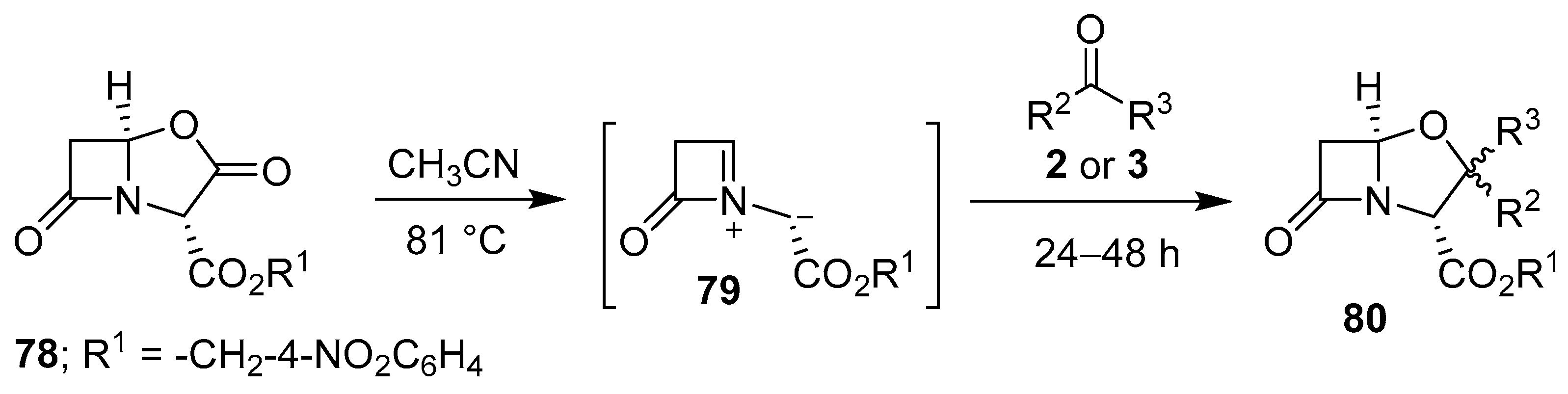
Scheme 18.
Cycloaddition of azomethine ylide 79 to aldehydes 2 or ketones 3.
Scheme 18.
Cycloaddition of azomethine ylide 79 to aldehydes 2 or ketones 3.
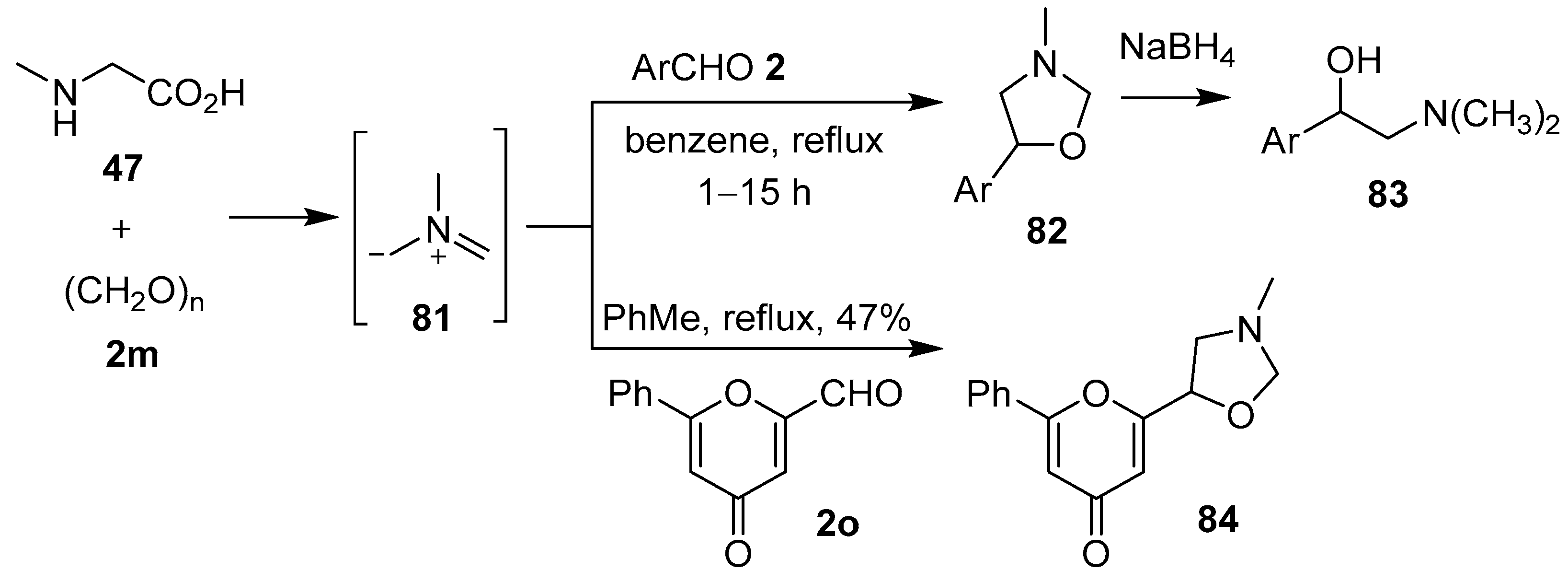
Scheme 19.
Cycloaddition of azomethine ylide 81 to aromatic aldehydes 2 or pyrone aldehyde 2o.
Scheme 19.
Cycloaddition of azomethine ylide 81 to aromatic aldehydes 2 or pyrone aldehyde 2o.
Scheme 20.
Cycloaddition of azomethine ylide 81 to aromatic aldehydes 2, and the subsequent products derived from ring-opening reactions of 5-aryloxazolidine 82.
Scheme 20.
Cycloaddition of azomethine ylide 81 to aromatic aldehydes 2, and the subsequent products derived from ring-opening reactions of 5-aryloxazolidine 82.
Scheme 21.
Cycloaddition of azomethine ylide 81 to aldehyde 2p and ketones 3, and the subsequent oxazolidine ring-opened products.
Scheme 21.
Cycloaddition of azomethine ylide 81 to aldehyde 2p and ketones 3, and the subsequent oxazolidine ring-opened products.
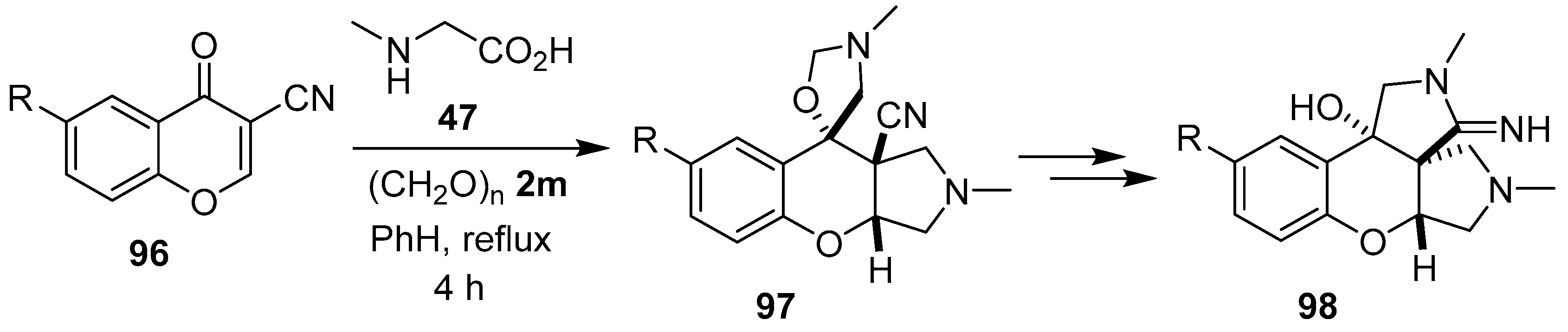
Scheme 22.
Dual cycloaddition of azomethine ylide 81 to 3-cyanochromones 96.
Scheme 22.
Dual cycloaddition of azomethine ylide 81 to 3-cyanochromones 96.
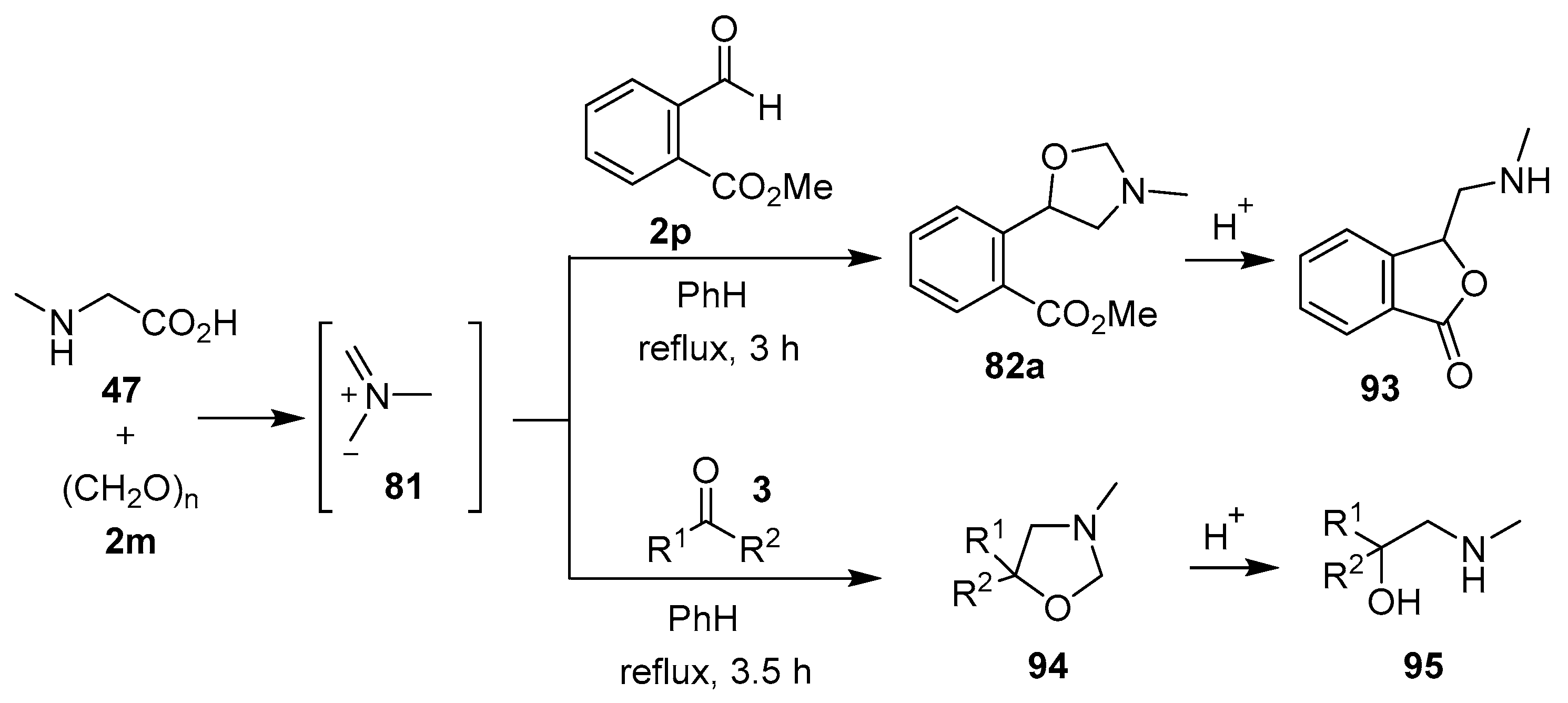
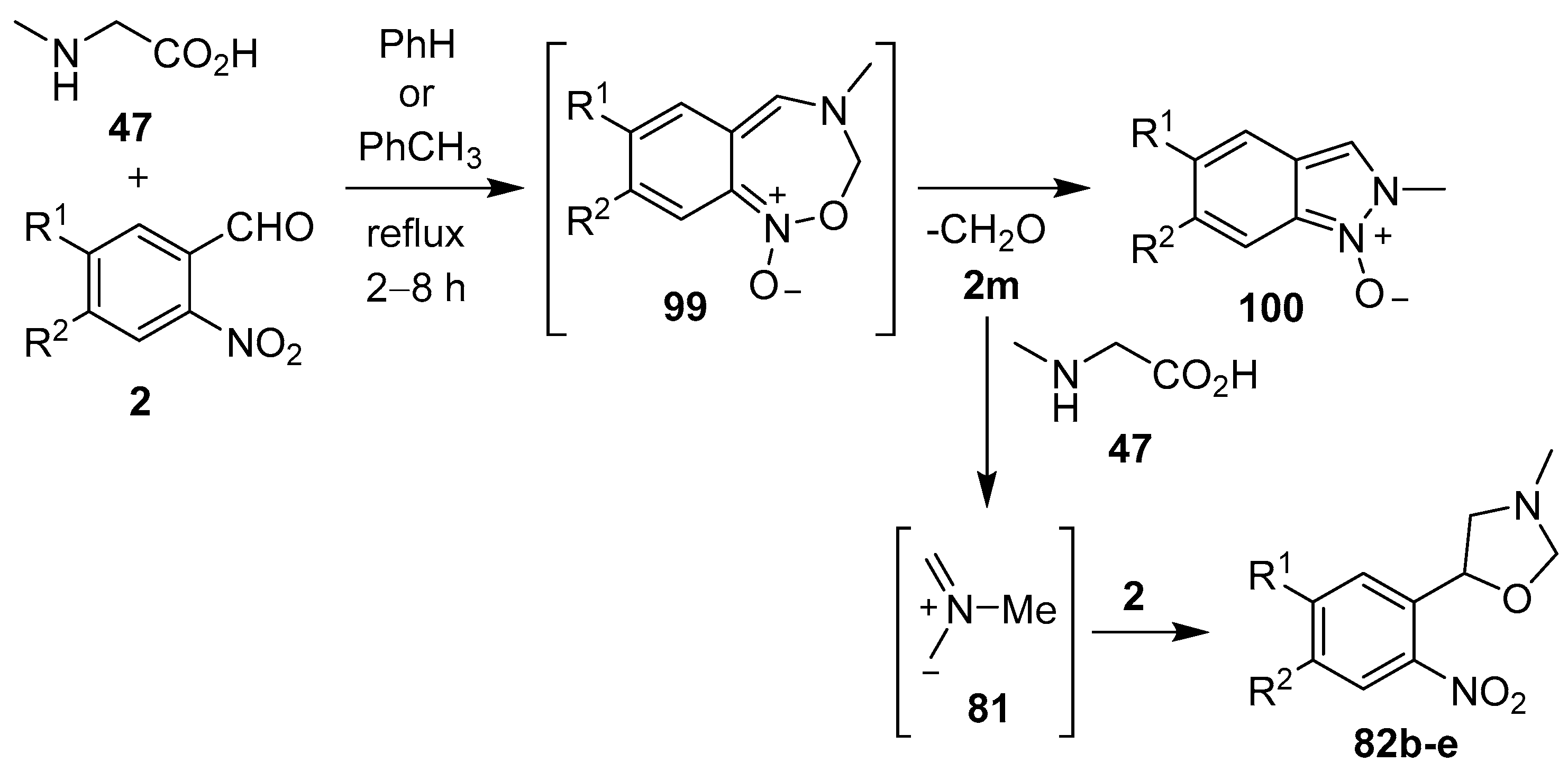
Scheme 23.
Reaction of sarcosine (47) with 2-nitrobenzaldehydes 2.
Scheme 23.
Reaction of sarcosine (47) with 2-nitrobenzaldehydes 2.
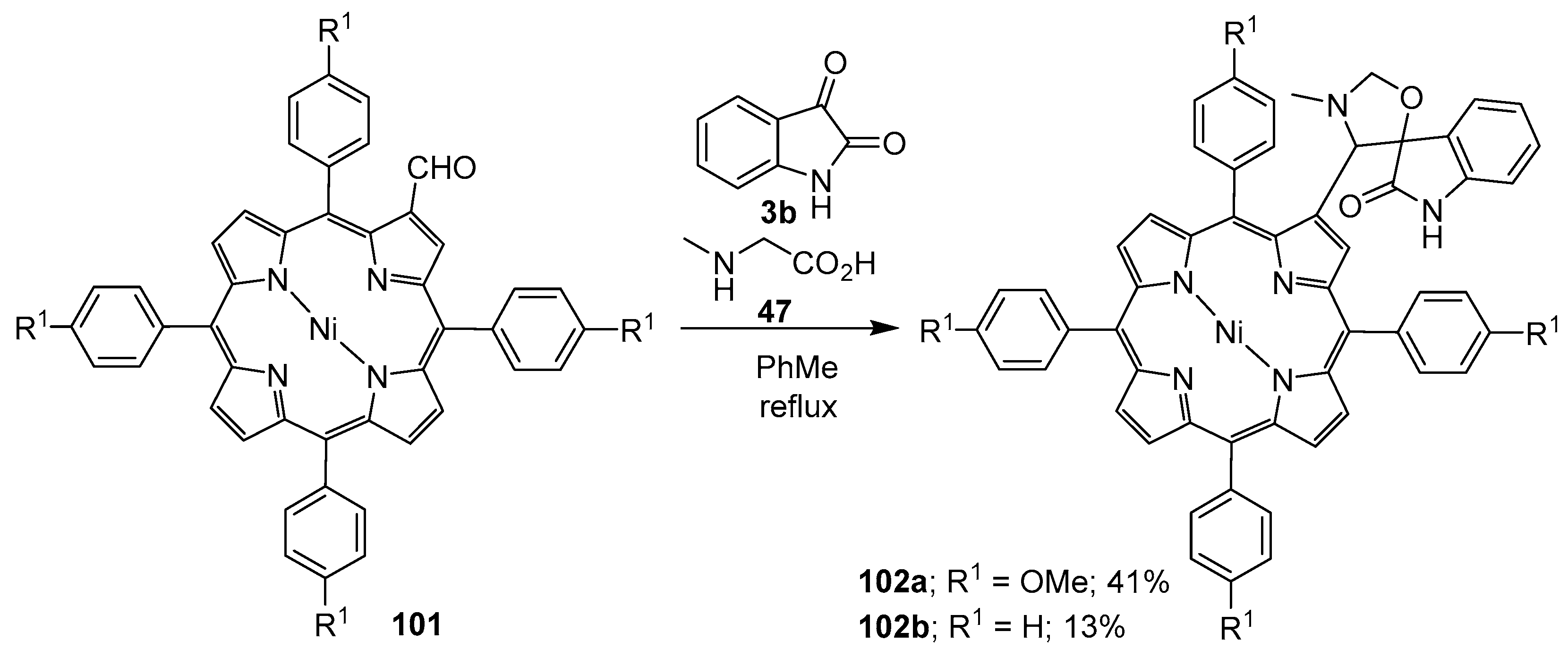
Scheme 24.
Cycloaddition of azomethine ylides generated from sarcosine (47) and porphyrins 101 to isatin (3b).
Scheme 24.
Cycloaddition of azomethine ylides generated from sarcosine (47) and porphyrins 101 to isatin (3b).
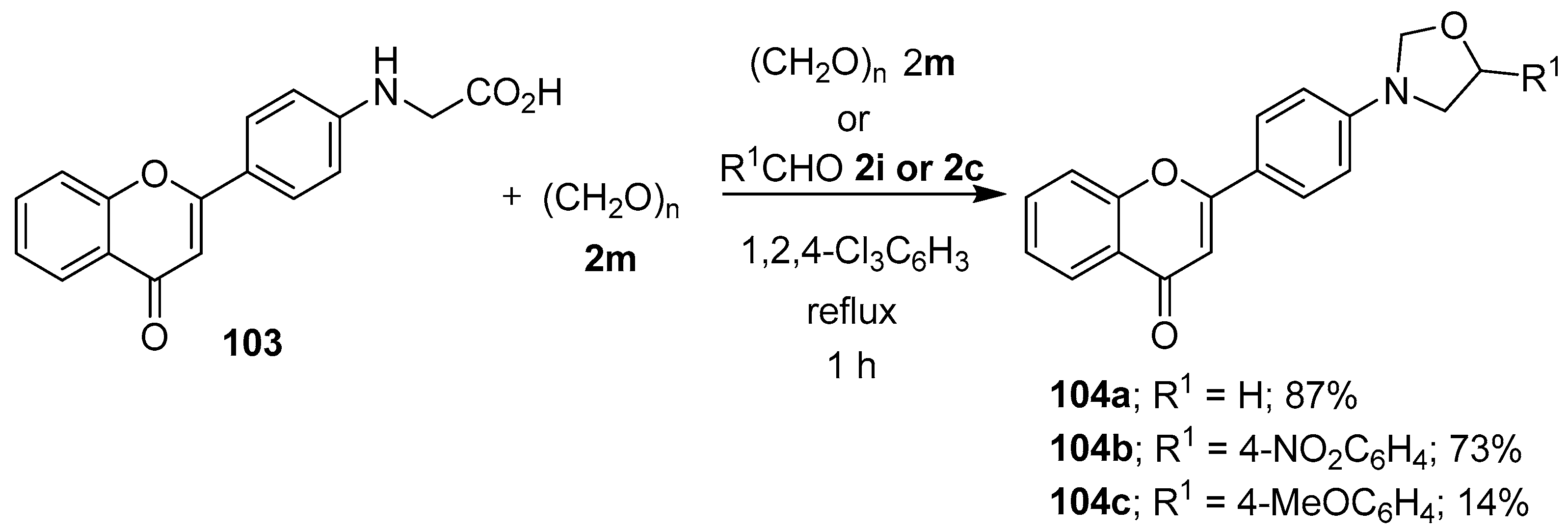
Scheme 25.
Cycloaddition of the azomethine ylide generated from N-[4-(chromon-2-yl)phenyl]glycine (103) and paraformaldehyde (2m) to a second molecule of paraformaldehyde (2m) or to aldehydes 2i or 2c.
Scheme 25.
Cycloaddition of the azomethine ylide generated from N-[4-(chromon-2-yl)phenyl]glycine (103) and paraformaldehyde (2m) to a second molecule of paraformaldehyde (2m) or to aldehydes 2i or 2c.
