
LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in Vivo Metabolism of (+)-[18F]Flubatine—A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors

 and
and
Abstract
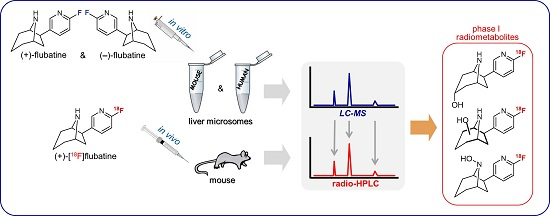
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
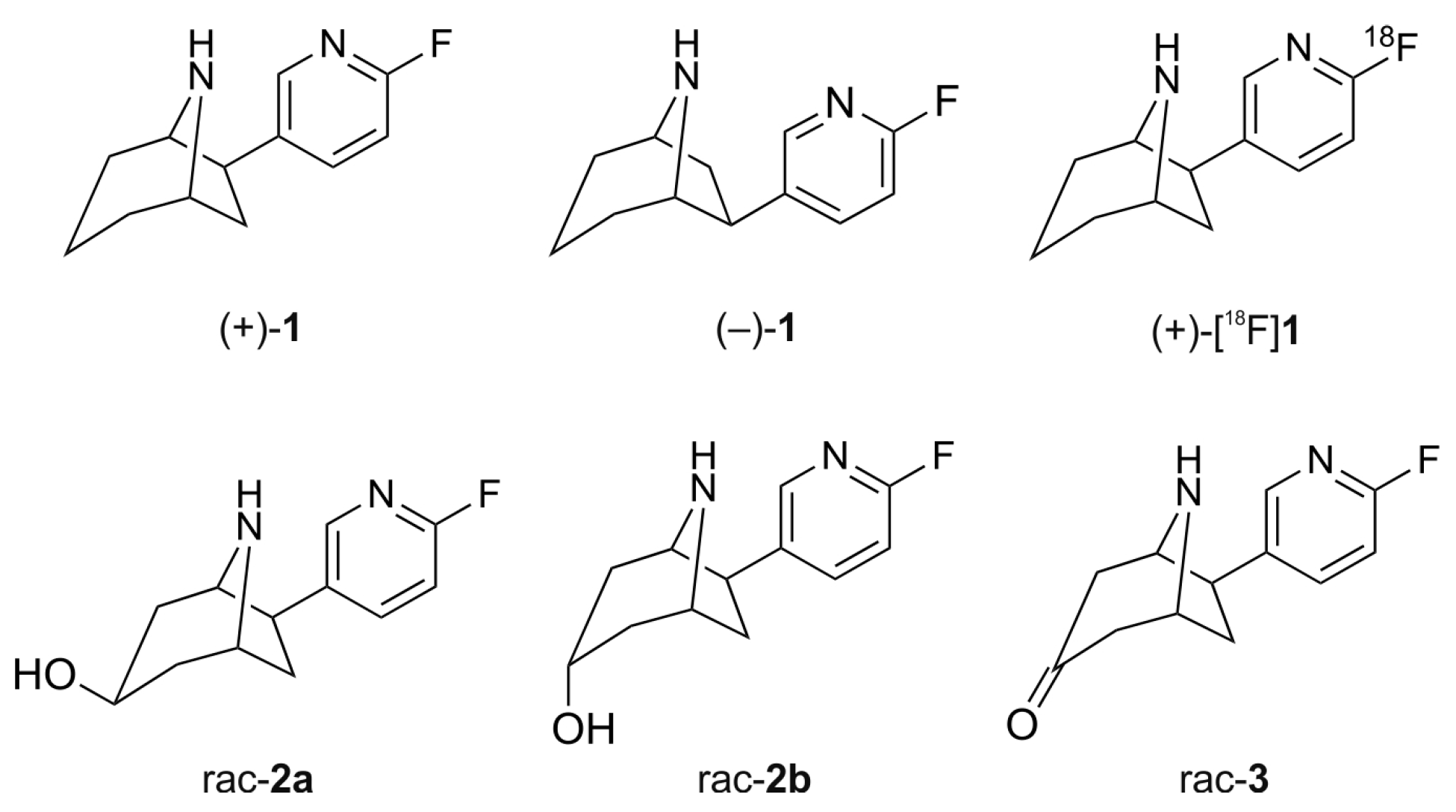
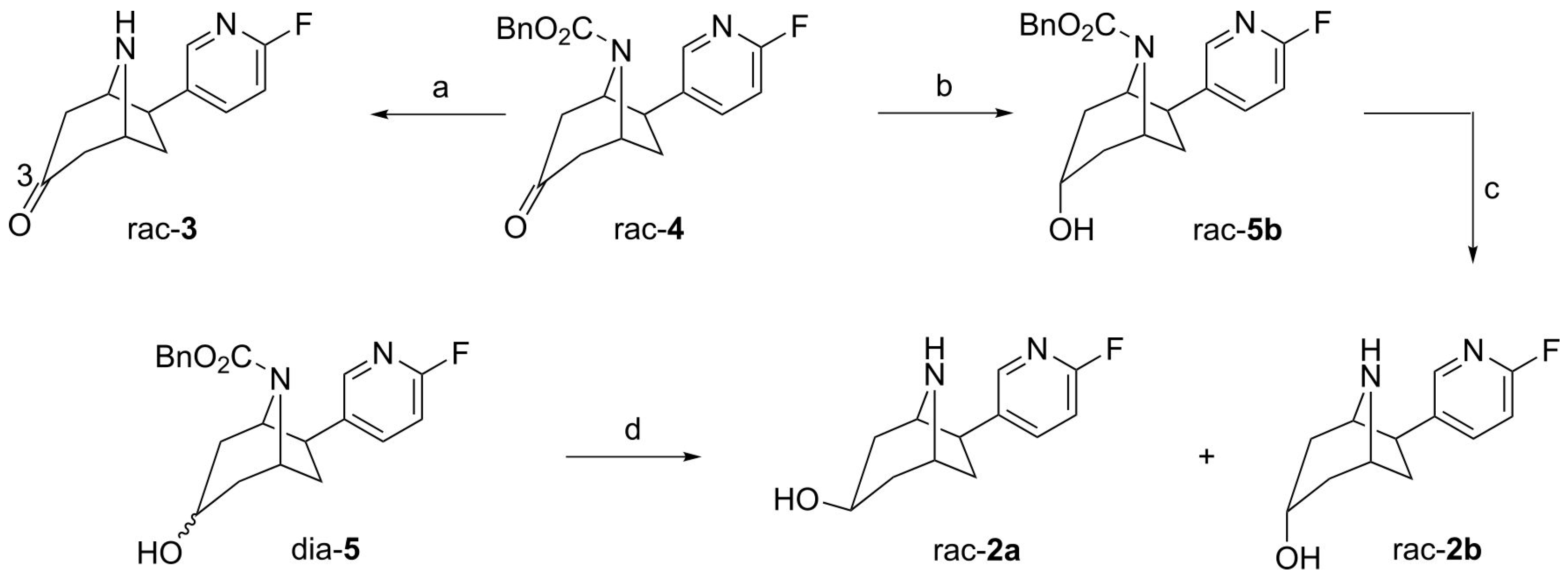
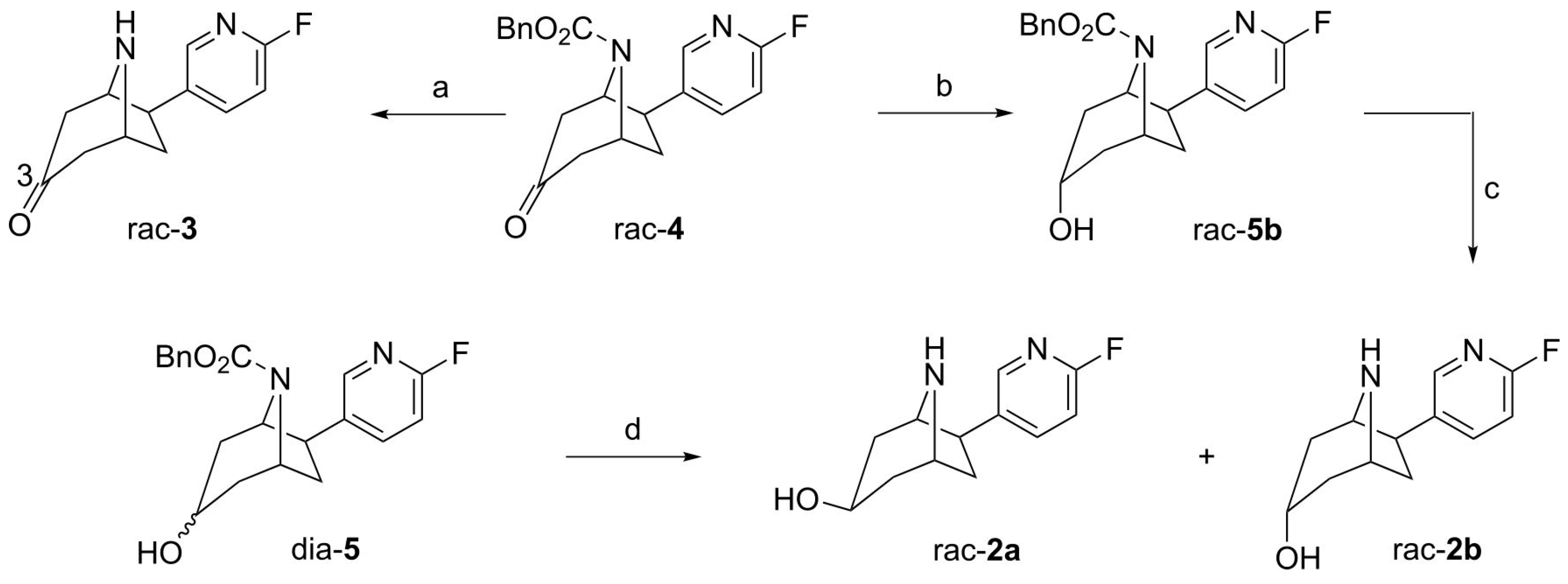