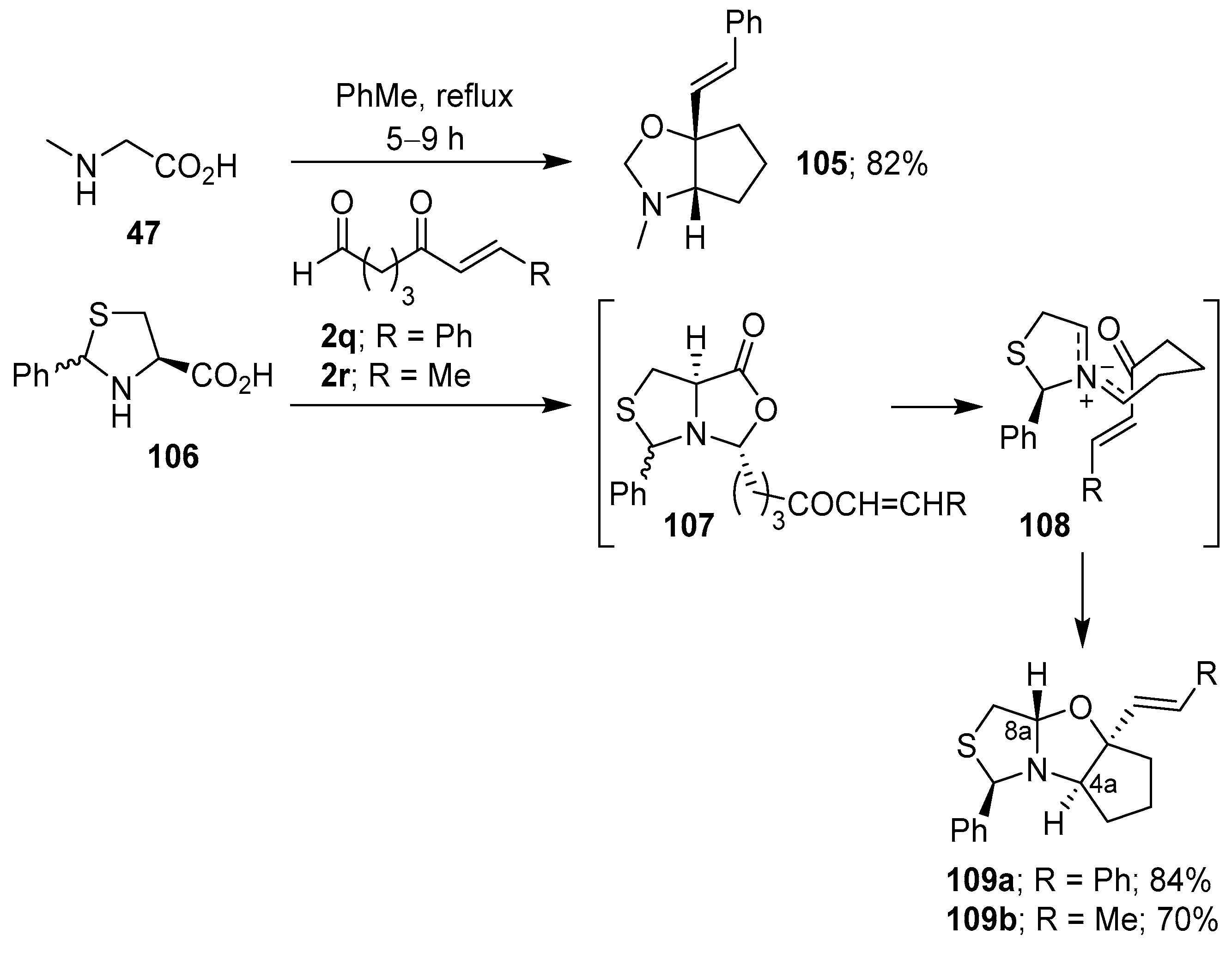
Scheme 26.
Intramolecular cycloaddition of the non-stabilized azomethine ylide generated from the condensation of α-amino acids with aldehydes bearing a tethered aldehyde.
Scheme 26.
Intramolecular cycloaddition of the non-stabilized azomethine ylide generated from the condensation of α-amino acids with aldehydes bearing a tethered aldehyde.
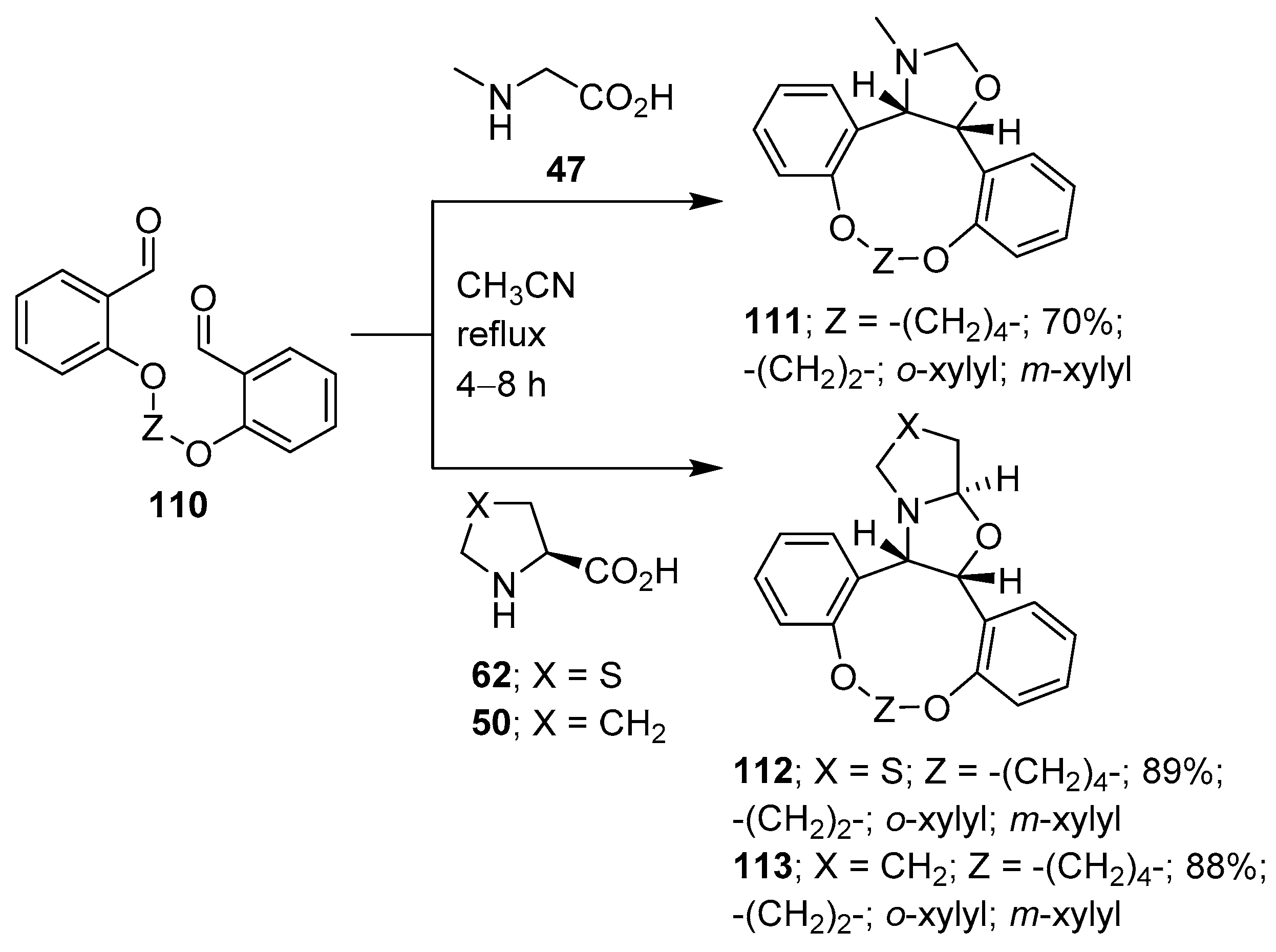
Scheme 27.
Intramolecular cycloaddition of azomethine ylides generated from dialdehyde 110 and sarcosine (47), thiazolidine-4-carboxylic acid (62) or L-proline (50).
Scheme 27.
Intramolecular cycloaddition of azomethine ylides generated from dialdehyde 110 and sarcosine (47), thiazolidine-4-carboxylic acid (62) or L-proline (50).
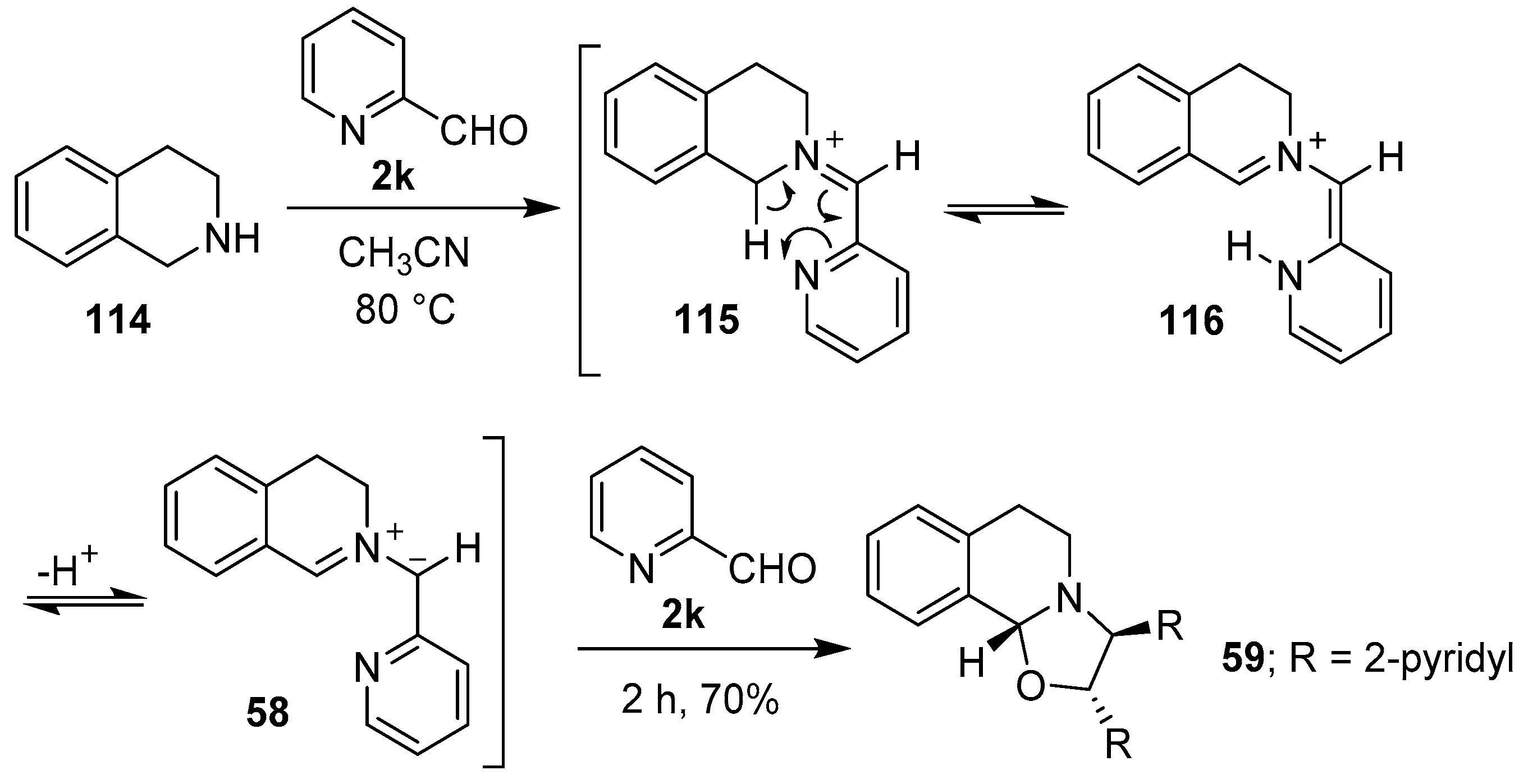
Scheme 28.
Cycloaddition of azomethine ylide 58 to 2-pyridaldehyde (2k).
Scheme 28.
Cycloaddition of azomethine ylide 58 to 2-pyridaldehyde (2k).
Scheme 29.
Cycloaddition of N-metalated azomethine ylides 118, generated from α-iminoesters 117, to benzaldehyde (2a).
Scheme 29.
Cycloaddition of N-metalated azomethine ylides 118, generated from α-iminoesters 117, to benzaldehyde (2a).
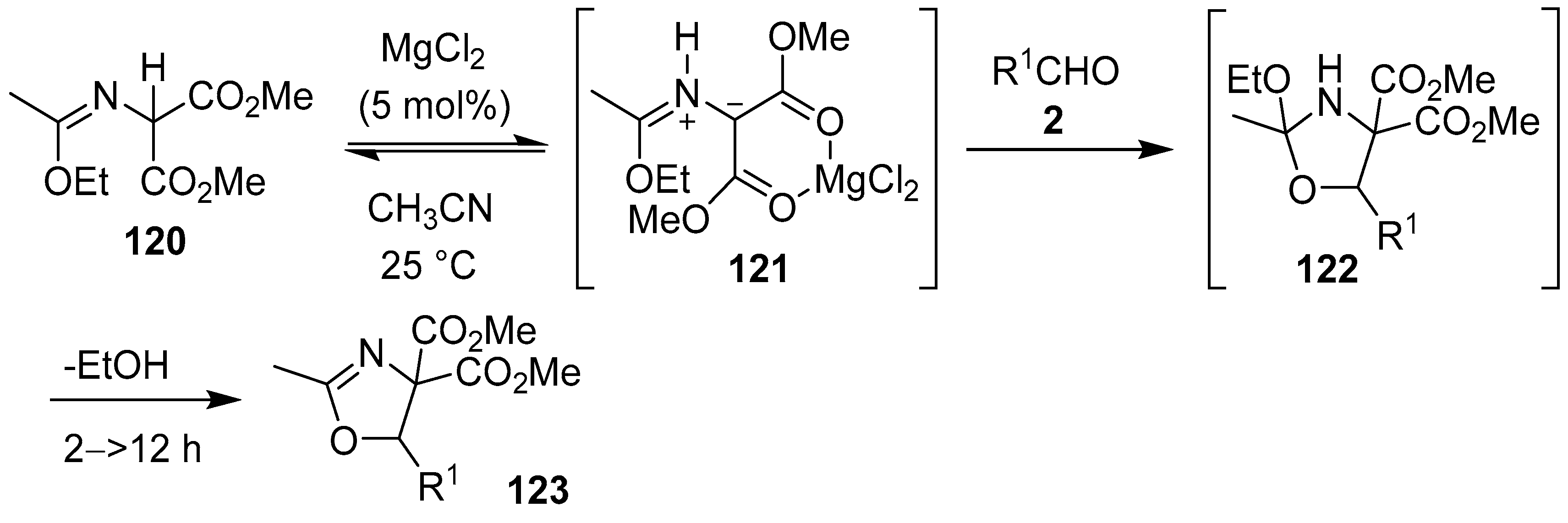
Scheme 30.
Cycloaddition of azomethine ylide 121, generated from malonic imidate 120 via 1,2-prototropy, to aldehydes 2.
Scheme 30.
Cycloaddition of azomethine ylide 121, generated from malonic imidate 120 via 1,2-prototropy, to aldehydes 2.
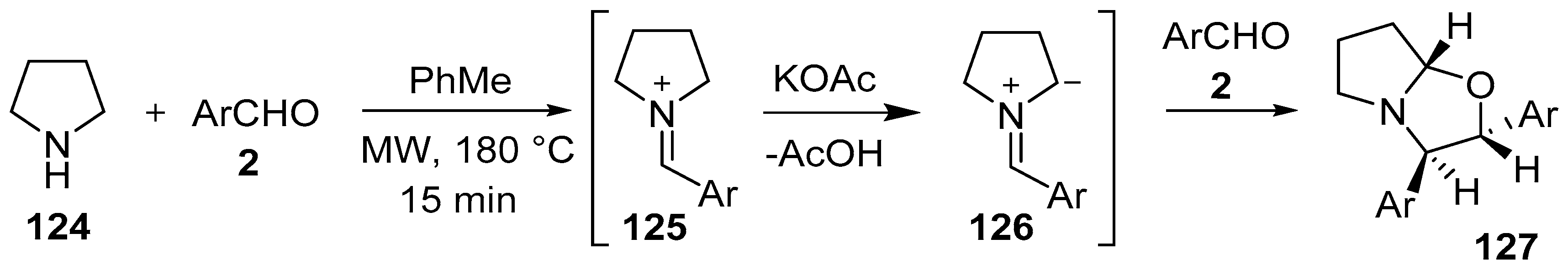
Scheme 31.
Cycloaddition of azomethine ylide 126, generated from pyrrolidine (124) and aromatic aldehydes 2, to a second molecule of aldehyde 2.
Scheme 31.
Cycloaddition of azomethine ylide 126, generated from pyrrolidine (124) and aromatic aldehydes 2, to a second molecule of aldehyde 2.
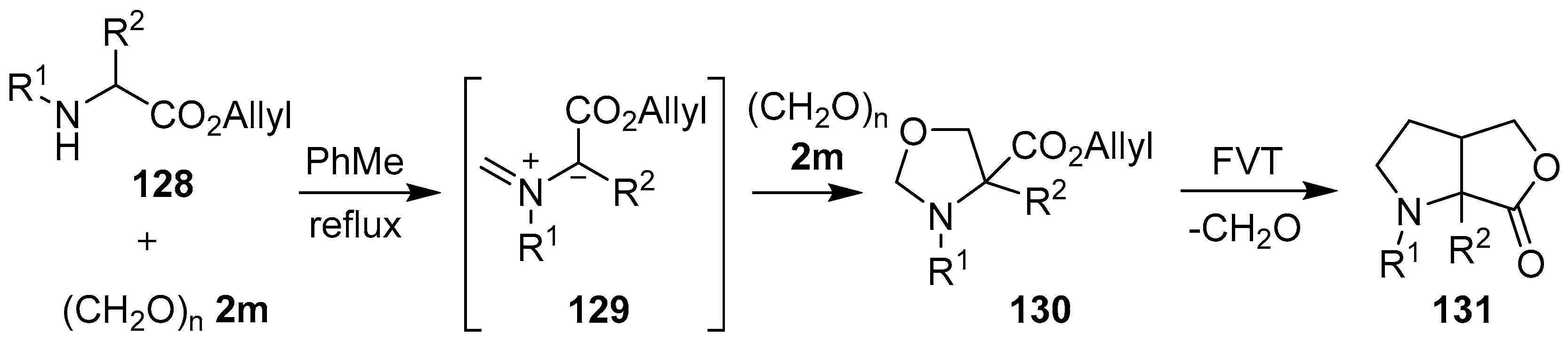
Scheme 32.
Cycloaddition of azomethine ylide 129, generated from allylic α-amino esters 128 and paraformaldehyde (2m), to a second molecule of paraformaldehyde (2m), and subsequent FVT-induced cycloreversion-intramolecular cycloaddition.
Scheme 32.
Cycloaddition of azomethine ylide 129, generated from allylic α-amino esters 128 and paraformaldehyde (2m), to a second molecule of paraformaldehyde (2m), and subsequent FVT-induced cycloreversion-intramolecular cycloaddition.
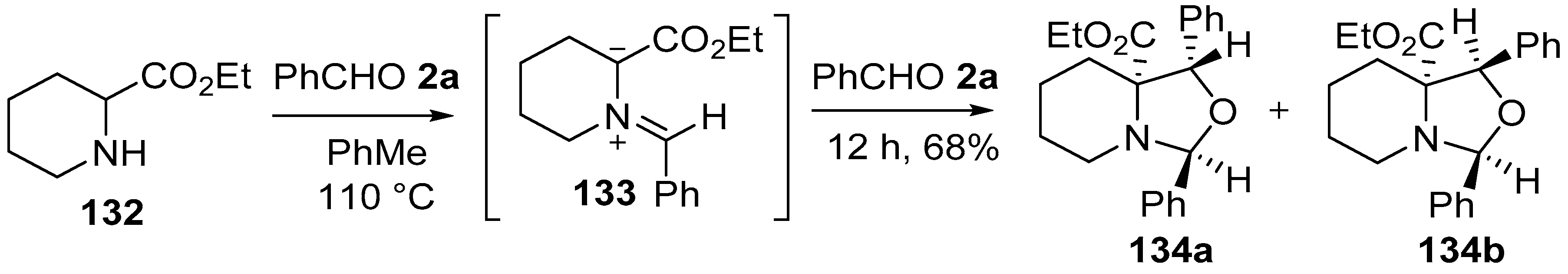
Scheme 33.
Cycloaddition of azomethine ylide 133, generated from pipecolic acid ethyl ester (132) and benzaldehyde (2a), to a second molecule of benzaldehyde (2a).
Scheme 33.
Cycloaddition of azomethine ylide 133, generated from pipecolic acid ethyl ester (132) and benzaldehyde (2a), to a second molecule of benzaldehyde (2a).
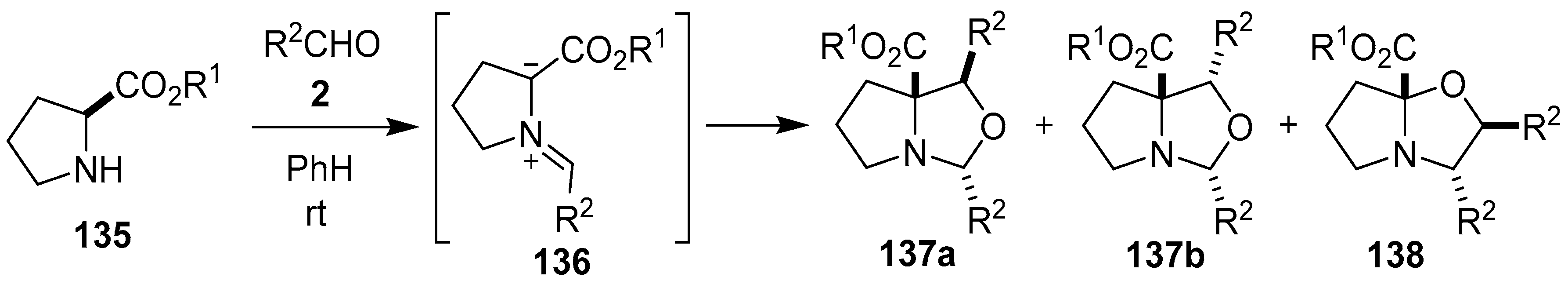
Scheme 34.
Cycloaddition of azomethine ylide 136, generated from L-proline alkyl esters 135 and aromatic aldehydes 2, to a second molecule of aldehyde 2.
Scheme 34.
Cycloaddition of azomethine ylide 136, generated from L-proline alkyl esters 135 and aromatic aldehydes 2, to a second molecule of aldehyde 2.
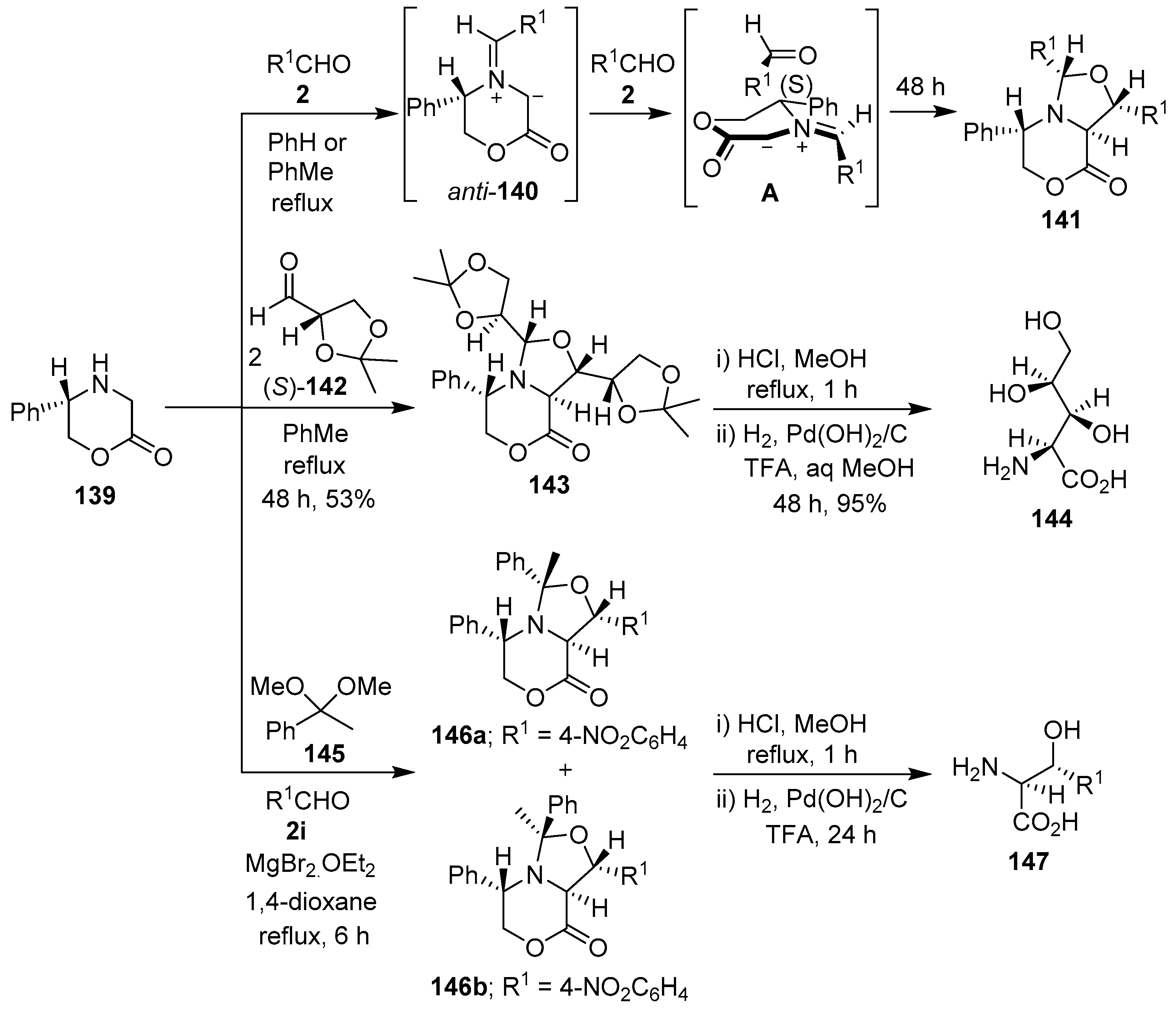
Scheme 35.