2.1. Synthesis of Reference Compounds
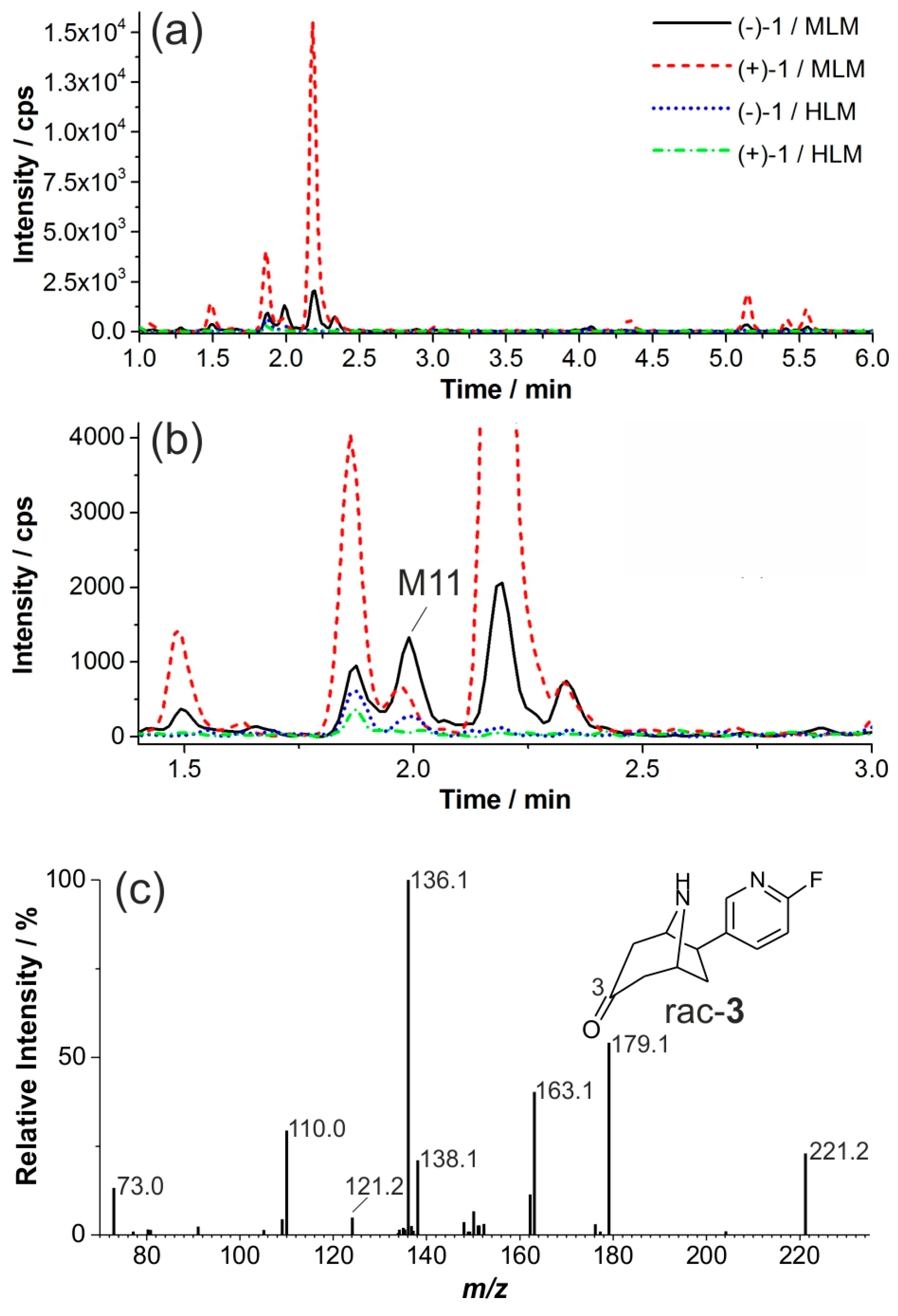
2.2. Investigation of Microsomal Incubations by LC-MS
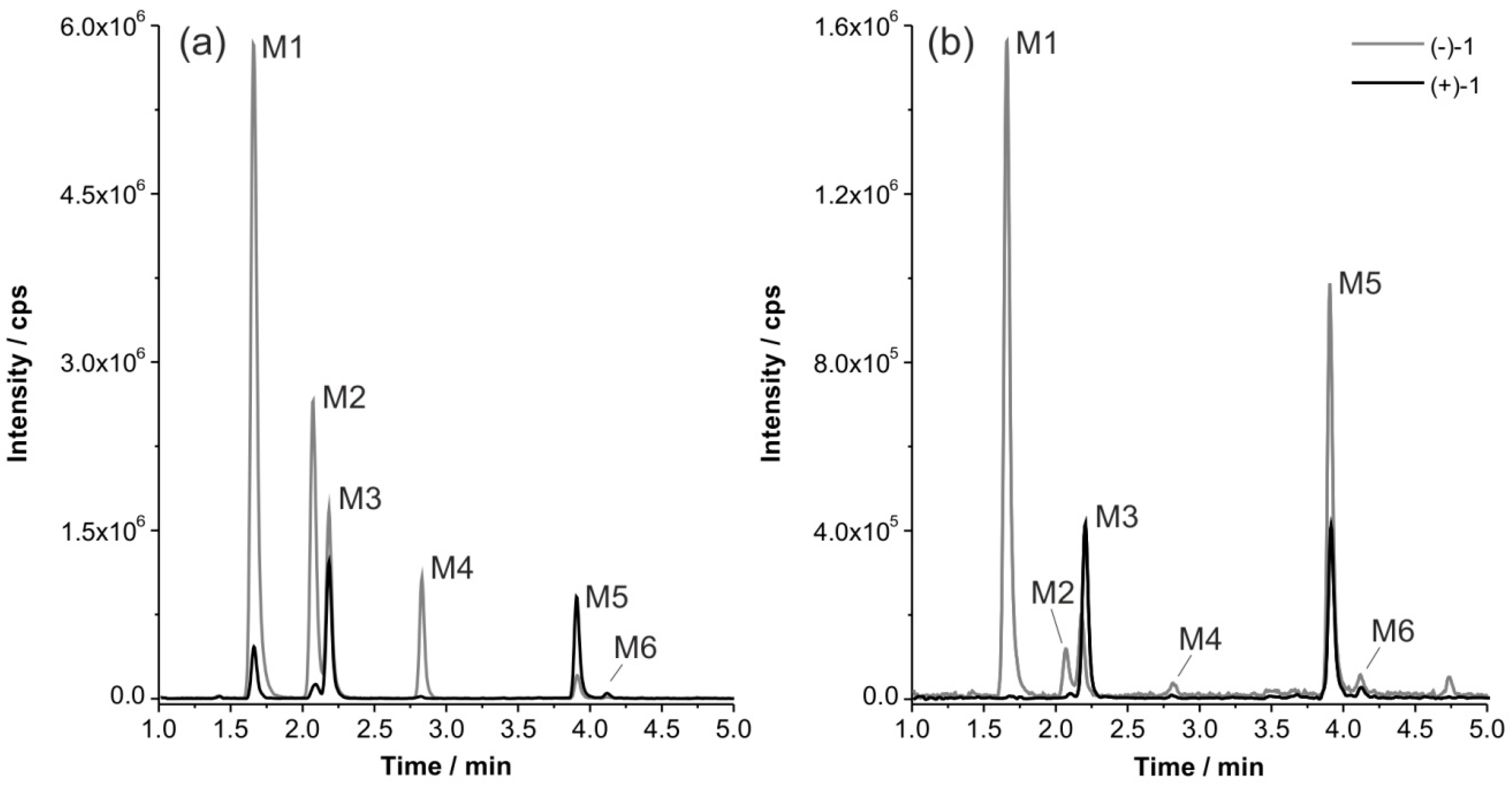