Synthesis of bicyclic oxazolidines via cycloaddition reactions of azomethine ylides, derived from 5-(S)-phenylmorpholine-2-one (139) and aldehydes 2, 142 or acetophenone dimethyl acetal (145), to a second equivalent of the same or a different aldehyde.
Scheme 35.
Synthesis of bicyclic oxazolidines via cycloaddition reactions of azomethine ylides, derived from 5-(S)-phenylmorpholine-2-one (139) and aldehydes 2, 142 or acetophenone dimethyl acetal (145), to a second equivalent of the same or a different aldehyde.
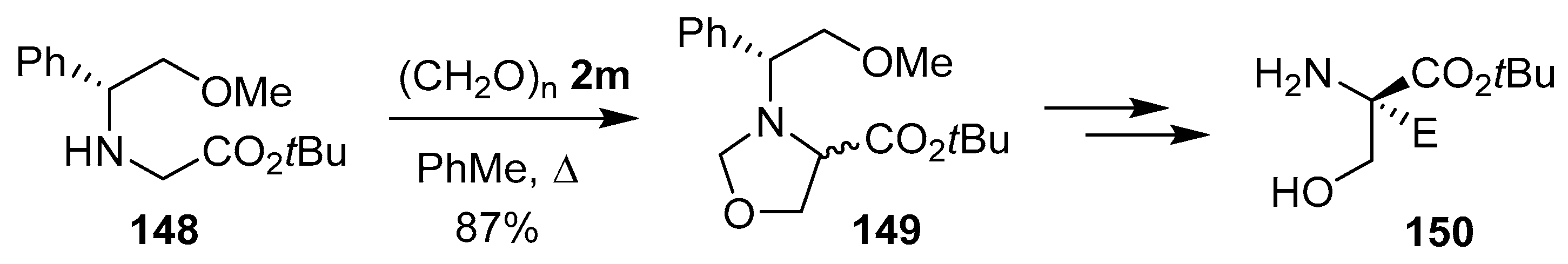
Scheme 36.
Reaction of glycine derivative 148 and paraformaldehyde (2m) to afford oxazolidine 149.
Scheme 36.
Reaction of glycine derivative 148 and paraformaldehyde (2m) to afford oxazolidine 149.
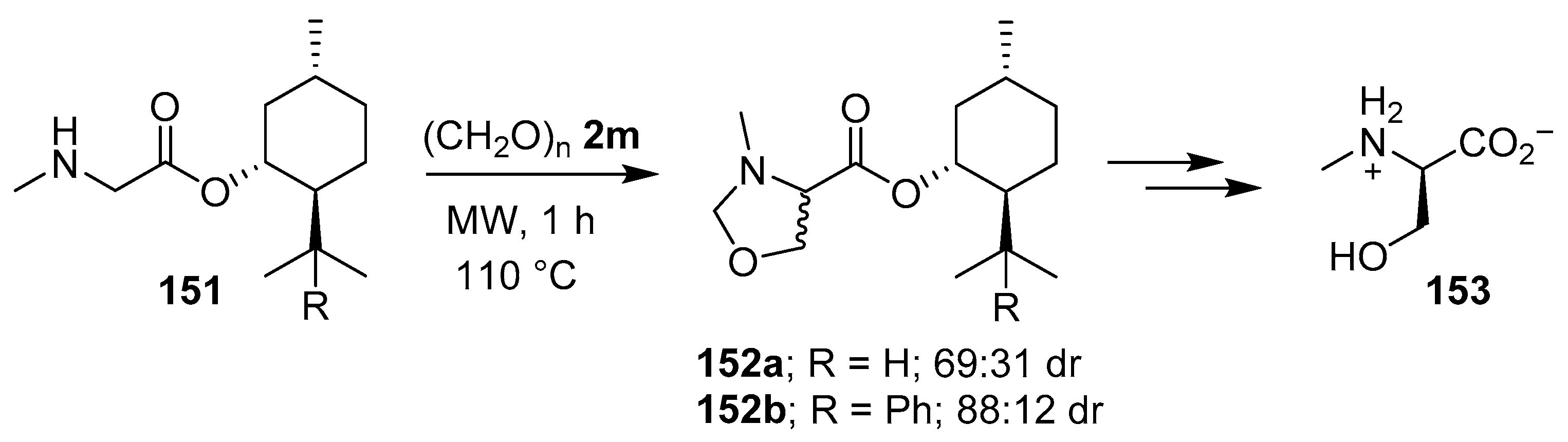
Scheme 37.
Reaction of sarcosine esters 151a and 151b with paraformaldehyde (2m) to afford oxazolidines 152a and 152b, respectively.
Scheme 37.
Reaction of sarcosine esters 151a and 151b with paraformaldehyde (2m) to afford oxazolidines 152a and 152b, respectively.
Scheme 38.
Cycloaddition of an azomethine ylide, generated from desilylation of a N-(trimethylsilylmethyl)-immonium ion, with 3-nitrobenzaldehyde (2h) (Ar = 3-NO2-C6H3).
Scheme 38.
Cycloaddition of an azomethine ylide, generated from desilylation of a N-(trimethylsilylmethyl)-immonium ion, with 3-nitrobenzaldehyde (2h) (Ar = 3-NO2-C6H3).
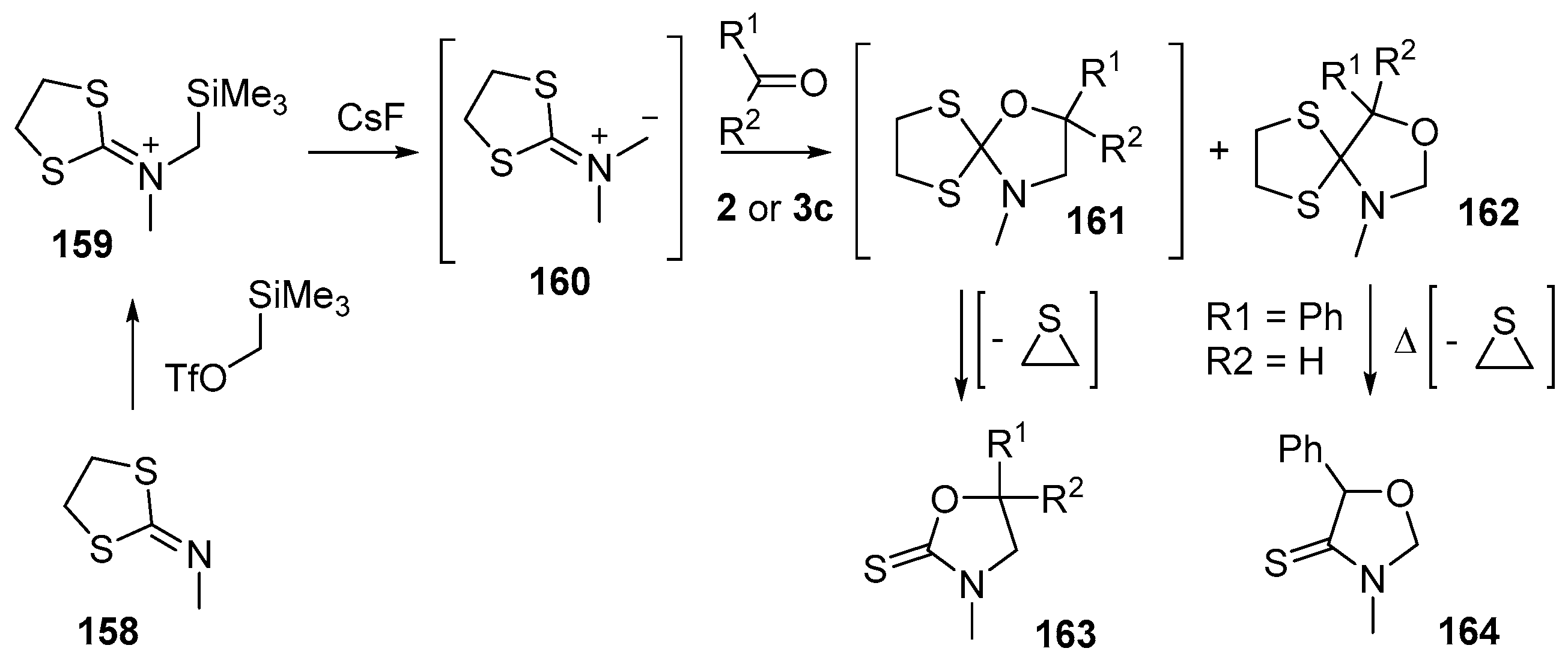
Scheme 39.
Cycloaddition of dithiolane-isocyanate immimium methylides 160 with aldehydes 2 and benzophenone (3c).
Scheme 39.
Cycloaddition of dithiolane-isocyanate immimium methylides 160 with aldehydes 2 and benzophenone (3c).
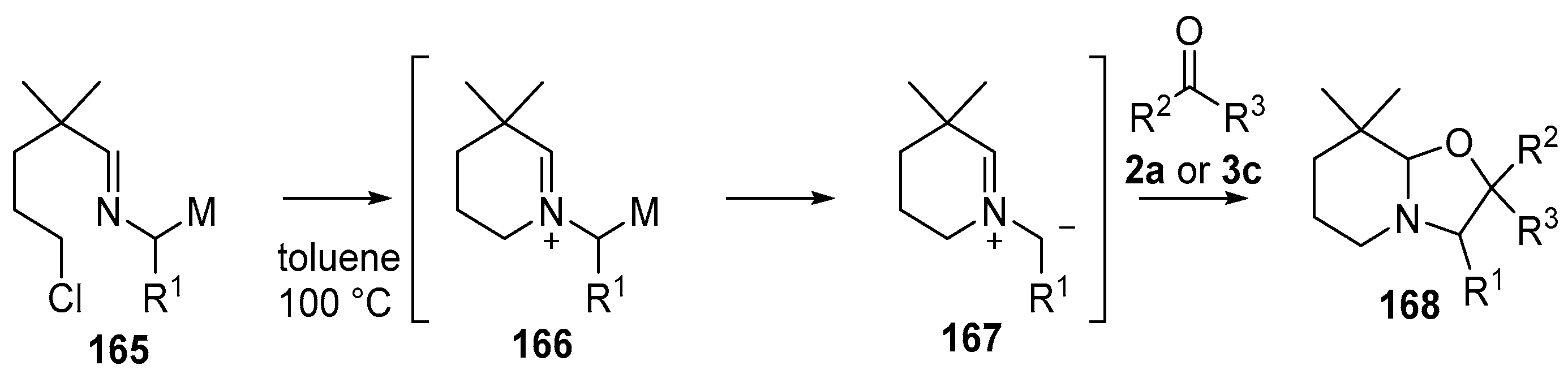
Scheme 40.
Cycloaddition of azomethine ylides, generated by an N-alkylation/demtallation ccascade, to aldehyde (2a) and benzophenone (3c).
Scheme 40.
Cycloaddition of azomethine ylides, generated by an N-alkylation/demtallation ccascade, to aldehyde (2a) and benzophenone (3c).
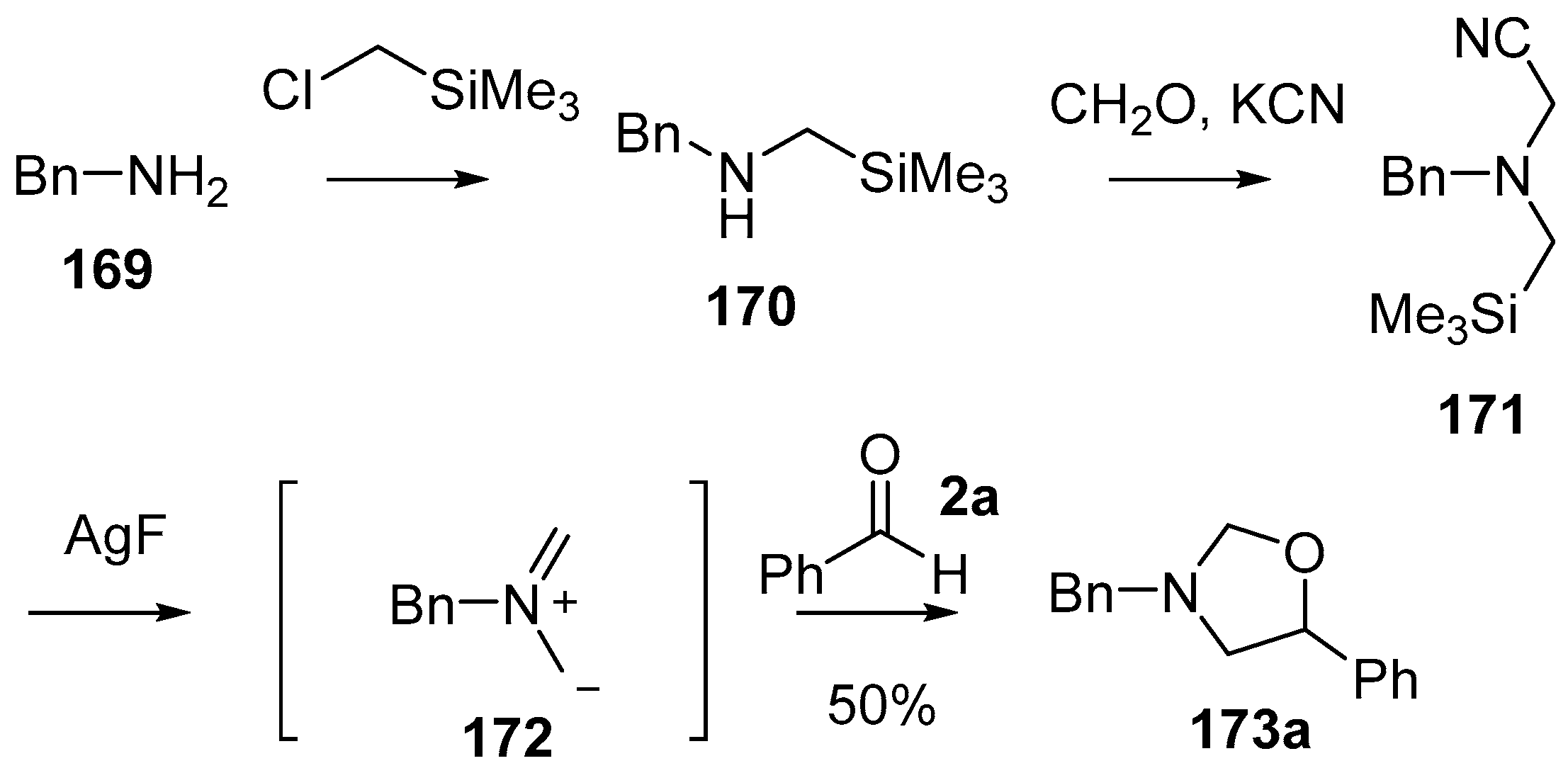
Scheme 41.
Cycloaddition of a non-stabilized ylide, derived from cyanomethylamine reagent 171, with benzaldehyde (2a).
Scheme 41.
Cycloaddition of a non-stabilized ylide, derived from cyanomethylamine reagent 171, with benzaldehyde (2a).
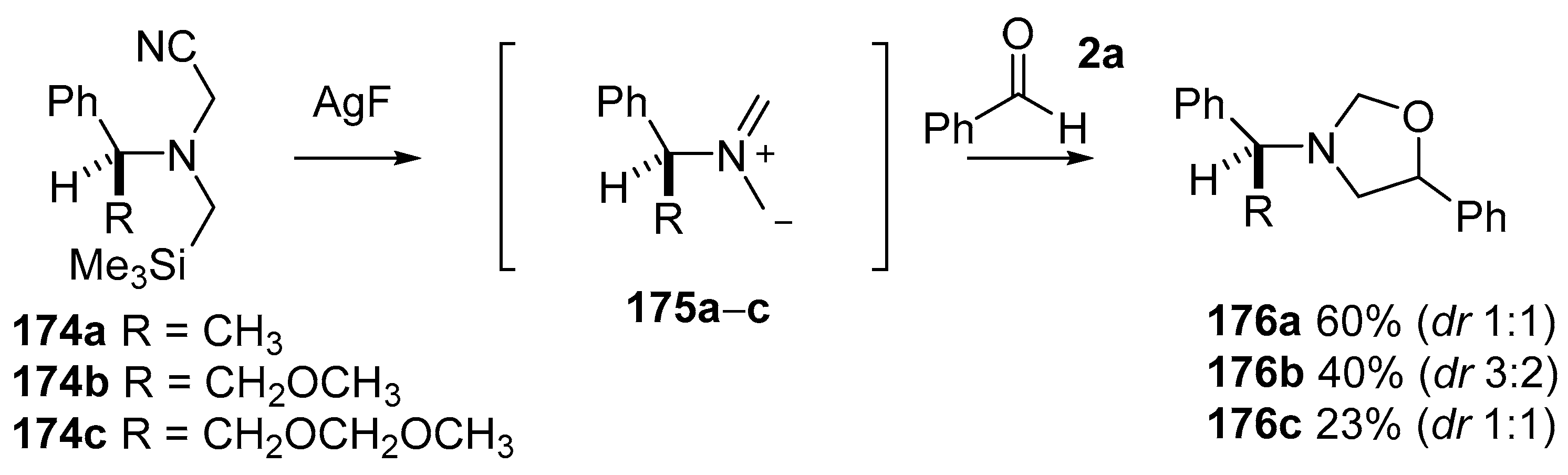
Scheme 42.
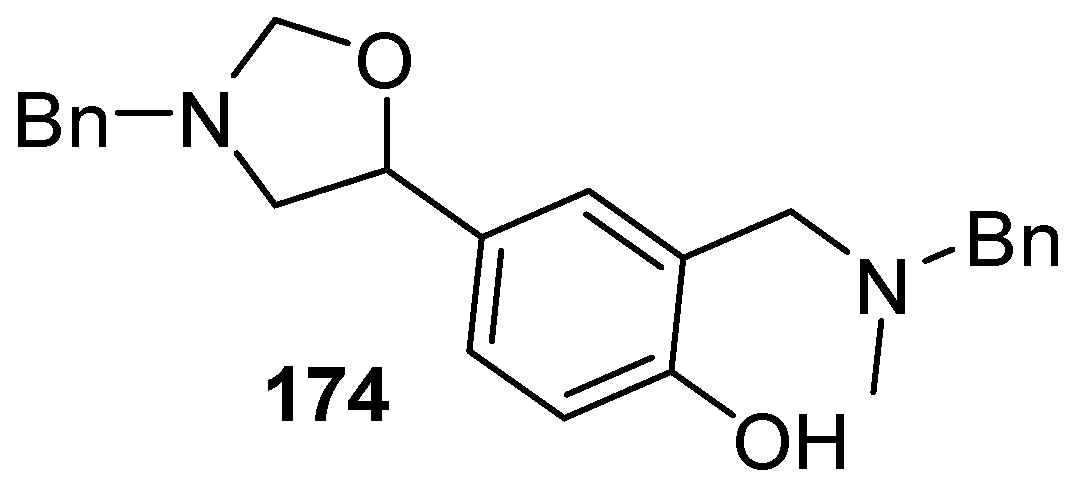
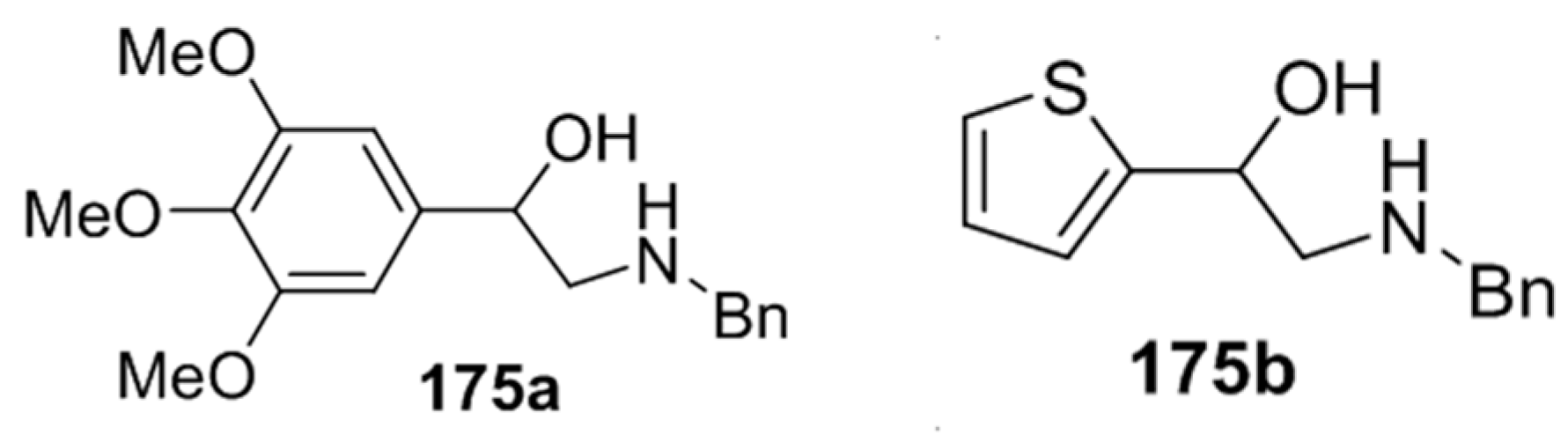
Cycloaddition of chiral non-stabilized ylides, derived from cyanomethylamine reagents 175, with benzaldehyde (2a).
Scheme 42.
Cycloaddition of chiral non-stabilized ylides, derived from cyanomethylamine reagents 175, with benzaldehyde (2a).
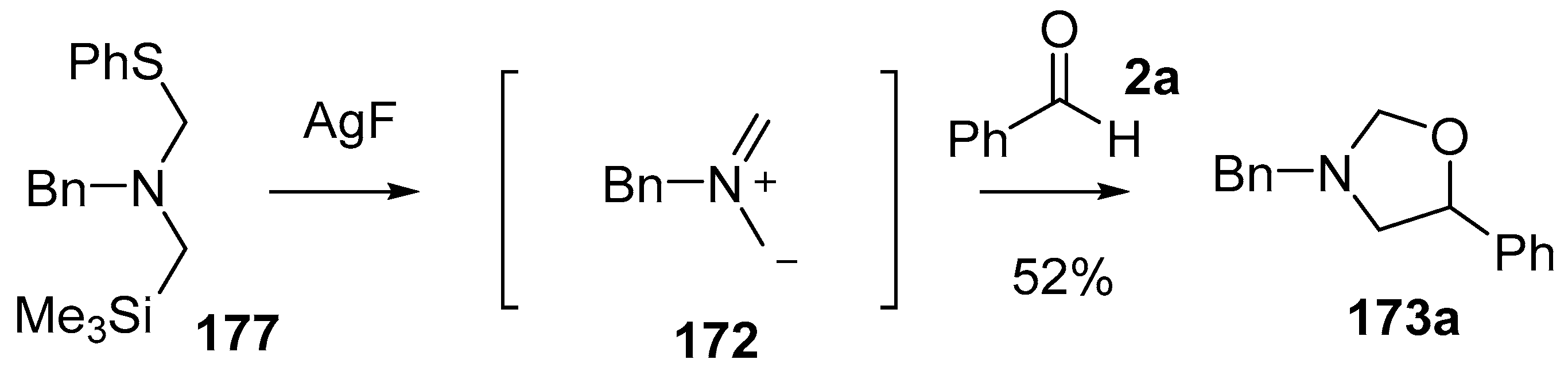
Scheme 43.
Cycloaddition of non-stabilized ylide 172, derived from phenylthiomethylamine reagent 177, with benzaldehyde (2a).
Scheme 43.
Cycloaddition of non-stabilized ylide 172, derived from phenylthiomethylamine reagent 177, with benzaldehyde (2a).
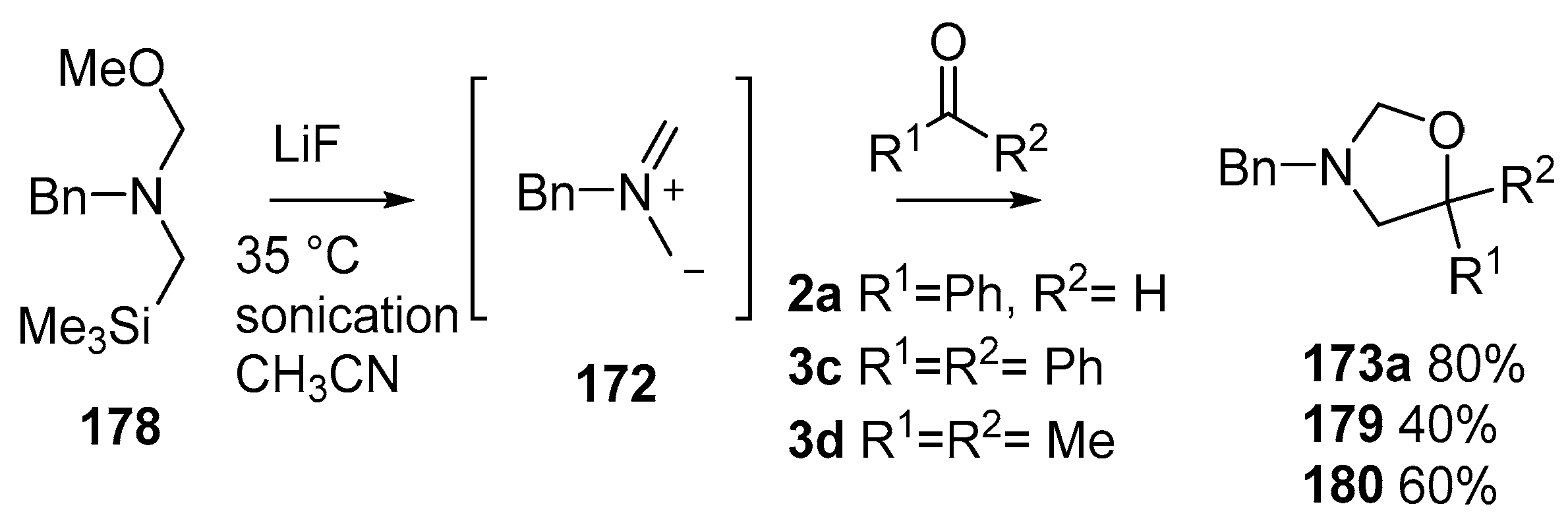
Scheme 44.
Cycloaddition of non-stabilized ylide 172, derived from methoxymethylamine reagent 178, with benzaldehyde (2a) and ketones 3c and 3d.
Scheme 44.
Cycloaddition of non-stabilized ylide 172, derived from methoxymethylamine reagent 178, with benzaldehyde (2a) and ketones 3c and 3d.
Scheme 45.
Cycloaddition of non-stabilized ylide 172, generated by CF3CO2H-catalyzed decomposition of methoxymethylamine reagent 178, with aromatic aldehydes 2.
Scheme 45.
Cycloaddition of non-stabilized ylide 172, generated by CF3CO2H-catalyzed decomposition of methoxymethylamine reagent 178, with aromatic aldehydes 2.
Figure 2.
The
bis adduct isolated from the reaction of 4-hydroxybenzaldehyde (
2t,
Table 23, entry 16).
Figure 2.
The
bis adduct isolated from the reaction of 4-hydroxybenzaldehyde (
2t,
Table 23, entry 16).
Figure 3.
Products from hydrazinolysis of oxazolidines 173j and 173r.
Figure 3.
Products from hydrazinolysis of oxazolidines 173j and 173r.
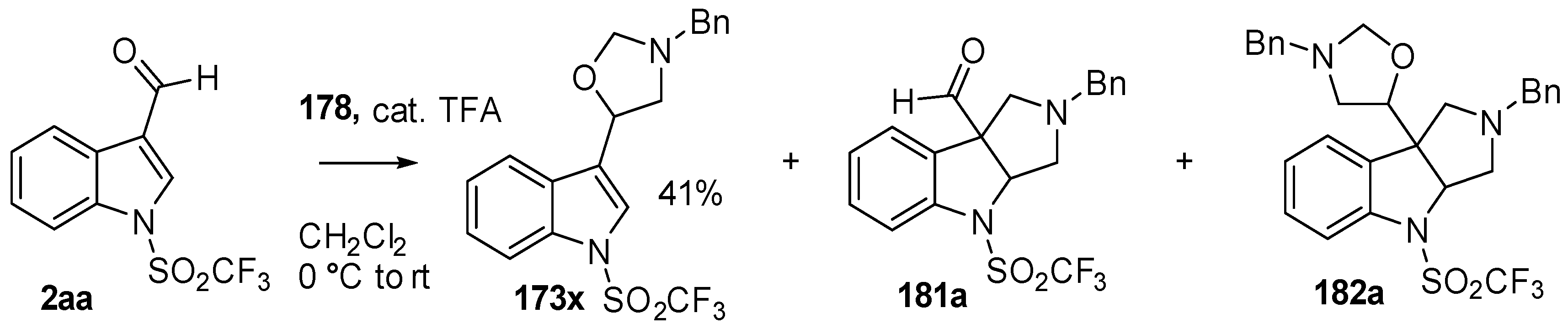
Scheme 46.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with indole-3-carboxaldehyde 2aa.
Scheme 46.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with indole-3-carboxaldehyde 2aa.
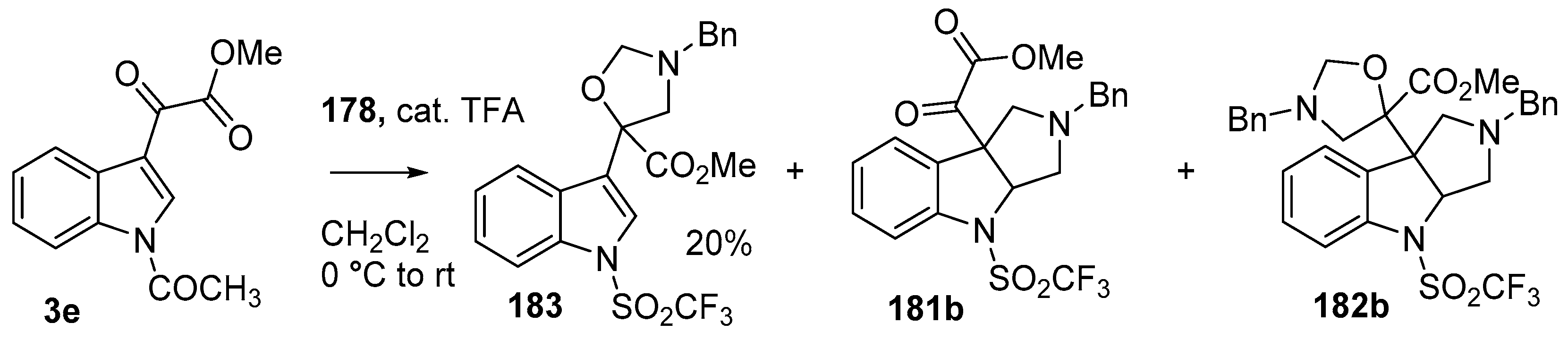
Scheme 47.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with indole-3-pyruvate 3e.
Scheme 47.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with indole-3-pyruvate 3e.
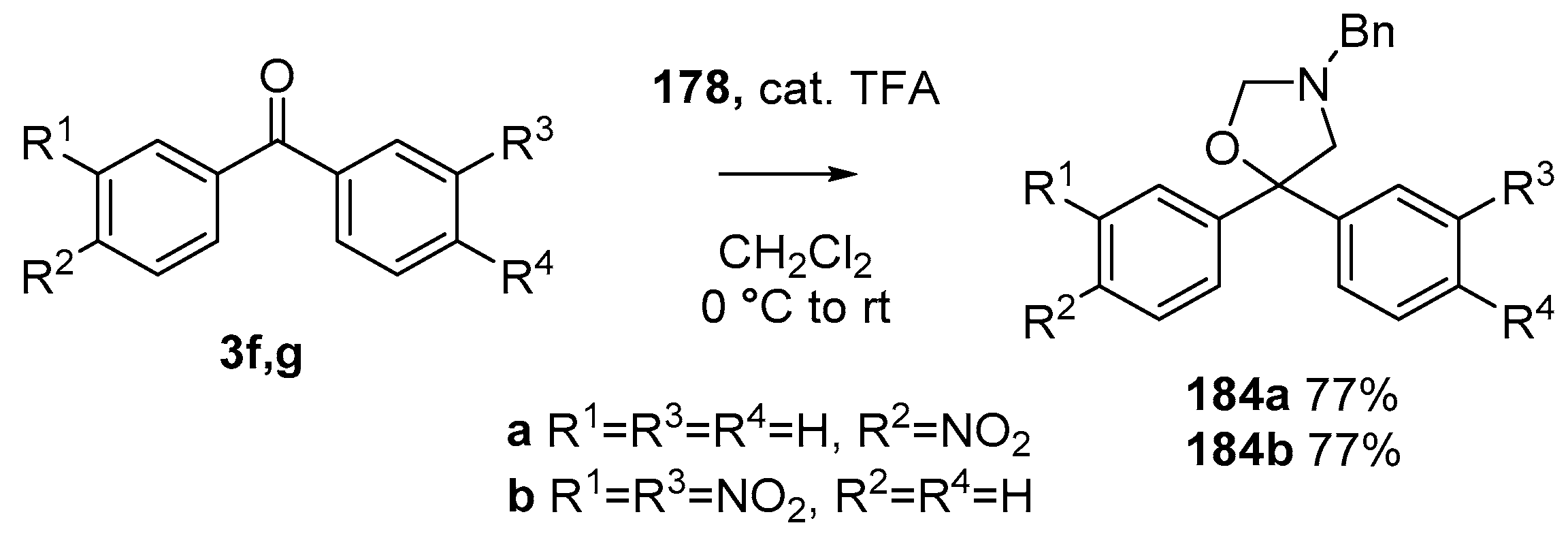
Scheme 48.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with benzophenones 3f and 3g.
Scheme 48.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with benzophenones 3f and 3g.
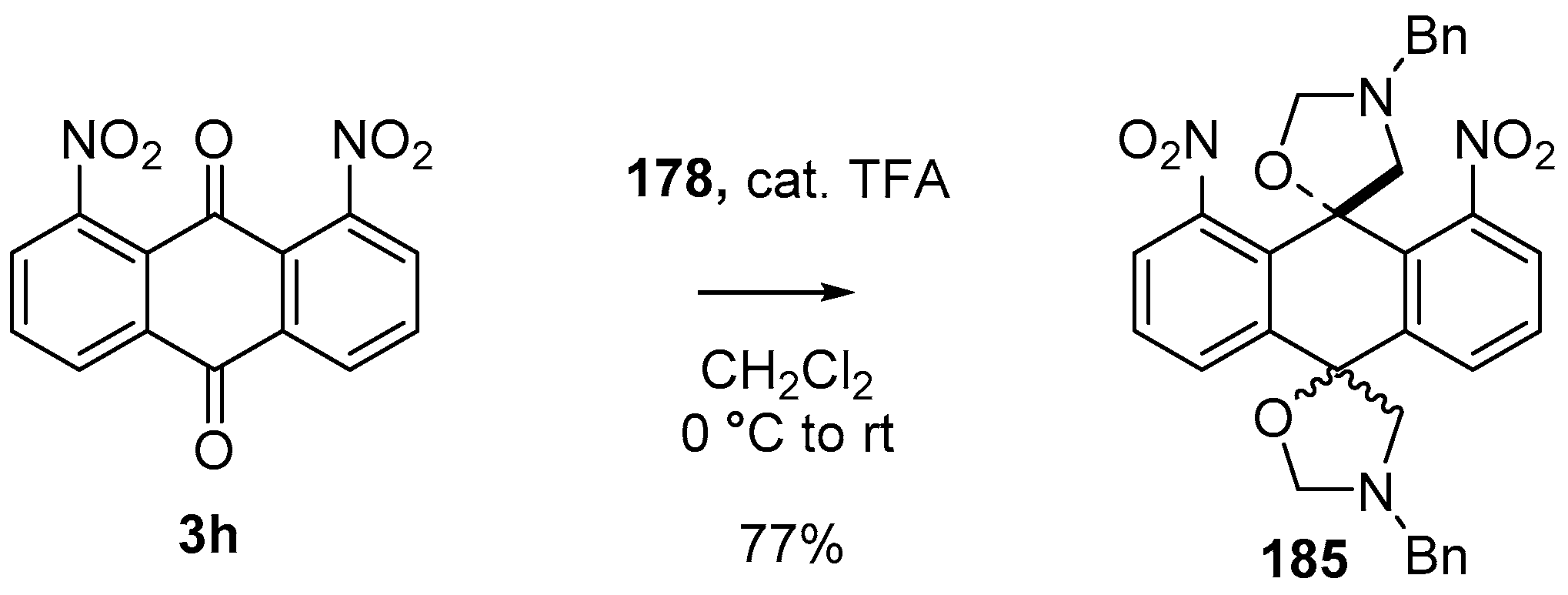
Scheme 49.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with anthroquinone 3h.
Scheme 49.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with anthroquinone 3h.
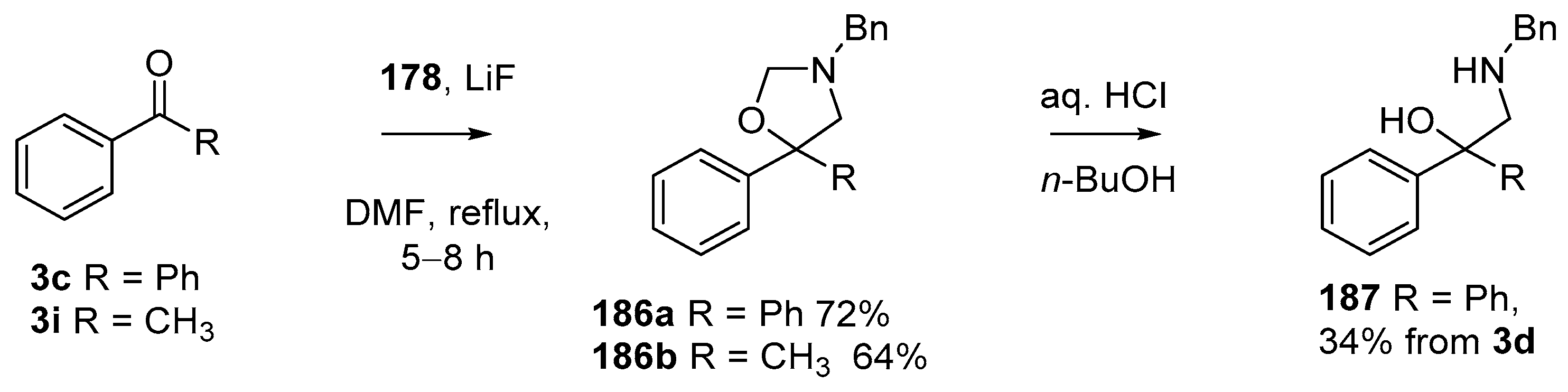
Scheme 50.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with aromatic ketones 3c and 3i.
Scheme 50.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with aromatic ketones 3c and 3i.
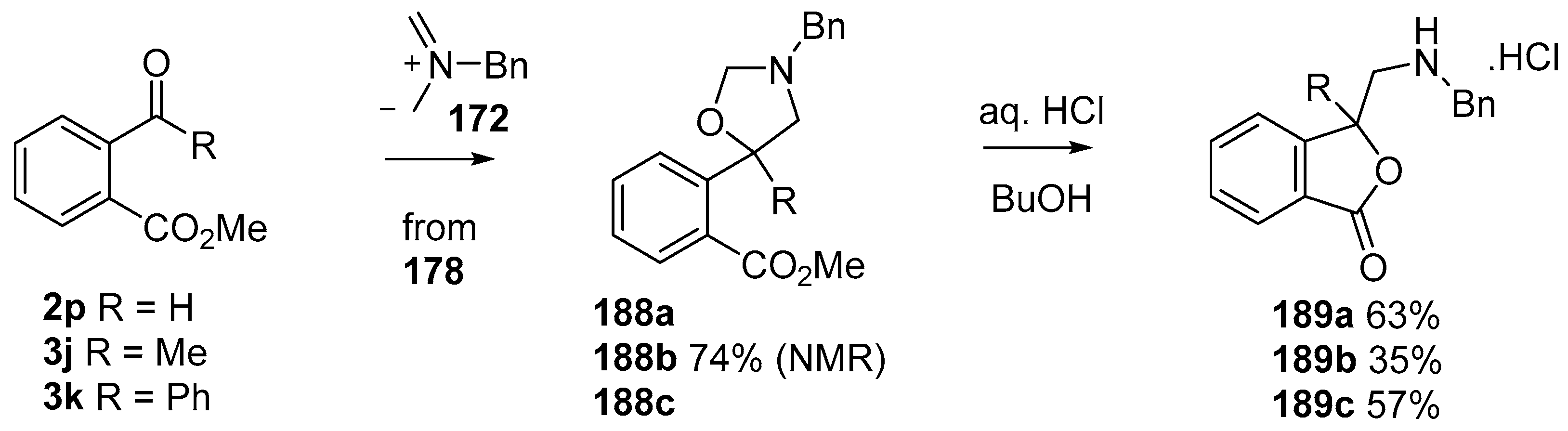
Scheme 51.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with aromatic aldehyde 2p and aromatic ketones 3j and 3k.
Scheme 51.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with aromatic aldehyde 2p and aromatic ketones 3j and 3k.
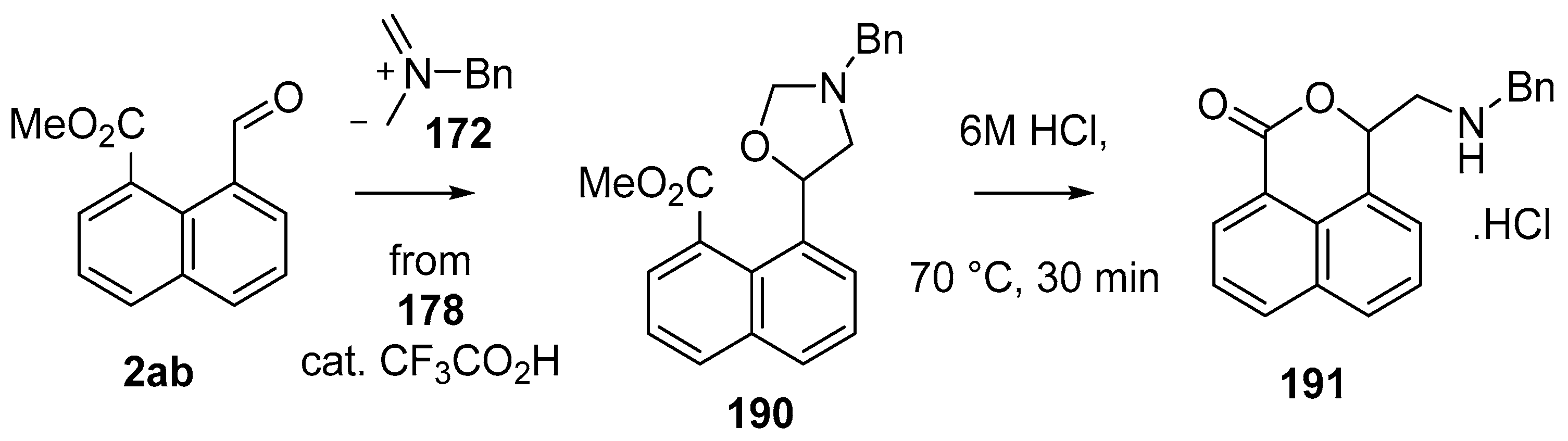
Scheme 52.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with aromatic aldehyde 2ab.
Scheme 52.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with aromatic aldehyde 2ab.
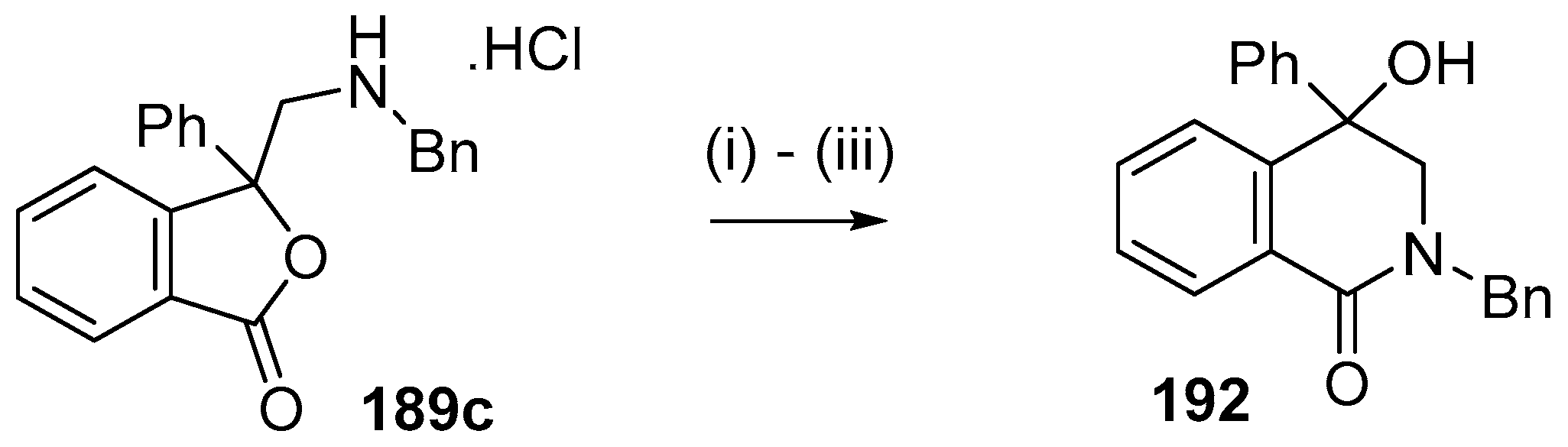
Scheme 53.
Lactamization of isobenzofuranone 189c. Reaction Conditions: (i) NaHCO3; (ii) NaOH; then NH4Cl; (iii) n-BuOH, reflux.
Scheme 53.
Lactamization of isobenzofuranone 189c. Reaction Conditions: (i) NaHCO3; (ii) NaOH; then NH4Cl; (iii) n-BuOH, reflux.
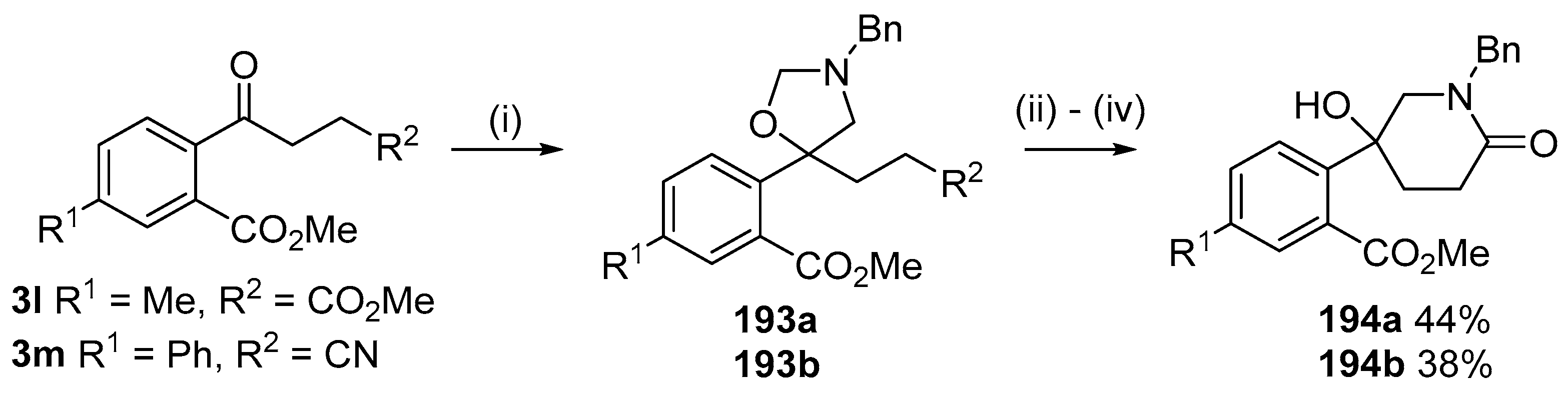
Scheme 54.
Cycloaddition of ylide 172 with aromatic ketones 3l and 3m followed by hydrolysis and condensation reactions to afford piperidones 194. Reaction Conditions: (i) 178 (2.5 equiv.), LiF, DMF, reflux, 5 h; (ii) aq. HCl, n-BuOH, 90 °C; (iii) NH4Cl;(iv) n-BuOH, reflux.
Scheme 54.
Cycloaddition of ylide 172 with aromatic ketones 3l and 3m followed by hydrolysis and condensation reactions to afford piperidones 194. Reaction Conditions: (i) 178 (2.5 equiv.), LiF, DMF, reflux, 5 h; (ii) aq. HCl, n-BuOH, 90 °C; (iii) NH4Cl;(iv) n-BuOH, reflux.
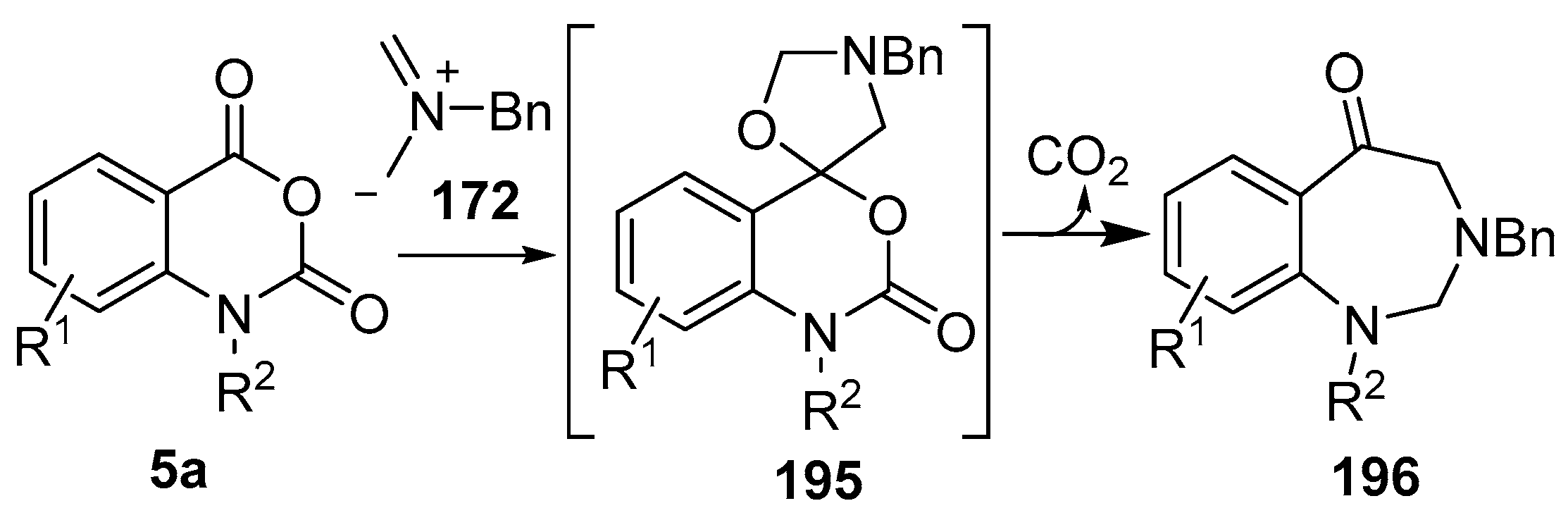
Scheme 55.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with isatoic anhydrides 5a, followed by conversion of the oxazolidines 195 to 1,3-benzodiazepin-5-ones 196.
Scheme 55.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with isatoic anhydrides 5a, followed by conversion of the oxazolidines 195 to 1,3-benzodiazepin-5-ones 196.
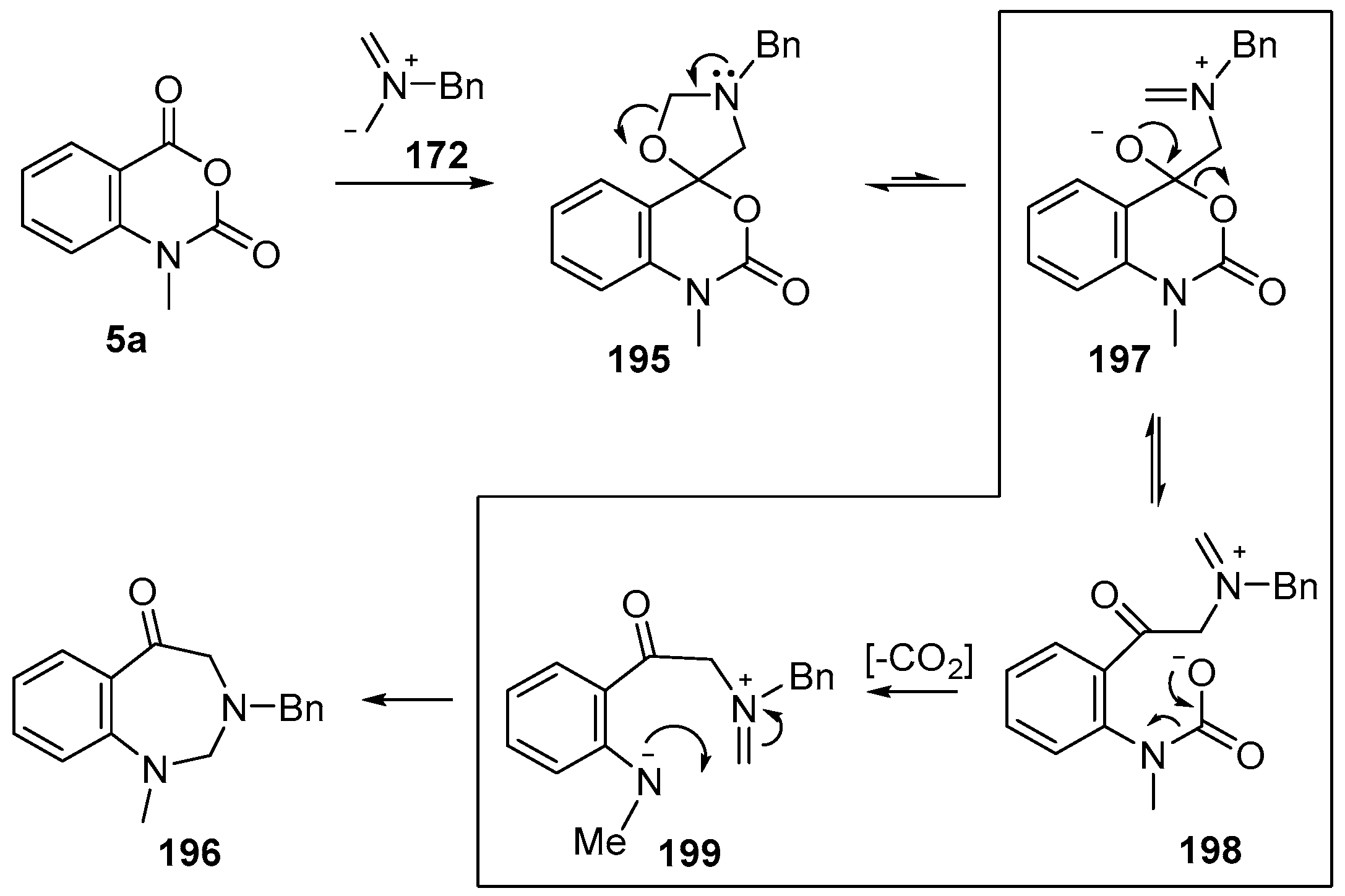
Scheme 56.
Proposed mechanism for the conversion of isatoic anhydrides 5a into benzodiazepinones 196.
Scheme 56.
Proposed mechanism for the conversion of isatoic anhydrides 5a into benzodiazepinones 196.
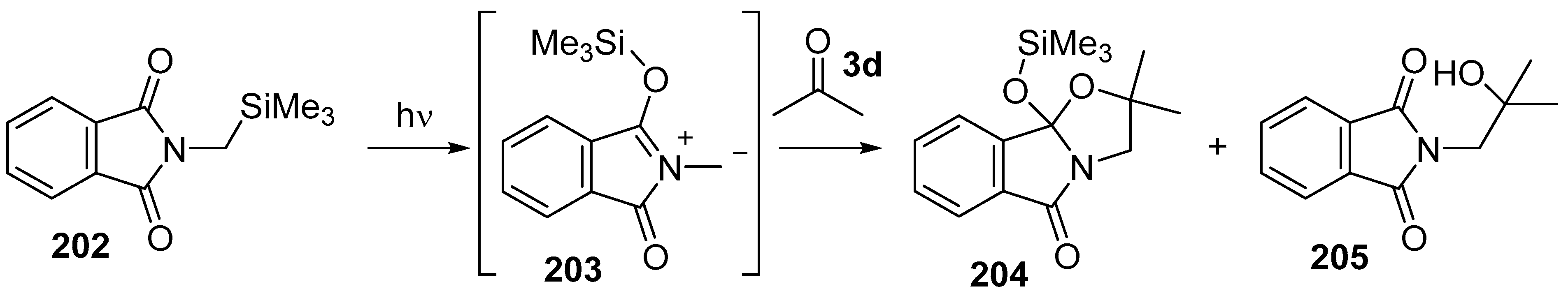
Scheme 57.
Cycloaddition of azomethine ylide 203, generated by photochemical rearrangement of N-(silylmethyl)phthalimide (202), with acetone (3d).
Scheme 57.
Cycloaddition of azomethine ylide 203, generated by photochemical rearrangement of N-(silylmethyl)phthalimide (202), with acetone (3d).
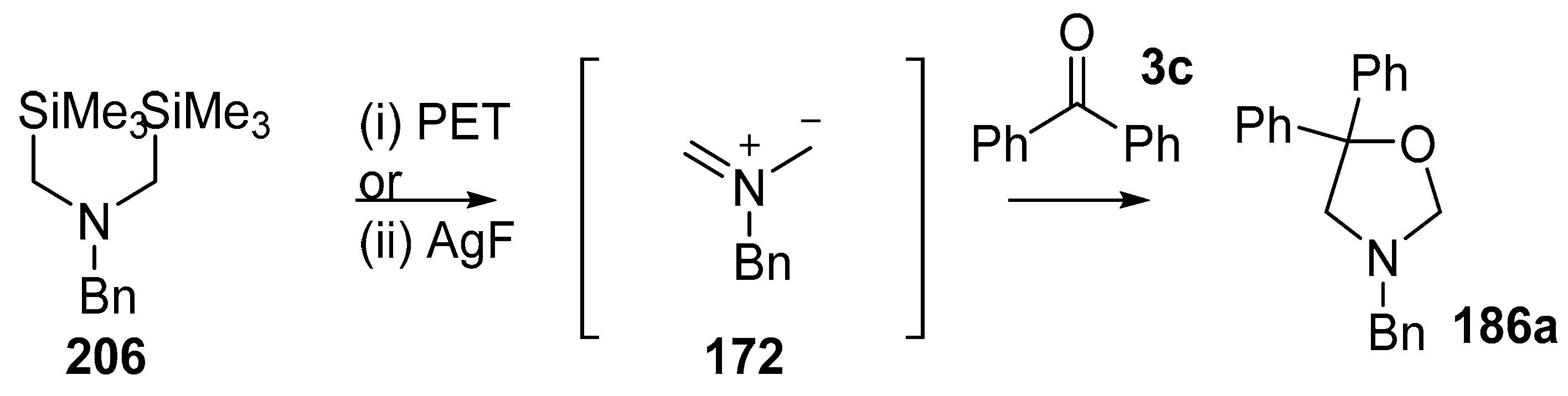
Scheme 58.
Cycloaddition of 172, formed by electrochemical oxidation of 206, with benzophenone 3c. Reaction conditions: (i) 206, 1,4-dicyanonaphthalene (0.20 equiv.), 3d (1.6 equiv.), 80%; (ii) 206, AgF (2 equiv.), 3d (1.2 equiv.), CH3CN, 62%.
Scheme 58.
Cycloaddition of 172, formed by electrochemical oxidation of 206, with benzophenone 3c. Reaction conditions: (i) 206, 1,4-dicyanonaphthalene (0.20 equiv.), 3d (1.6 equiv.), 80%; (ii) 206, AgF (2 equiv.), 3d (1.2 equiv.), CH3CN, 62%.
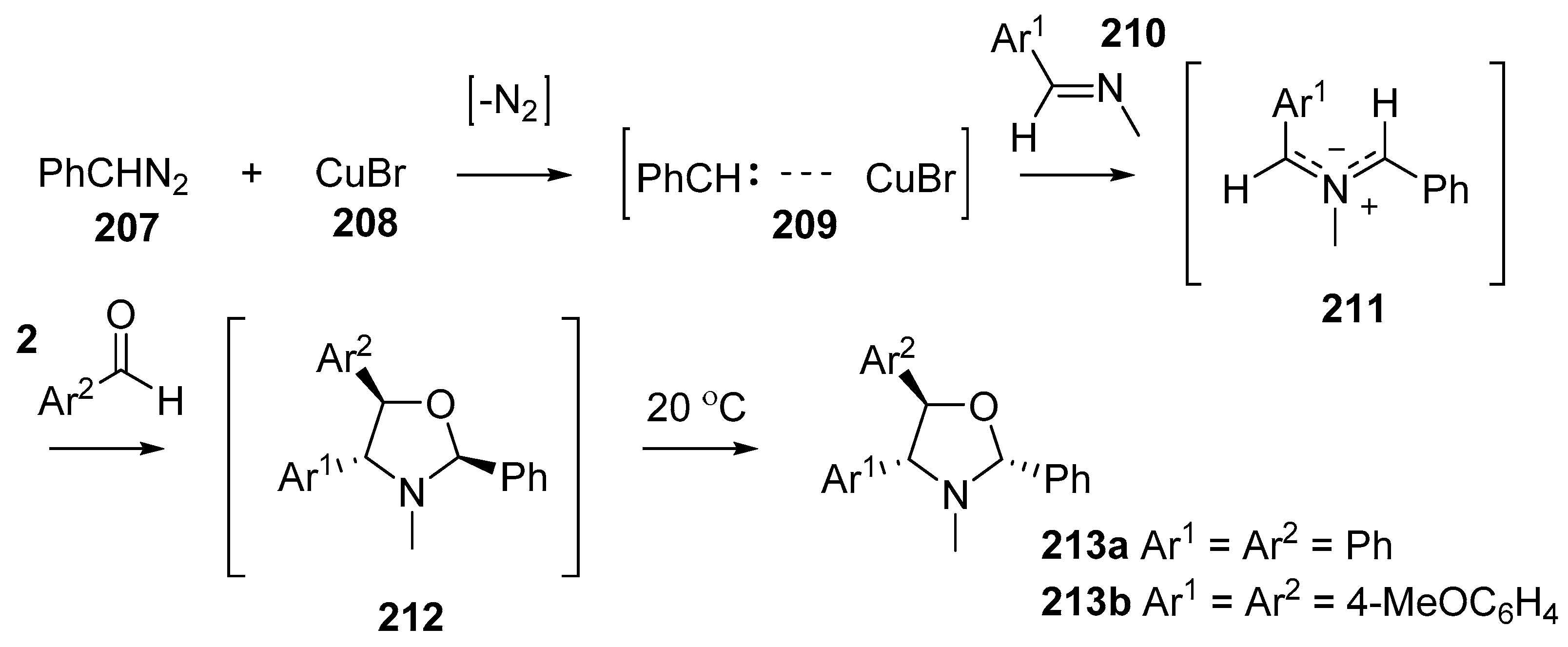
Scheme 59.
Cycloaddition of azomethines 211, generated from CuBr-catalyzed decomposition of phenyldiazomethane (207) in the presence of imines 210, with aromatic aldehydes 2.
Scheme 59.
Cycloaddition of azomethines 211, generated from CuBr-catalyzed decomposition of phenyldiazomethane (207) in the presence of imines 210, with aromatic aldehydes 2.
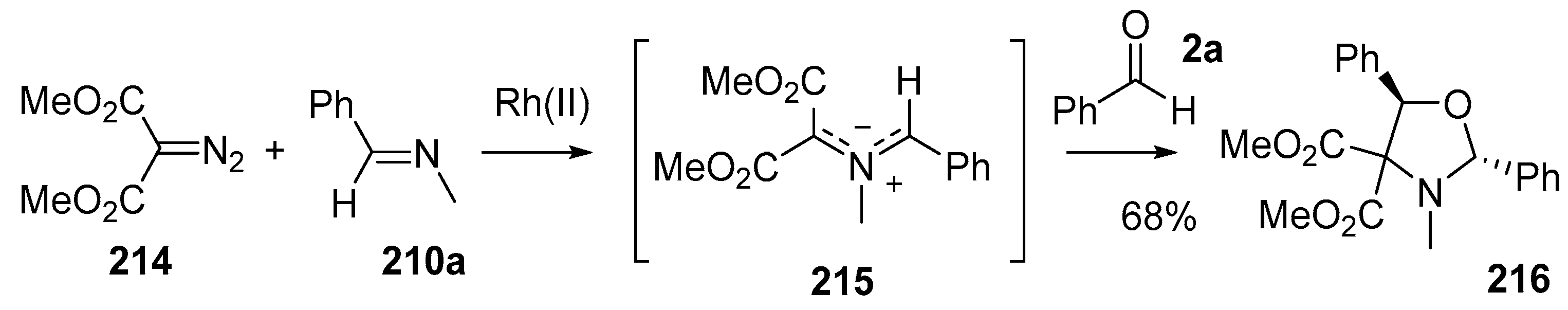
Scheme 60.
Cycloaddition of azomethine ylide 215, generated from Rh(II)-catalyzed decomposition of diazomalonate 214 in the presence of imine 210a, and benzaldehyde (2c).
Scheme 60.
Cycloaddition of azomethine ylide 215, generated from Rh(II)-catalyzed decomposition of diazomalonate 214 in the presence of imine 210a, and benzaldehyde (2c).
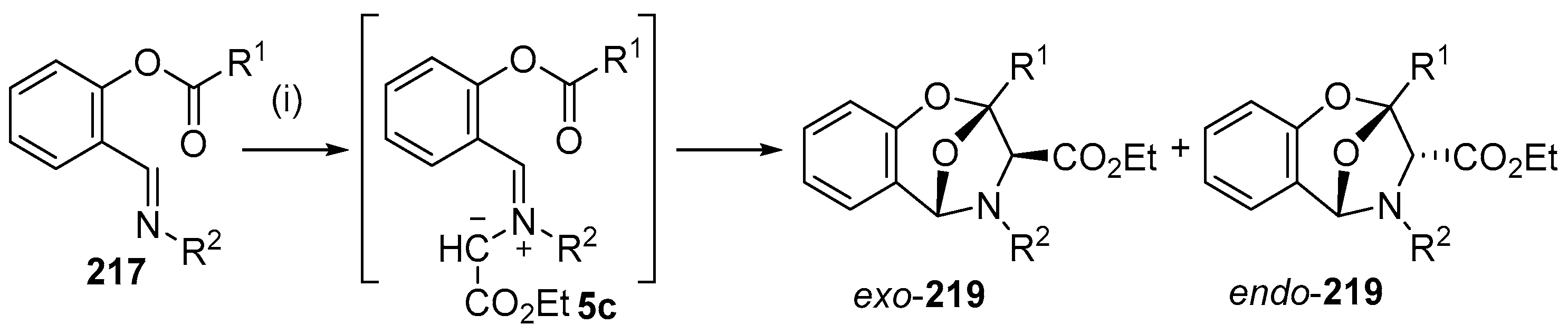
Scheme 61.
Intramolecular cycloaddition of azomethine ylide and ester carbonyl of intermediate 5c. Reagents and Conditions: (i) N2CCHCO2Et 218; Cu(tfacac)2, CH2Cl2, 40 °C.
Scheme 61.
Intramolecular cycloaddition of azomethine ylide and ester carbonyl of intermediate 5c. Reagents and Conditions: (i) N2CCHCO2Et 218; Cu(tfacac)2, CH2Cl2, 40 °C.
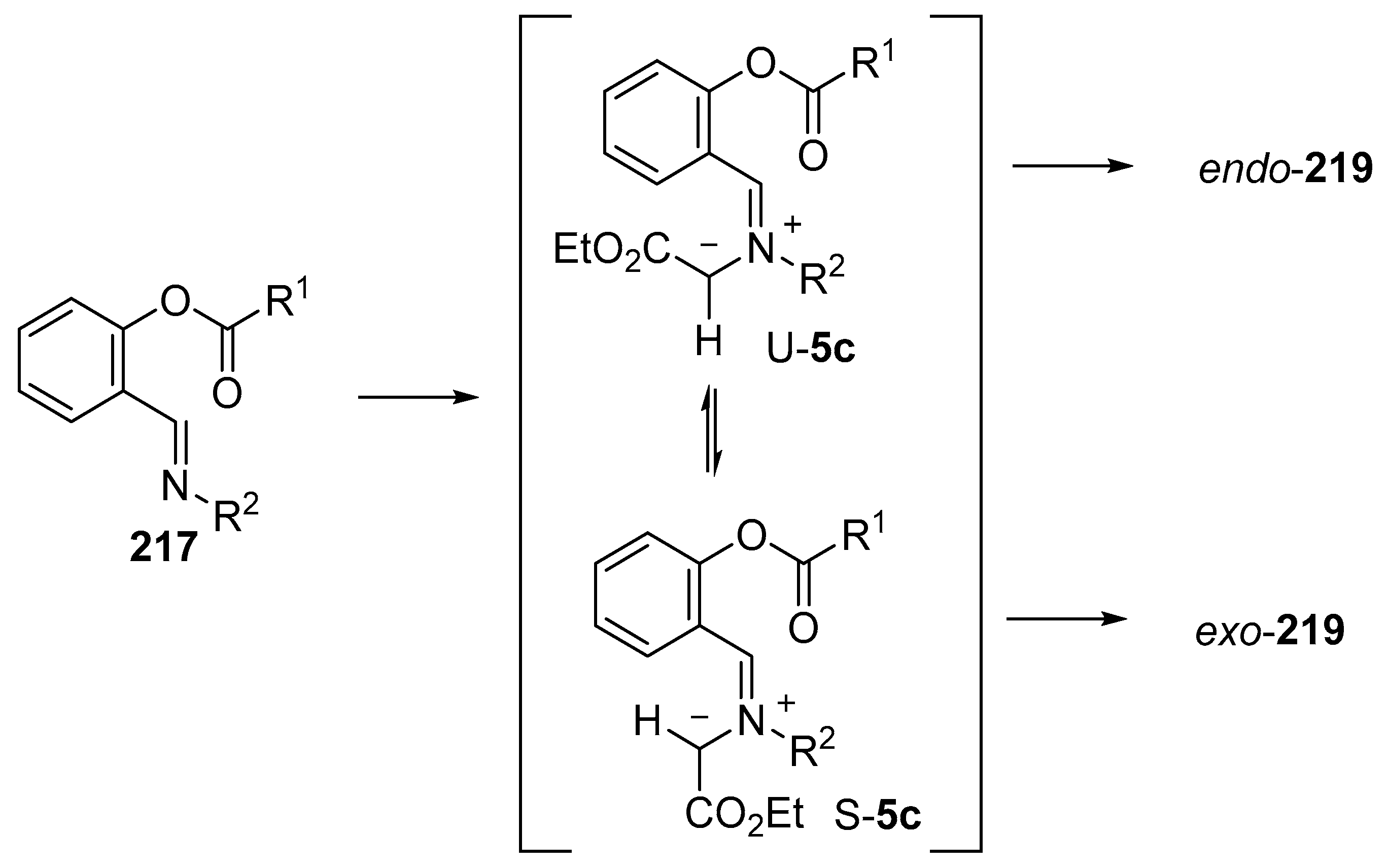
Scheme 62.
Mechanistic rationale for the formation of endo- and exo-cycloadducts 219.
Scheme 62.
Mechanistic rationale for the formation of endo- and exo-cycloadducts 219.
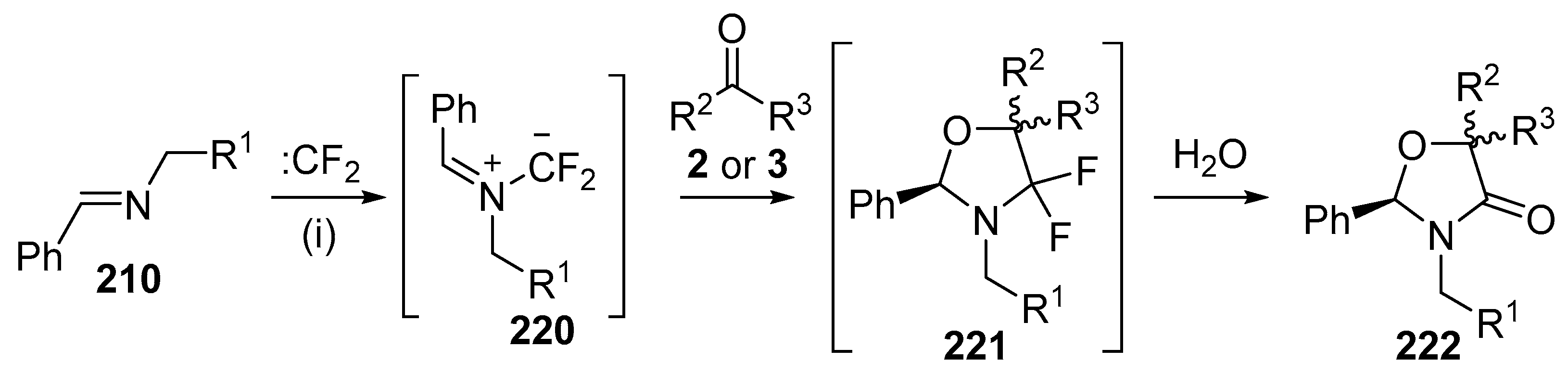
Scheme 63.
Cycloaddition of azomethine ylides 220 with aldehydes 2 or ketones 3. Reaction conditions: (i) imine 210, CBr2CF2, Pb powder, TBAF, carbonyl compound, CH2Cl2.
Scheme 63.
Cycloaddition of azomethine ylides 220 with aldehydes 2 or ketones 3. Reaction conditions: (i) imine 210, CBr2CF2, Pb powder, TBAF, carbonyl compound, CH2Cl2.
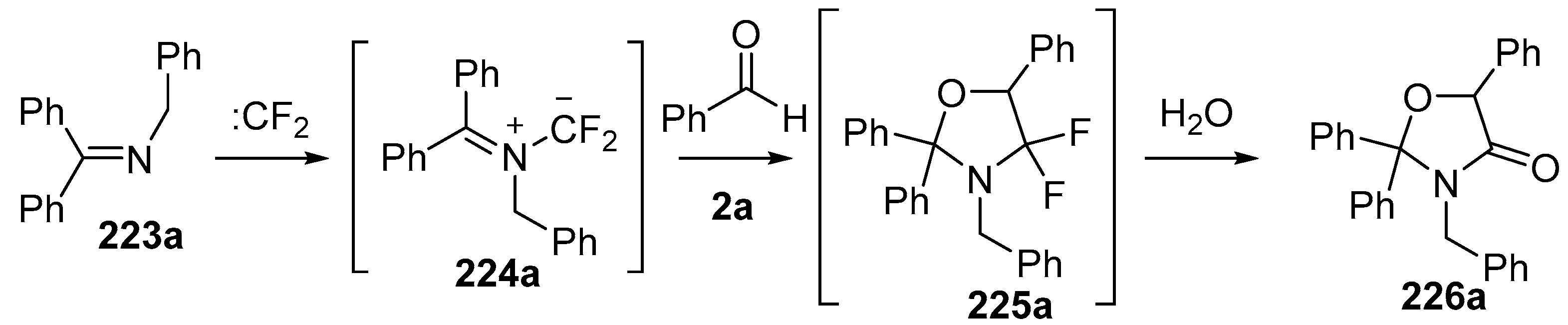
Scheme 64.
Cycloaddition of azomethine ylide 224a, generated from difluorocarbene and imine 223a, with benzaldehyde (2a).
Scheme 64.
Cycloaddition of azomethine ylide 224a, generated from difluorocarbene and imine 223a, with benzaldehyde (2a).
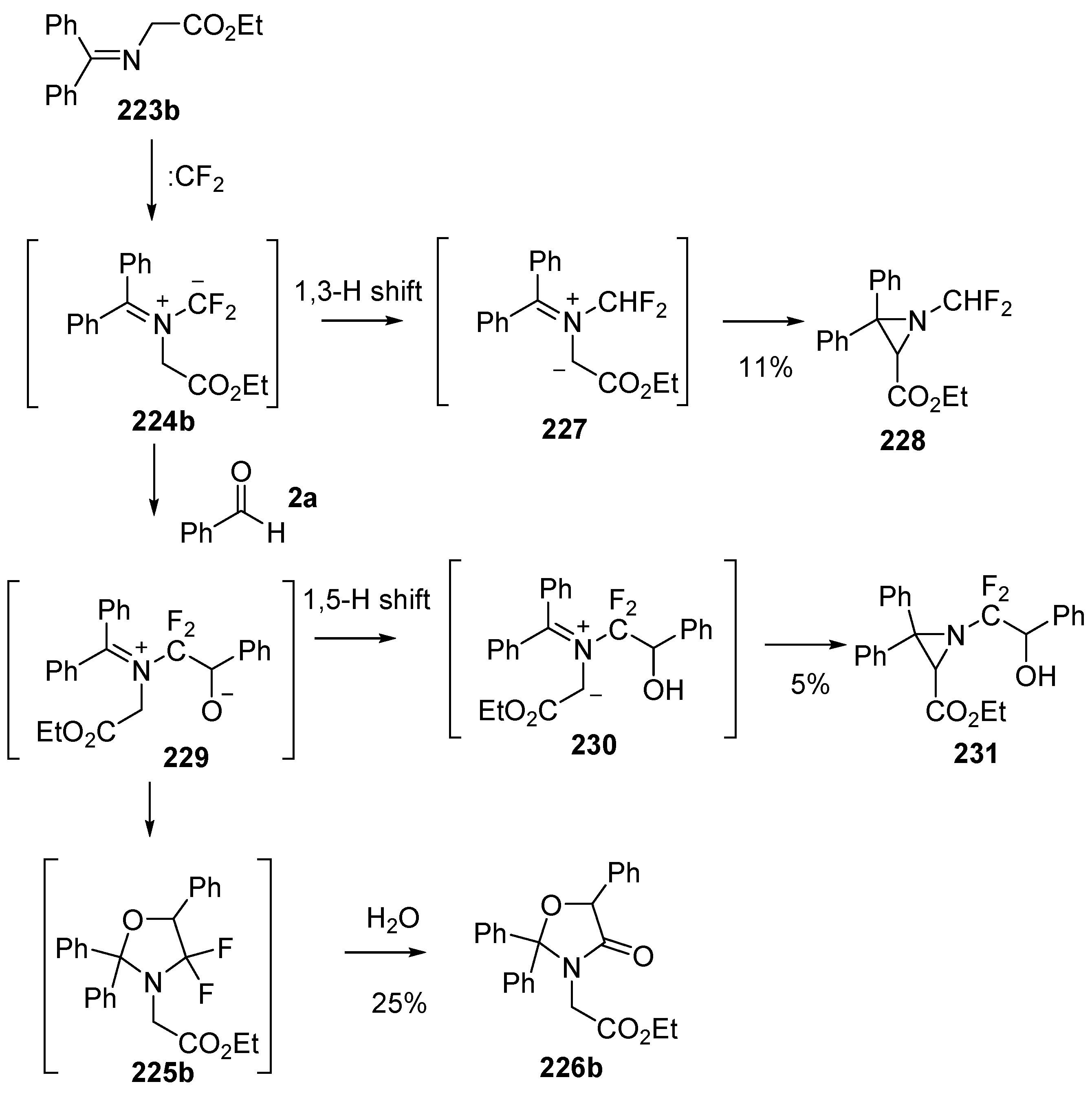
Scheme 65.
Cycloaddition of azomethine ylide 224b, generated from difluorocarbene and imine 223b, with benzaldehyde (2a).
Scheme 65.
Cycloaddition of azomethine ylide 224b, generated from difluorocarbene and imine 223b, with benzaldehyde (2a).
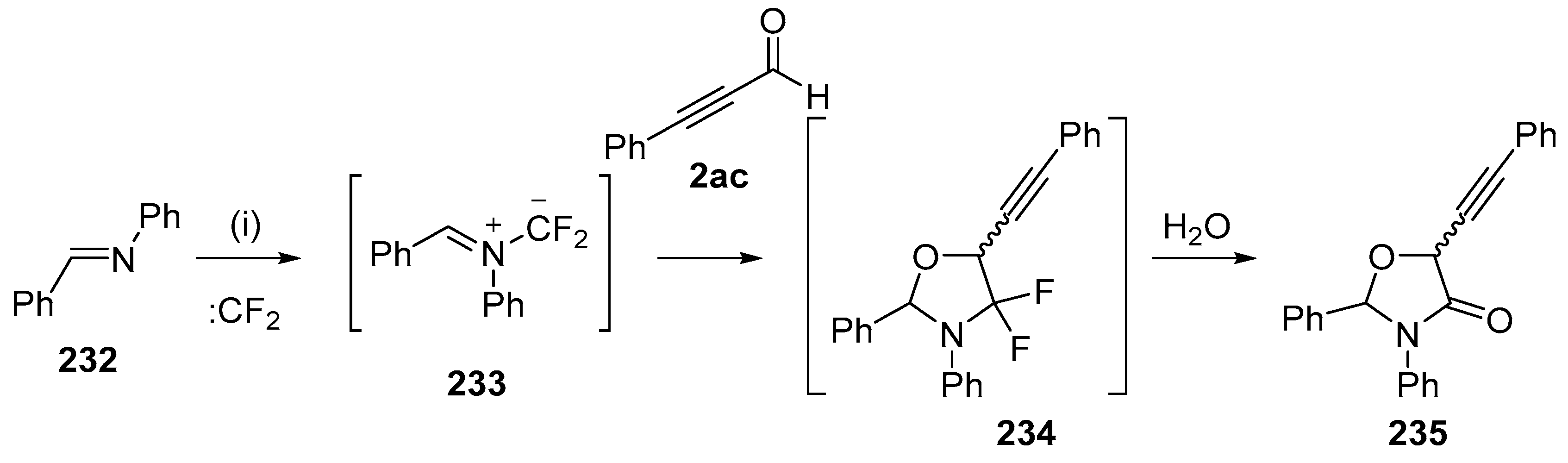
Scheme 66.
Regioselective cycloaddition of azomethine ylide 233 to the aldehyde of 2ac. Reaction conditions: (i) imine 232, CBr2CF2, Pb powder, TBAF, 2ac, CH2Cl2.
Scheme 66.
Regioselective cycloaddition of azomethine ylide 233 to the aldehyde of 2ac. Reaction conditions: (i) imine 232, CBr2CF2, Pb powder, TBAF, 2ac, CH2Cl2.
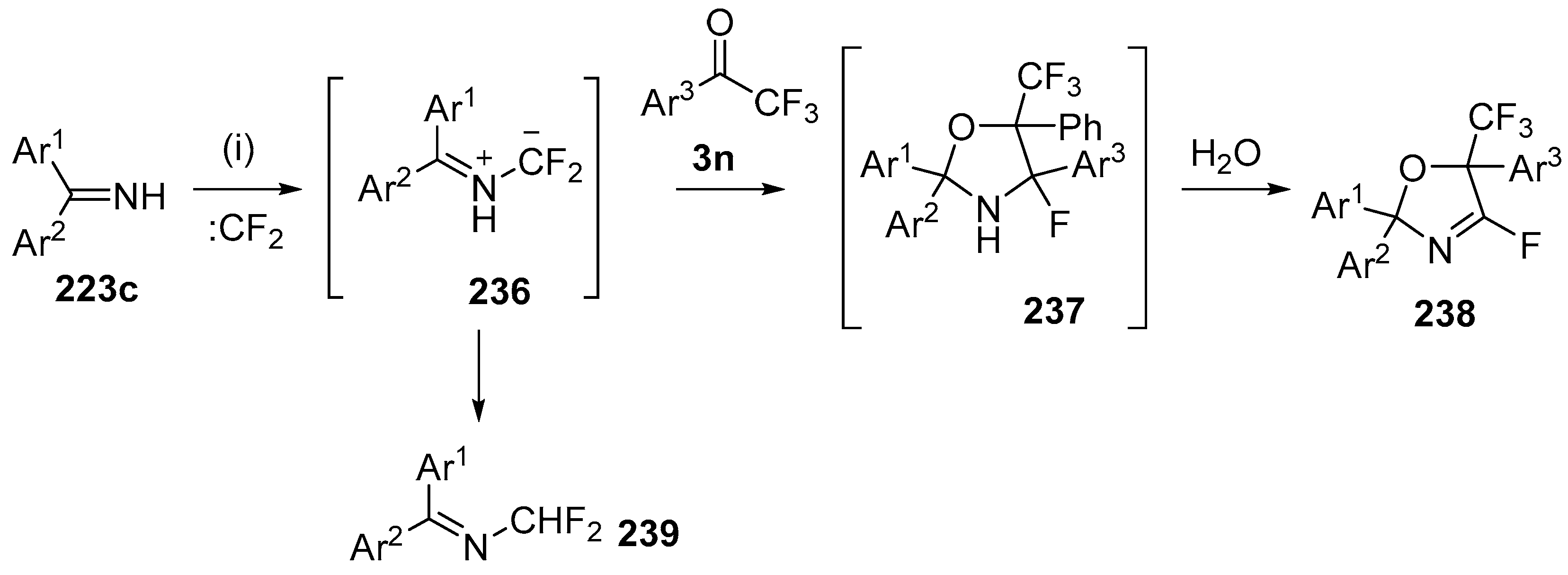
Scheme 67.
Cycloaddition of azomethine ylides 236 to trifluoroacetophenones 3n. Reagants and Conditions: (i): imine 223c, Pb (3 equiv.), Bu4NBr (3 equiv.), CF2Br2 (3 equiv.), Ar1COCF3 3 (3 equiv.), CH2Cl2, ultrasound.
Scheme 67.
Cycloaddition of azomethine ylides 236 to trifluoroacetophenones 3n. Reagants and Conditions: (i): imine 223c, Pb (3 equiv.), Bu4NBr (3 equiv.), CF2Br2 (3 equiv.), Ar1COCF3 3 (3 equiv.), CH2Cl2, ultrasound.
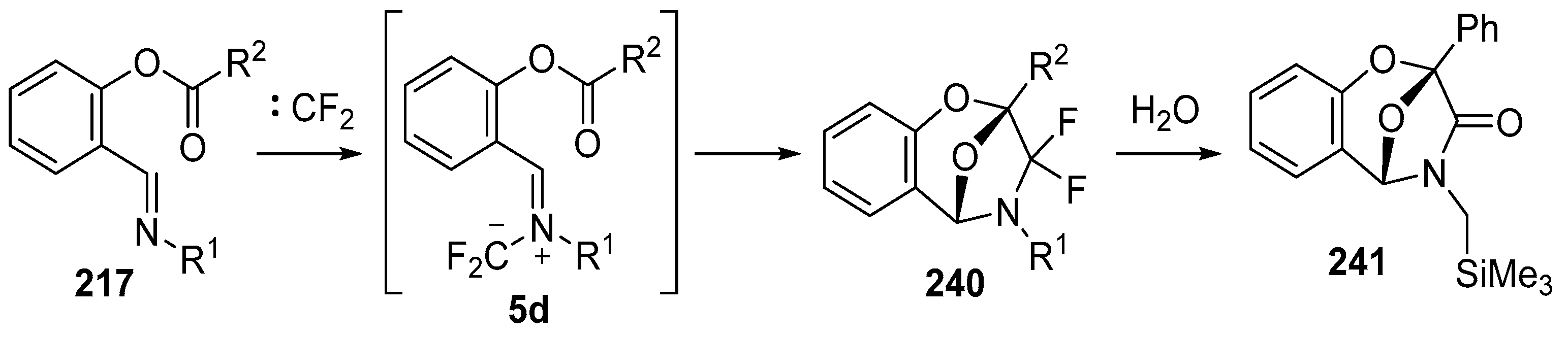
Scheme 68.
Intramolecular cycloaddition of the azomethine ylide and ester moieties of intermediate 5d.
Scheme 68.
Intramolecular cycloaddition of the azomethine ylide and ester moieties of intermediate 5d.
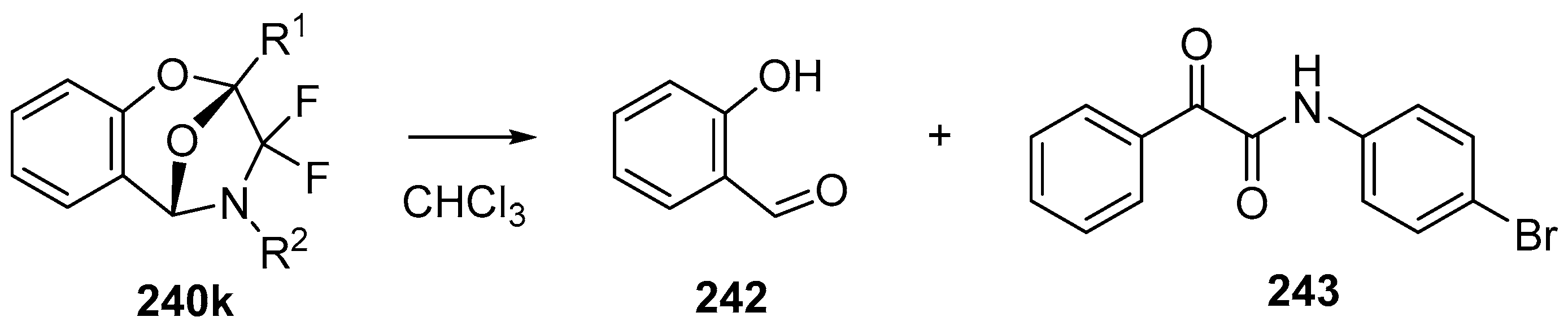
Scheme 69.
Decomposition of cycloadduct 240k in chloroform.
Scheme 69.
Decomposition of cycloadduct 240k in chloroform.
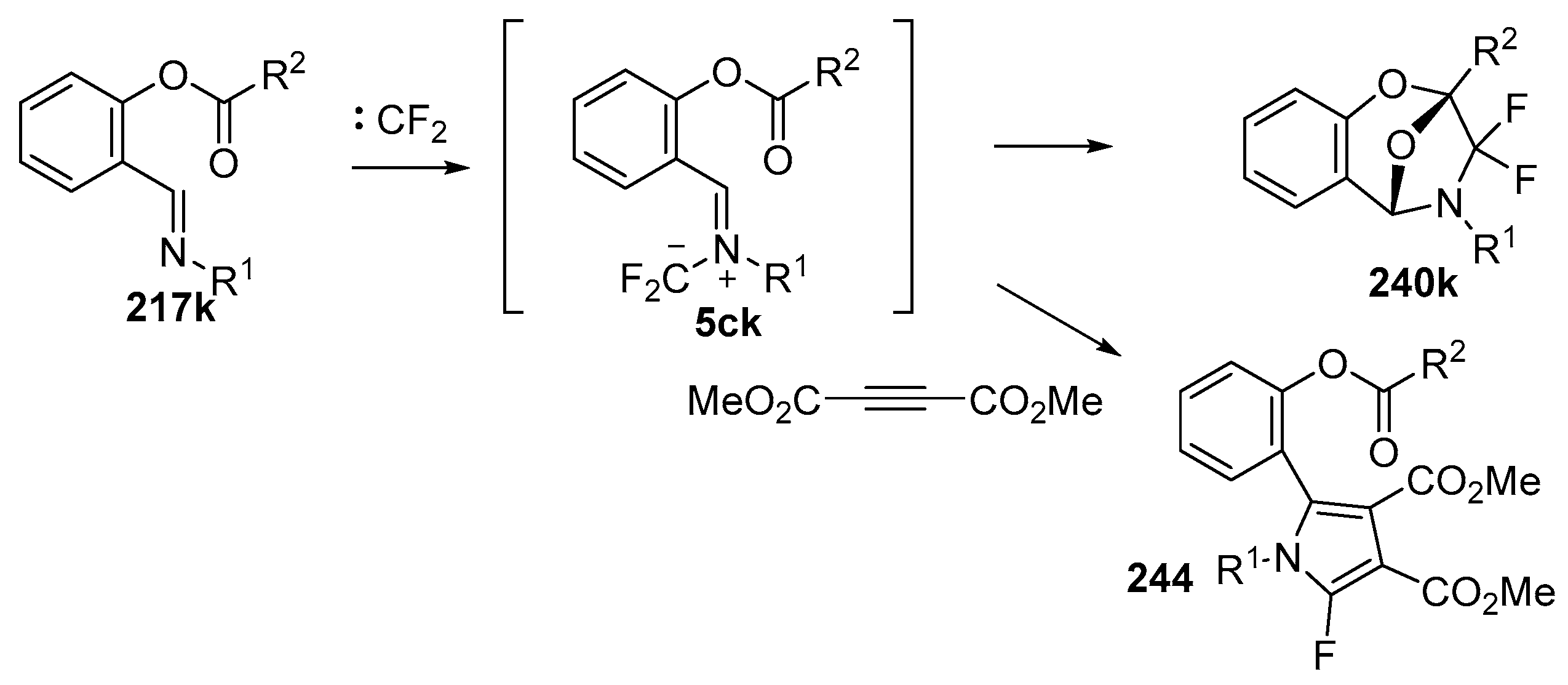
Scheme 70.
Intramolecular versus intermolecule competition reactions of azomethine ylides 5ck.
Scheme 70.
Intramolecular versus intermolecule competition reactions of azomethine ylides 5ck.
Figure 4.
Analogous azomethine ylide that fails to undergo intramolecular cycloaddition.
Figure 4.
Analogous azomethine ylide that fails to undergo intramolecular cycloaddition.
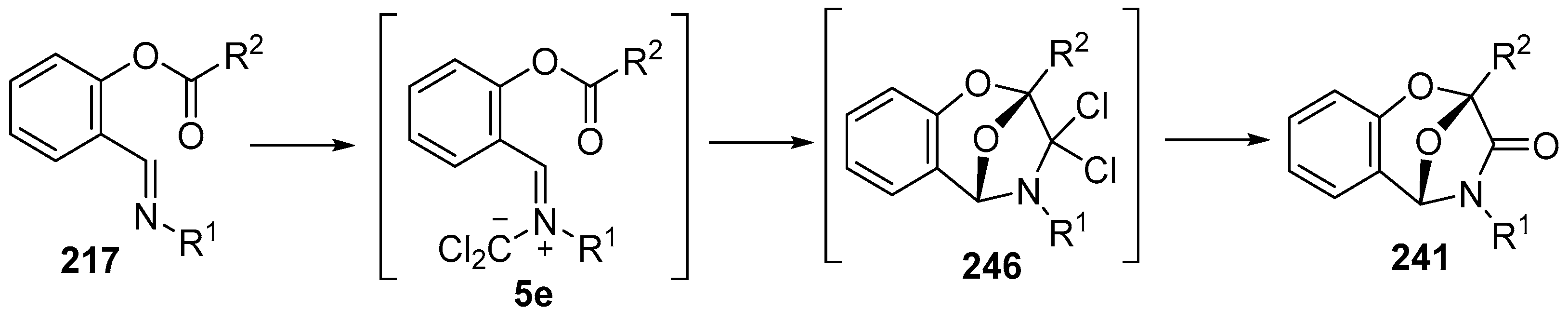
Scheme 71.
Intramolecular cycloadditions of the azomethine ylide 5e.
Scheme 71.
Intramolecular cycloadditions of the azomethine ylide 5e.
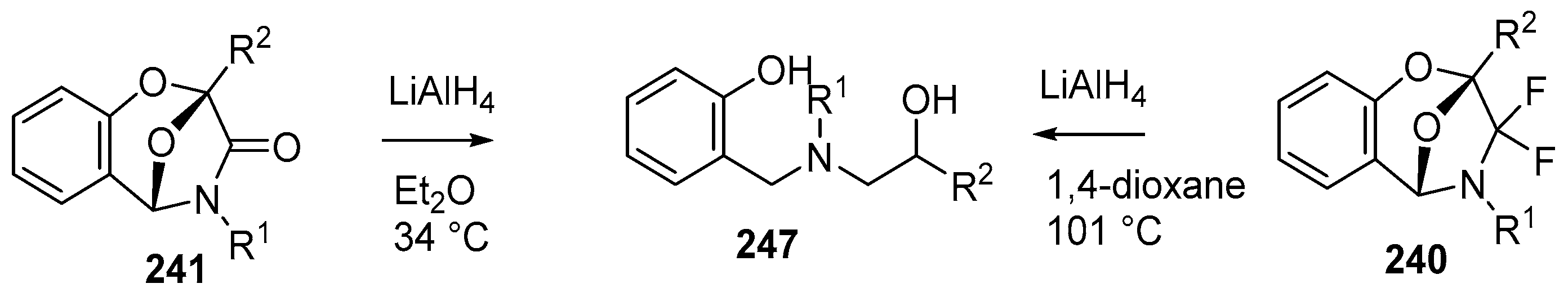
Scheme 72.
Reduction of cycloadducts 240 and 241.
Scheme 72.
Reduction of cycloadducts 240 and 241.
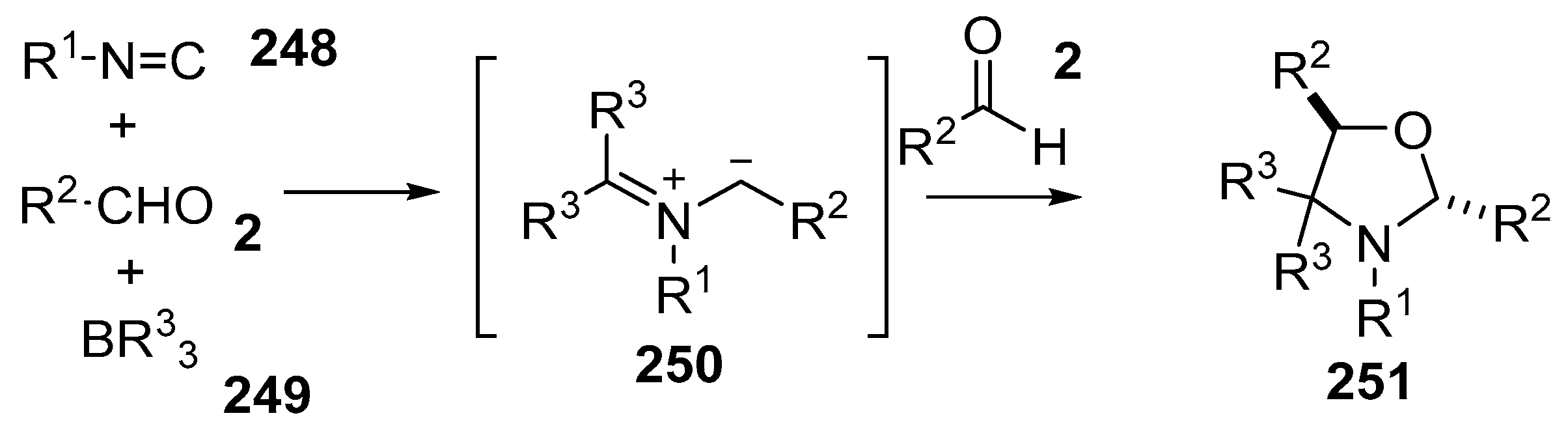
Scheme 73.
Cycloaddition of azomethine ylides, generated by the reaction of aldehydes 2, isocyanides 248 and organoboron compounds 249.
Scheme 73.
Cycloaddition of azomethine ylides, generated by the reaction of aldehydes 2, isocyanides 248 and organoboron compounds 249.
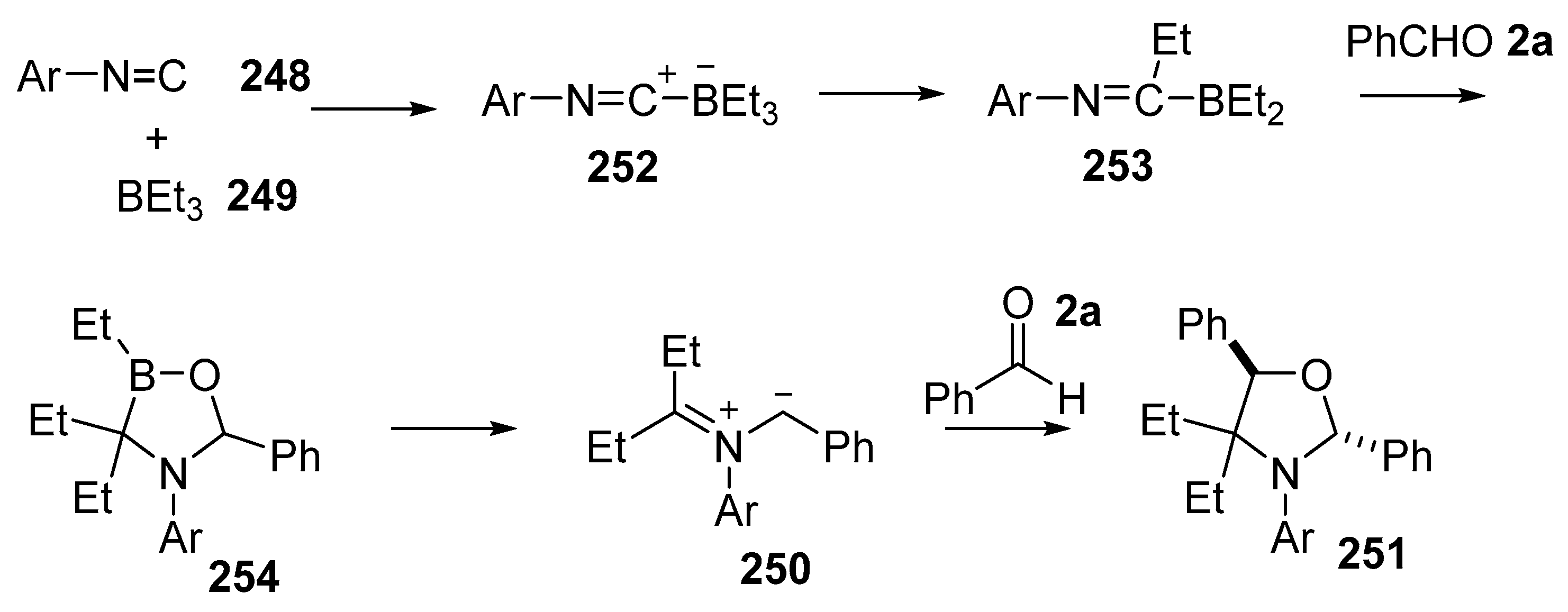
Scheme 74.
Proposed mechanism for the formation of oxazolidines 251.
Scheme 74.
Proposed mechanism for the formation of oxazolidines 251.
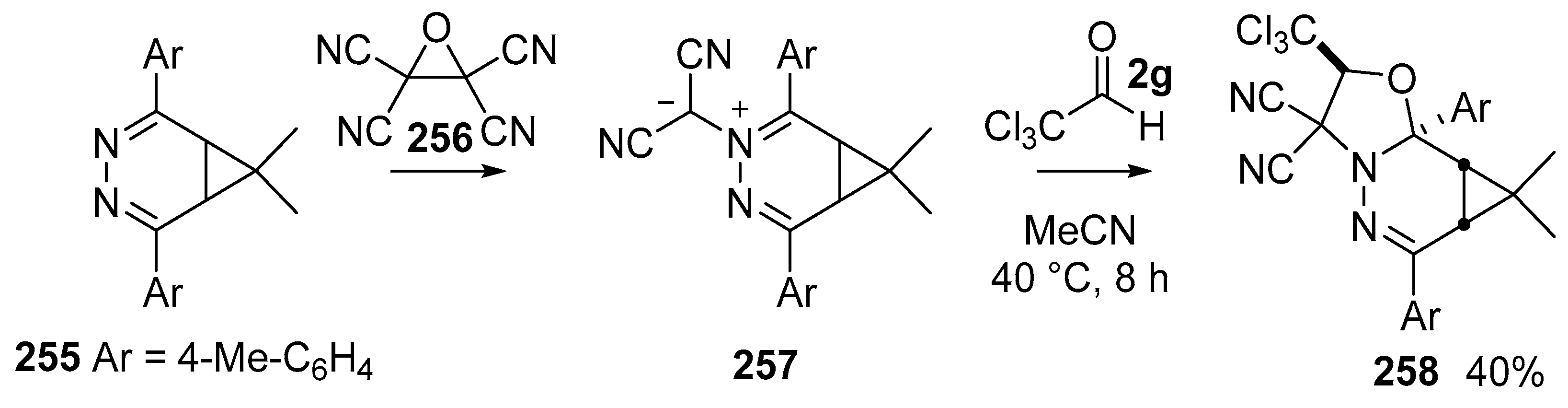
Scheme 75.
Cycloaddition of azomethine ylides 257 with trichloroacetaldehyde 2g.
Scheme 75.
Cycloaddition of azomethine ylides 257 with trichloroacetaldehyde 2g.
Table 1.
Cycloaddition of azomethine ylide
19 to aldehydes
2a–
f (
Scheme 3).
Table 1.
Cycloaddition of azomethine ylide 19 to aldehydes 2a–f (Scheme 3).
| Entry | R1 | Product 20 (% Yield) |
|---|
| 1 | Ph | 20a (100) |
| 2 | CH3 | 20b (60) |
| 3 | 4-OMe-C6H4 | 20c (100) |
| 4 | PhCH=CH | 20d (100) |
| 5 | i-Pr | 20e (45) |
| 6 | Ph2CH | 20f (80) |
Table 2.
Cycloaddition of
trans-azomethine ylide
24 to aldehydes
2 (
Scheme 4).
Table 2.
Cycloaddition of trans-azomethine ylide 24 to aldehydes 2 (Scheme 4).
| Entry | R1 | R2 | R3 | R4 | Oxazolidines 25 and 26
(4,5-trans:cis 1, % Yield) |
|---|
| 1 | 3-NO2-C6H4 | i-Pr | Ph | 4-Cl-3-NO2-C6H3 | a (61:39, 56) |
| 2 | 4-NO2-C6H4 | C6H11 | Ph | 4-Cl-3-NO2-C6H3 | b (75:25, 44) |
| 3 | Ph | i-Pr | Ph | 4-NO2-C6H4 | c (100:0, 21) |
| 4 | 3-NO2-C6H4 | i-Pr | Ph | 4-NO2-C6H4 | d (45:55, 71) |
| 5 | 4-NO2-C6H4 | C6H11 | Ph | 4-NO2-C6H4 | e (100:0, 25) |
| 6 | 3-NO2-C6H4 | i-Pr | Ph | 2,4-(NO2)2-C6H3 | f (0:100, 65) |
| 7 | 3-NO2-C6H4 | i-Pr | Ph | Ph | g (36:64, 48) |
| 8 | Ph | C6H11 | Ph | 4-NO2-C6H4 | h (100:0, 45) |
| 9 | 3-NO2-C6H4 | C6H11 | Ph | Ph | i (42:58, 44) |
| 10 | 3-NO2-C6H4 | C6H11 | Ph | 4-NO2-C6H4 | j (32:68, 57) |
| 11 | Ph | i-Pr | Ph | CCl3 | k (100:0, 84) |
| 12 | 3-NO2-C6H4 | i-Pr | Ph | CCl3 | l (100:0, 65) |
| 13 | 4-NO2-C6H4 | C6H11 | Ph | CCl3 | m (100:0, 78) |
| 14 | Ph | Bn | 4-MeC6H4 | CCl3 | n (100:0, 38) |
| 15 | 3-NO2-C6H4 | C6H11 | Ph | CCl3 | o (100:0, 72) |
Table 3.
Cycloaddition of the azomethine ylides derived from aziridines
23 to diphenylcyclopropenone (
3a) (
Scheme 5).
Table 3.
Cycloaddition of the azomethine ylides derived from aziridines 23 to diphenylcyclopropenone (3a) (Scheme 5).
| Entry | R1 | R2 | R3 | Product 29 (% Yield) |
|---|
| 1 | Ph | Me | Ph | a (20) 1 |
| 2 | Ph | i-Pr | Ph | b (24) 1 |
| 3 | Ph | i-Pr | 4-NO2-C6H4 | c (24) 1 |
| 4 | Ph | C6H11 | Ph | d (65) |
| 5 | 4-NO2-C6H4 | C6H11 | Ph | e (23) 1 |
| 6 | 3-NO2-C6H4 | C6H11 | Ph | f (27) 1 |
| 7 | 4-NO2-C6H4 | C6H11 | Ph | g (64) |
| 8 | 4-Ph-C6H4 | C6H11 | Ph | h (100) |
| 9 | Ph | C6H11 | 4-OMe-C6H4 | i (79) |
| 10 | Ph | C6H11 | 4-Me-C6H4 | j (20) 1 |
| 11 | Ph | C6H11 | 4-NO2-C6H4 | k (23) |
| 12 | Ph | C6H11 | CH3 | l (84) |
| 13 | 4-Ph-C6H4 | C6H11 | CH3 | m (59) |
Table 4.
Cycloaddition of azomethine ylide
37 to aromatic aldehydes
2a and
2i (
Scheme 7).
Table 4.
Cycloaddition of azomethine ylide 37 to aromatic aldehydes 2a and 2i (Scheme 7).
| Entry | R1 = R2 | R3 | R4 | Product 38 (% Yield) |
|---|
| 1 | H | 4-NO2-C6H4 | 4-NO2-C6H4 | a (80) |
| 2 | H | Ph | Ph | b (29) |
| 3 | H | 4-NO2-C6H4 | Ph | c (59) |
| 4 | Me | 4-NO2-C6H4 | 4-NO2-C6H4 | d (39) |
Table 5.
Cycloaddition of azomethine ylides
45 to aldehydes
2 catalyzed by nickel(II) perchlorate (
Scheme 9).
Table 5.
Cycloaddition of azomethine ylides 45 to aldehydes 2 catalyzed by nickel(II) perchlorate (Scheme 9).
| Entry | Ar | R1 | R2 | % Yield 46 |
|---|
| 1 | Ph | Me | Ph | 67 |
| 2 | Ph | Me | 3-Me-C6H4 | 64 |
| 3 | Ph | Me | 4-i-Pr-C6H4 | 72 |
| 4 | Ph | Me | 4-OMe-C6H4 | 92 |
| 5 | Ph | Me | 2-OMe-C6H4 | 55 |
| 6 | Ph | Me | 4-Cl-C6H4 | 50 |
| 7 | Ph | Me | 4-Br-C6H4 | 55 |
| 8 | Ph | Me | 2-furyl | 80 |
| 9 | Ph | Me | 1-napthyl | 70 |
| 10 | Ph | Me | (E)-Ph-CH=CH | 87 |
| 11 | Ph | Me | 3,4,5-(OMe)3-C6H2 | 89 |
| 12 | 4-NO2-C6H4 | Me | 3,4,5-(OMe)3-C6H2 | 99 |
| 13 | 4-Cl-C6H4 | Me | 3,4,5-(OMe)3-C6H2 | 83 |
| 14 | 4-Br-C6H4 | Me | 3,4,5-(OMe)3-C6H2 | 97 |
| 15 | 4-Me-C6H4 | Me | 3,4,5-(OMe)3-C6H2 | 84 |
| 16 | 4-i-Pr-C6H4 | Me | 3,4,5-(OMe)3-C6H2 | 84 |
| 17 | 3-Me-C6H4 | Me | 3,4,5-(OMe)3-C6H2 | 88 |
| 18 | 2-Br-C6H4 | Me | 3,4,5-(OMe)3-C6H2 | 90 |
| 19 | Ph | Et | 3,4,5-(OMe)3-C6H2 | 82 |
| 20 | Ph | i-Pr | 3,4,5-(OMe)3-C6H2 | 90 |
| 21 | 4-i-Pr-C6H4 | i-Pr | 3,4,5-(OMe)3-C6H2 | 90 |
| 22 | 4-NO2-C6H4 | Me | Ph | 84 |
| 23 | 4-NO2-C6H4 | Me | 4-OMe-C6H4 | 96 |
| 24 | 4-NO2-C6H4 | Me | 4-Cl-C6H4 | 70 |
| 25 | Ph | Me | 3,4,5-(OMe)3-C6H2 | 82 |
Table 6.
Zinc triflate-catalyzed cycloaddition of azomethine ylide
45 to aldehydes
2 (
Scheme 9).
Table 6.
Zinc triflate-catalyzed cycloaddition of azomethine ylide 45 to aldehydes 2 (Scheme 9).
| Entry | Ar | R1 | R2 | % Yield 46 |
|---|
| 1 | Ph | Me | Ph | 65 |
| 2 | Ph | Me | 2-Br-C6H4 | 51 1 |
| 3 | Ph | Me | 4-Br-C6H4 | 62 |
| 4 | Ph | Me | 2-OMe-C6H4 | 81 |
| 5 | Ph | Me | 3-OMe-C6H4 | 77 |
| 6 | Ph | Me | 4-OMe-C6H4 | 84 |
| 7 | Ph | Me | 4-Me-C6H4 | 85 |
| 8 | Ph | Me | 4-NO2-C6H4 | 52 |
| 9 | 2-Br-C6H4 | Me | 4-Me-C6H4 | 87 |
| 10 | 2-Br-C6H4 | Me | 4-OMe-C6H4 | 74 |
| 11 | 3-Br-C6H4 | Me | 4-Me-C6H4 | 97 |
| 12 | 3-Br-C6H4 | Me | 4-OMe-C6H4 | 97 |
| 13 | 3-Br-C6H4 | Me | 4-Br-C6H4 | 94 |
| 14 | 4-Me-C6H4 | Me | 4-OMe-C6H4 | 84 |
| 15 | Ph | Et | Ph | 69 |
| 16 | Ph | Et | 4-Me-C6H4 | 74 |
| 17 | Ph | Et | 4-OMe-C6H4 | 72 |
| 18 | Ph | Me | 1-napthyl | 73 |
| 19 | Ph | Me | (E)-Ph-C=CH | 78 |
| 20 | 3-Br-C6H4 | Me | 2-furyl | 63 |
Table 7.
Cycloaddition of azomethine ylide
51 to aromatic aldehydes
2 (
Scheme 12).
Table 7.
Cycloaddition of azomethine ylide 51 to aromatic aldehydes 2 (Scheme 12).
| Entry | R1 | 2,3-trans:cis 1 | 52/53 (% yield) |
|---|
| 1 | Ph | 100:0 2 | 52a (63%) 2 |
| 2 | 4-NO2-C6H4 | 66:34 | 52b (60%); 53b (30%) |
| 3 | 4-OMeC6H4 | 100:0 | 52c (37%) |
| 4 | 4-pyridyl | 80:20 | 52d (35%) 3 |
| 5 | PhCO | 100:0 | 52e (85%) |
Table 8.
Reaction between glycine derivatives
47 or
54 and aromatic aldehydes
2a or
2i (
Scheme 13).
Table 8.
Reaction between glycine derivatives 47 or 54 and aromatic aldehydes 2a or 2i (Scheme 13).
| Entry | R1 | R2 | Product Ratio 1 | Product 55 (% Yield) |
|---|
| 1 | Me | 4-NO2-C6H4 | 55a (48):55b (19):56a (17):56b (17) | 55a (35%), 55b (19%) 2 |
| 2 | Me | Ph | 56a (33):56b (24) | 3 |
| 3 | Bn | 4-NO2-C6H4 | 55a (38):55b (23):56a (19):56b (21) | 55a (35%), 55b (19%) 4 |
| 4 | Bn | Ph | 5 | 55a (6%) |
Table 9.
Domino decarboxylative condensation, [3 + 2] cycloaddition and ring-opening aromatization process to give
N-β-hydroxyethyl pyrroles
70 (
Scheme 16).
Table 9.
Domino decarboxylative condensation, [3 + 2] cycloaddition and ring-opening aromatization process to give N-β-hydroxyethyl pyrroles 70 (Scheme 16).
| Entry | R1 | Product 70
% Yield (dr) 1 |
|---|
| 1 | 3-NO2-C6H4 | 95 (70:30) |
| 2 | 2-NO2-C6H4 | 73 (53:47) |
| 3 | 4-NO2-C6H4 | 85 (65:35) |
| 4 | 3-CN-C6H4 | 68 (65:35) |
| 5 | 4-CN-C6H4 | 75 (65:35) |
| 6 | 4-CF3-C6H4 | 45 (60:40) |
| 7 | 3-NO2-4-Cl-C6H3 | 83 (67:33) |
| 8 | 3-NO2-4-OMe-C6H3 | 85 (73:27) |
| 9 | 2-Cl-5-NO2-C6H3 | 87 (73:27) |
| 10 | 3-CN-5-F-C6H4 | 72 (72:28) |
| 11 | 3-pyridyl | 82 (57:43) |
| 12 | CO2Et | 43 (63:37) |
Table 10.
Cycloaddition of azomethine ylide
79 to aldehydes
2 or ketones
3 (
Scheme 18).
Table 10.
Cycloaddition of azomethine ylide 79 to aldehydes 2 or ketones 3 (Scheme 18).
| Entry | R2 | R3 | Oxapenams 80
% Yield (exo:endo) 1 |
|---|
| 1 | Ph | H | 19 (1:1) |
| 2 | 4-NO2-C6H4 | H | 21 (1:1) |
| 3 | 4-Cl-C6H4 | H | 26 (1:1) |
| 4 | 4-CF3-C6H4 | H | 21 (1:1) |
| 5 | C6F5 | H | 40 (1:1.5) |
| 6 | CF3 | CF3 | 56 |
| 7 | fluorenyl | 25 |
Table 11.
Cycloaddition of azomethine ylide
81 to aldehydes
2 (
Scheme 19).
Table 11.
Cycloaddition of azomethine ylide 81 to aldehydes 2 (Scheme 19).
| Entry | Ar | Oxazolidines 82
% Yield |
|---|
| 1 | 2-NO2-C6H4 | 95 |
| 2 | 3-NO2-C6H4 | 98 |
| 3 | 4-NO2-C6H4 | 92 |
| 4 | 4-Cl-C6H4 | 87 |
| 5 | 2,4-(Cl)2-C6H3 | 91 |
| 6 | 2-Br-C6H4 | 86 |
| 7 | 3,4-(OCH2O)-C6H3 | 58 |
| 8 | 1-naphthyl | 72 |
| 9 | 2-pyridyl | 77 |
| 10 | 2-thienyl | 87 |
Table 12.
Cycloaddition of azomethine ylide
81 to ketones
3 (
Scheme 21).
Table 13.
Cycloaddition of excess azomethine ylide
81 to 3-cyanochromones
96 (
Scheme 22).
Table 13.
Cycloaddition of excess azomethine ylide 81 to 3-cyanochromones 96 (Scheme 22).
| Entry | R | Oxazolidines 97
% Yield 1 |
|---|
| 1 | H | quant. |
| 2 | Me | quant. |
| 3 | Cl | quant. (32) 2 |
Table 14.
Ring-contraction of benz-1,2,6-oxadiazepines
99 to indazole-
N-oxides
100 and formation of 3-methyl-5-aryloxazolidines
82b–
e (
Scheme 23).
Table 14.
Ring-contraction of benz-1,2,6-oxadiazepines 99 to indazole-N-oxides 100 and formation of 3-methyl-5-aryloxazolidines 82b–e (Scheme 23).
| Entry | R1 | R2 | Indazole-N-oxides 100 % Yield | Oxazolidines 82
% Yield |
|---|
| 1 | H | H | 40 | 43 |
| 2 | ~OCH2O~ | 32 | 30 |
| 3 | OMe | OMe | 34 | 38 |
| 4 | Br | H | 37 | 33 |
Table 15.
Cycloaddition of
N-metalated azomethine ylide
118 to benzaldehyde (
2a) (
Scheme 29)
1.
Table 15.
Cycloaddition of N-metalated azomethine ylide 118 to benzaldehyde (2a) (Scheme 29) 1.
| Entry | R1 | R2 | Oxazolidines 119
% Conversion 2 (trans:cis) 3 |
|---|
| 1 | Ph | Me | 56 (1.6:1) |
| 2 | 4-F-C6H4 | Et | 63 (1.7:1) |
| 3 | 4-F-C6H4 | Et | 82 (1.7:1) |
| 4 | 4-CN-C6H4 | Et | 90 (2.3:1) |
| 5 | 4-CF3-C6H4 | Et | 96 (2.3:1) |
| 6 | 4-CN-C6H4 | Et | 80 (1.8:1) 4 |
| 7 | 4-CF3-C6H4 | Et | 93 (2.0:1) 5 |
| 8 | 4-CF3-C6H4 | Me | 84 (2.5:1) |
| 9 | 4-CF3-C6H4 | Me | 78 (2.0:1) 6 |
Table 16.
Cycloaddition of azomethine ylide 121 to aldehydes 2 (
Scheme 30).
Table 16.
Cycloaddition of azomethine ylide 121 to aldehydes 2 (Scheme 30).
| Entry | R1 | Oxazolines 123
% Yield |
|---|
| 1 | Ph | 89 |
| 2 | E-Ph-CH=CH | 84 |
| 3 | 2-pyridyl | 90 |
| 4 | 2-furyl | 91 |
| 5 | i-Pr | 80 |
| 6 | Et | 95 |
| 7 1 | CO2Et | 72 |
Table 17.
Cycloaddition of azomethine ylide
126 to aromatic aldehydes
2 (
Scheme 31).
Table 17.
Cycloaddition of azomethine ylide 126 to aromatic aldehydes 2 (Scheme 31).
| Entry | Ar | Oxazolidines 127
% Yield |
|---|
| 1 | 4-Cl-C6H4 | 72 |
| 2 | Ph | 72 |
| 3 | 4-Me-C6H4 | 74 |
| 4 | 4-OMe-C6H4 | 75 |
| 5 | 4-SMe-C6H4 | 70 |
| 6 | 3,4-OCH2O-C6H3 | 68 |
| 7 | 4-NMe2-C6H4 | 61 |
| 8 | 4-F-C6H4 | 70 |
| 9 | 4-Br-C6H4 | 65 |
| 10 | 2-Cl-C6H4 | 60 |
| 11 | 3-NO2-C6H4 | 78 |
| 12 | 4-NO2-C6H4 | 77 |
| 13 | 2-thienyl | 62 |
Table 18.
Cycloaddition of azomethine ylide
129 to formaldehyde (
2m), and subsequent FVT-induced cycloreversion-intramolecular cycloaddition (
Scheme 32).
Table 18.
Cycloaddition of azomethine ylide 129 to formaldehyde (2m), and subsequent FVT-induced cycloreversion-intramolecular cycloaddition (Scheme 32).
| Entry | R1 | R2 | Oxazolidines 130
% Yield | Pyrrolidines 131
% Yield |
|---|
| 1 | Me | H | 88 | 72 |
| 2 | (CH2)2CO2Me | H | 82 | 82 |
| 3 | -(CH2)3- | 60 | 98 |
Table 19.
Cycloaddition of azomethine ylide
136 to aldehydes
2 (
Scheme 34).
Table 19.
Cycloaddition of azomethine ylide 136 to aldehydes 2 (Scheme 34).
| Entry | R1 | R2 | Oxazolidines 137a:137b:138c 1 |
|---|
| 1 | Me | Ph | 86:12:2 2 |
| 2 | Me | 4-OMe-C6H4 | 65:35:0 |
| 3 | Me | 4-NO2-C6H4 | 65 3 |
| 4 | t-Bu | Ph | 50:50:0 |
Table 20.
Cycloaddition of azomethine ylide
140 to aldehydes
2 (
Scheme 35).
Table 20.
Cycloaddition of azomethine ylide 140 to aldehydes 2 (Scheme 35).
| Entry | R1 | Oxazolidines 141
% Yield |
|---|
| 1 | H | quant. |
| 2 | Ph | 69 |
| 3 | 4-F-C6H4 | 61 |
| 4 | 4-NO2-C6H4 | 45 |
| 5 | 4-OMe-C6H4 | 50 |
| 6 | 2-furyl | 51 |
| 7 | n-Pr | 86 |
| 8 | n-Bu | 80 |
| 9 | c-C6H11 | 80 |
Table 21.
Cycloaddition of azomethine ylide
160 to aldehydes and a ketone (
Scheme 39).
Table 21.
Cycloaddition of azomethine ylide 160 to aldehydes and a ketone (Scheme 39).
| Entry | R1 | R2 | Product (% Yield) |
|---|
| 1 | Ph | H | 163a (59) + 162a (11) |
| 2 | 4-NO2-C6H4 | H | 163b (51) + 162b (7) |
| 3 | 2-naphthyl | H | 163c (28) + 162c (3) |
| 4 | 4-OMe-C6H4 | H | 163d (52) + 162d (7) |
| 5 | Ph | Ph | 163e (47) |
| 6 | E-Ph-CH=CH | H | 163f (59) + 162f (11) |
| 7 | 2-pyridyl | H | 163g (63) |
Table 22.
Cycloaddition of azomethine ylides
167 to aldehyde 2
a and ketone
3c (
Scheme 40).
Table 22.
Cycloaddition of azomethine ylides 167 to aldehyde 2a and ketone 3c (Scheme 40).
| Entry | 165 | Carbonyl Compound | Product 168 | Yield, ds Ratio |
|---|
| 1 | a (R1 = H, M = SnBu3) | PhCHO 2a | a | 63%, 1.4:1 |
| 2 | b (R1 = i-Pr, M = SnBu3) | PhCHO 2a | b | 56%, 4:1.9:1 1,2 |
| 3 | a (R1 = H, M = SnBu3) | Ph2CO 3c | c | 65% |
| 4 | c (R1 = H, M = SiMe3) | Ph2CO 3c | c | 81% |
Table 23.
Cycloaddition of azomethine ylide
172, formed from silylamine reagent
178, with aromatic aldehydes
2 (
Scheme 45).
Table 23.
Cycloaddition of azomethine ylide 172, formed from silylamine reagent 178, with aromatic aldehydes 2 (Scheme 45).
| Entry | Ar | Product | % Yield |
|---|
| 1 | Ph | 173a | 95 |
| 2 | 2-NO2-C6H4 | 173b | 75 |
| 3 | 4-F-C6H4 | 173c | 89 |
| 4 | 2,4-(F)2-C6H3 | 173d | 80 |
| 5 | 2-Br-C6H4 | 173e | 76 |
| 6 | 4-Br-C6H4 | 173f | 86 |
| 7 | 4-CN-C6H4 | 173g | 82 |
| 8 | 4-NMe2-C6H4 | 173h | 78 |
| 9 | 4-OMe-C6H4 | 173i | 93 |
| 10 | 3,4,5-(OMe)3-C6H2 | 173j | ~100 |
| 11 | 4-NHAc-C6H4 | 173k | 78 |
| 12 | 2,4,6-(Me)3-C6H2 | 173l | 80 |
| 13 | 4-CO2H-CC6H4 | 173m | 59 |
| 14 | 2-OH-C6H4 | 173n | 54 |
| 15 | 3-OH-C6H4 | 173o | ‒ |
| 16 | 4-OH-C6H4 | 173p | ‒ |
Table 24.
Cycloaddition of azomethine ylide
172, formed from silylamine reagent
178, with heteroaromatic aldehydes
2 (
Scheme 45).
Table 25.
Transformation of isatoic anhydride and N-functionalised isatoic anhydrides 5a into 1,3-benzodiazepin-5-ones 196.
Table 25.
Transformation of isatoic anhydride and N-functionalised isatoic anhydrides 5a into 1,3-benzodiazepin-5-ones 196.
| Entry | R | Method 1 | Time (h) | % Yield 2 196 |
|---|
| 1 | H | A | 24 | 42 |
| 2 | Me | A | 36 | 92 |
| 3 | Et | A | 16 | 79 |
| 4 | Allyl | A | 16 | 71 |
| 5 | Bn | A | 40 | 77 |
| 6 | Ph | A | 16 | 80 |
| 7 | H | B | 3 | 0 |
| 8 | Me | B | 6 | 88 |
| 9 | Et | B | 2 | 96 |
| 10 | Allyl | B | 4 | 76 |
| 11 | Bn | B | 3 | 100 |
| 12 | Ph | B | 12 | 90 |
Table 26.
Transformation of benzo-substituted N-methyl isatoic anhydrides 5a into 1,3-benzo-diazepin-5-ones 196 1.
Table 26.
Transformation of benzo-substituted N-methyl isatoic anhydrides 5a into 1,3-benzo-diazepin-5-ones 196 1.
| Entry | R1 | R2 | R3 | R4 | Time (h) | % Yield 2 196 |
|---|
| 1 | H | H | H | H | 6 | 88 |
| 2 | OMe | H | H | H | 3 | 80 |
| 3 | H | F | H | H | 24 | 66 |
| 4 | H | CO2Me | H | H | 1.5 | 94 |
| 5 | H | OMe | H | H | 48 | - 3 |
| 6 | H | H | Me | H | 24 | 66 4 |
| 7 | H | H | Cl | H | 3 | 76 |
| 8 | H | H | Br | H | 2 | 63 |
| 9 | H | H | OMe | H | 41 | 46 5 |
| 10 | H | F | F | H | 6 | 93 |
| 11 | H | H | H | Me | 24 | - 6 |
| 12 | H | OMe | OMe | H | 56 | - 7 |
Table 27.
1,3-Dipolar cycloaddition reaction of phthalic anhydrides 5b with azomethine ylide 172, generated in situ from precursor 178 1.
Table 28.
Reductive ring-opening of oxazolidines 200 with NaBH4 1.
Table 29.
Reaction of imines
217, ethyl diazoacetate (
218) and Cu(tfacac)
2 in CH
2Cl
2 at 40 °C (
Scheme 61).
Table 29.
Reaction of imines 217, ethyl diazoacetate (218) and Cu(tfacac)2 in CH2Cl2 at 40 °C (Scheme 61).
| Entry | R1 | R2 | % Yield 219 Endo/Exo |
|---|
| 1 | 4-OMe-C6H4 | 4-Br-C6H4 | 41/0 |
| 2 | Ph | 4-Br-C6H4 | 39/0 |
| 3 | 4-OMe-C6H4 | 4-Cl-C6H4 | 27/0 |
| 4 | 4-Me-C6H4 | 4-Br-C6H4 | 40/0 |
| 5 | E-Ph-CH=CH | 4-Br-C6H4 | 18/0 |
| 6 | 4-CN-C6H4 | 4-Br-C6H4 | 17/20 |
| 7 | 4-NO2-C6H4 | 4-Br-C6H4 | 29/9 |
| 8 | 3-NO2-C6H4 | 4-Br-C6H4 | 11/9 |
| 9 | E-Ph-CH=CPh | Ph | 0/0 |
| 10 | 4-OMe-C6H4 | 4-OMe-C6H4 | 0/0 |
| 11 | 4-OMe-C6H4 | Ph | 0/0 |
| 12 | Ph | Ph | 0/0 |
| 13 | 4-NO2-C6H4 | 4-OMe-C6H4 | 0/0 |
Table 30.
Reaction of benzaldehyde-derived imines
210 with difluorocarbene and aldehydes
2 or ketones
3, yielding oxazolidinones
222 (
Scheme 63).
Table 30.
Reaction of benzaldehyde-derived imines 210 with difluorocarbene and aldehydes 2 or ketones 3, yielding oxazolidinones 222 (Scheme 63).
| Entry | R1 | R2 | R3 | Product | % Yield (dr) |
|---|
| 1 | Ph | Ph | H | 222a | 39 (25:14) |
| 2 | CO2Et | Ph | H | 222b | 25 (3:2) |
| 3 | SiMe3 | Ph | H | 222c | 37 (2:1) |
| 4 | SiMe3 | 2-(CHO)-C6H4 | H | 222d | 13 (10:3) |
| 5 | Ph | CH3 | H | 222e | 43 (1:1) |
| 6 | Ph | CH3 | CH3 | 222f | 3 |
| 7 | Ph | Ph | CH3 | 222g | - |
Table 31.
The reaction of imines
223c, difluorocarbene and trifluoromethylketones
3 (
Scheme 67).
Table 31.
The reaction of imines 223c, difluorocarbene and trifluoromethylketones 3 (Scheme 67).
| Entry | Ar1 | Ar2 | Ar3 | % Yield 238 |
|---|
| 1 | Ph | Ph | Ph | 66 |
| 2 | 4-Cl-C6H4 | 4-Cl-C6H4 | Ph | 61 |
| 3 | 4-Cl-C6H4 | 4-CN-C6H4 | Ph | 10 1 |
| 4 | 4-CF3-C6H4 | 4-CF3-C6H4 | Ph | 12 |
| 5 | 3-CF3-C6H4 | 3-CF3-C6H4 | Ph | 38 |
| 6 | Ph | Ph | 4-CH3-C6H4 | 63 |
| 7 | Ph | Ph | 4-Cl-C6H4 | 54 |
| 8 | Ph | Ph | 4-F-C6H4 | 76 |
| 9 | Ph | Ph | 3-CF3-C6H4 | 74 |
Table 32.
Intramolecular cycloaddition of ylide and carbonyl esters within
5d yielding epoxybenzodiazepines
240 (
Scheme 68)
1.
Table 32.
Intramolecular cycloaddition of ylide and carbonyl esters within 5d yielding epoxybenzodiazepines 240 (Scheme 68) 1.
| Entry | Imine 217 | R1 | R2 | Product | % Yield |
|---|
| 1 | a | Ph | Ph | 240a | 74 |
| 2 | b | Ph | 4-Ome-C6H4 | 240b | 92 |
| 3 | c | Ph | 4-CN-C6H4 | 240c | 70 |
| 4 | d | Ph | 2,4-(Cl)2-C6H3 | 240d | 73 |
| 5 | e | Ph | 1-naphthyl | 240e | 75 |
| 6 | f | Ph | CH2=Cme | 240f | 77 |
| 7 | g | Ph | (E)-Me-CH=CH | 240g | 68 |
| 8 | h | Ph | (E)-Ph-CH=CH | 240h | 56 |
| 9 | i | Ph | (E)-Ph-CH=CPh | 240i | 70 |
| 10 | j | 4-Br-C6H4 | 2-Furyl | 240j | 75 |
| 11 | k | 4-Br-C6H4 | Ph | 240k | 85 |
| 12 | l | 2,4-(Cl)2-C6H3 | Ph | 240l | 70 |
| 13 | m | (4-Cl-C6H4)2-CH | Ph | 240m | 88 |
| 14 | n | Me3SiCH2 | Ph | 241n | 67 |
Table 33.
Intramolecular cycloaddition of dichlorocarbene derived ylides and ester carbonyl
5e yielding epoxybenzodiazepinones
241 (
Scheme 71)
1.
Table 33.
Intramolecular cycloaddition of dichlorocarbene derived ylides and ester carbonyl 5e yielding epoxybenzodiazepinones 241 (Scheme 71) 1.
| Entry | Imine 217 | R1 | R2 | Method 1 | Product 241 | % Yield |
|---|
| 1 | b | Ph | 4-OMe-C6H4 | A | b | 82 |
| 2 | b | Ph | 4-OMe-C6H4 | B | b | 92 |
| 3 | o | 4-Br-C6H4 | 4-OMe-C6H4 | A | o | 97 |
| 4 | d | Ph | 2,4-(Cl)2-C6H3 | A | d | 56 |
| 5 | p | 4-Br-C6H4 | 4-CN-C6H4 | B | p | 74 |
| 6 | q | Ph | 2-furyl | B | q | 47 |
| 7 | i | Ph | (E)-Ph-CH=CPh | A | i | 100 |
| 8 | k | 4-Br-C6H4 | Ph | B | k | 60 |
Table 34.
Reduction of cycloadducts
241 and
240 with LiAlH
4 (
Scheme 72).
Table 34.
Reduction of cycloadducts 241 and 240 with LiAlH4 (Scheme 72).
| Entry | Starting Material | R1 | R2 | Method 1 | Product 247 | % Yield |
|---|
| 1 | 241b | Ph | 4-OMe-C6H4 | A | b | 77 |
| 2 | 241o | 4-Br-C6H4 | 4-OMe-C6H4 | A | o | 47 2 |
| 3 | 241i | Ph | (E)-Ph-CH=CPh | A | i | 68 |
| 4 | 240a | Ph | Ph | B | a | 65 |
| 5 | 240m | (4-Cl-C6H4)2CH | Ph | B | m | 70 |
Table 35.
Multicomponent reaction of aldehydes
2, isocyanides
248 and trialkylboron reagents
249 (
Scheme 73).
Table 35.
Multicomponent reaction of aldehydes 2, isocyanides 248 and trialkylboron reagents 249 (Scheme 73).
| Entry | R1 | R2 | R3 | Cycloadduct 251
% Yield |
|---|
| 1 | 4-OMe-C6H4 | 4-Cl-C6H4 | Et | 85 |
| 2 | 4-Me-C6H4 | 4-Cl-C6H4 | Et | 67 |
| 3 | Ph | 4-Cl-C6H4 | Et | 22 |
| 4 | 4-Cl-C6H4 | 4-Cl-C6H4 | Et | 65 |
| 5 | 4-F-C6H4 | 4-Cl-C6H4 | Et | 36 |
| 6 | cyclohexyl | 4-Cl-C6H4 | Et | - |
| 7 | 1-cyclohexenyl | 4-Cl-C6H4 | Et | - |
| 8 | PhCH2 | 4-Cl-C6H4 | Et | - |
| 9 | TsCH2 | 4-Cl-C6H4 | Et | - |
| 10 | 4-OMe-C6H4 | 4-OMe-C6H4 | Et | 50 |
| 11 | 4-OMe-C6H4 | 3-pyridyl | Et | 62 |
| 12 | 4-OMe-C6H4 | Me3C | Et | - |
| 13 | 4-OMe-C6H4 | EtO2C | Et | 7 |
| 14 | 4-Cl-C6H4 | 4-Cl-C6H4 | Bu | 40 |










































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
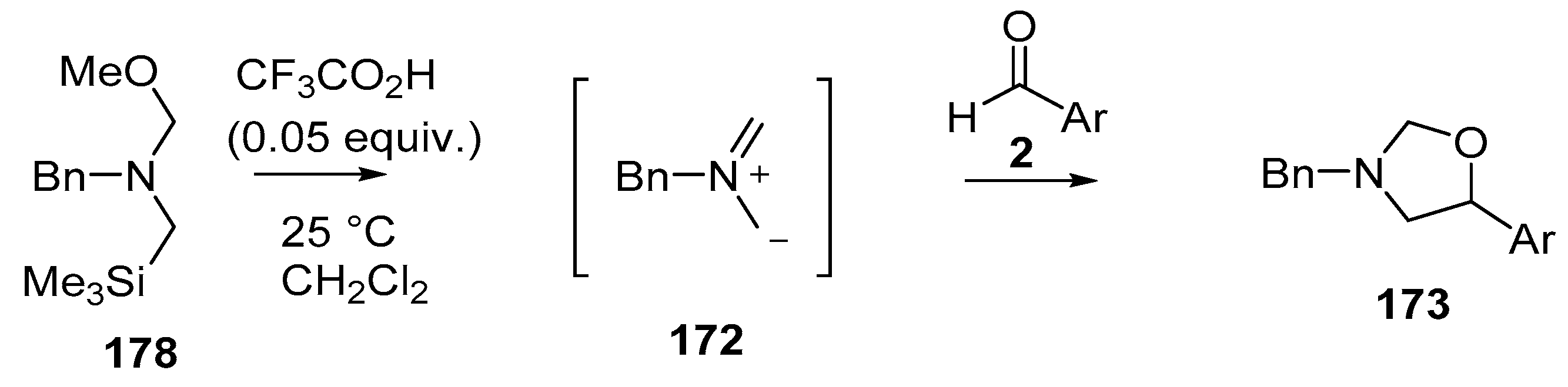{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





