2.3. In Vitro Formation of Monohydroxylated Metabolites of (+)-1 and (−)-1
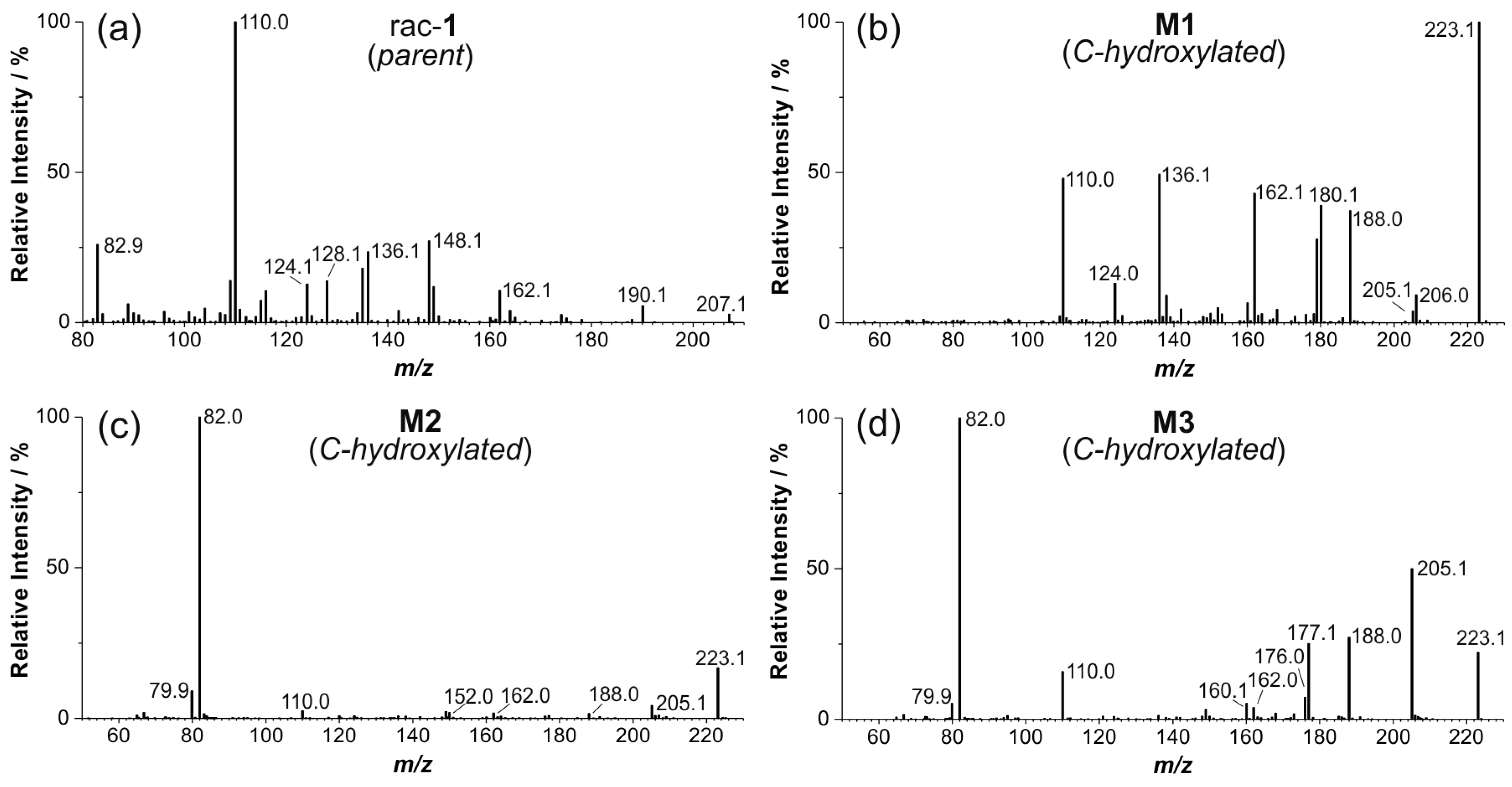
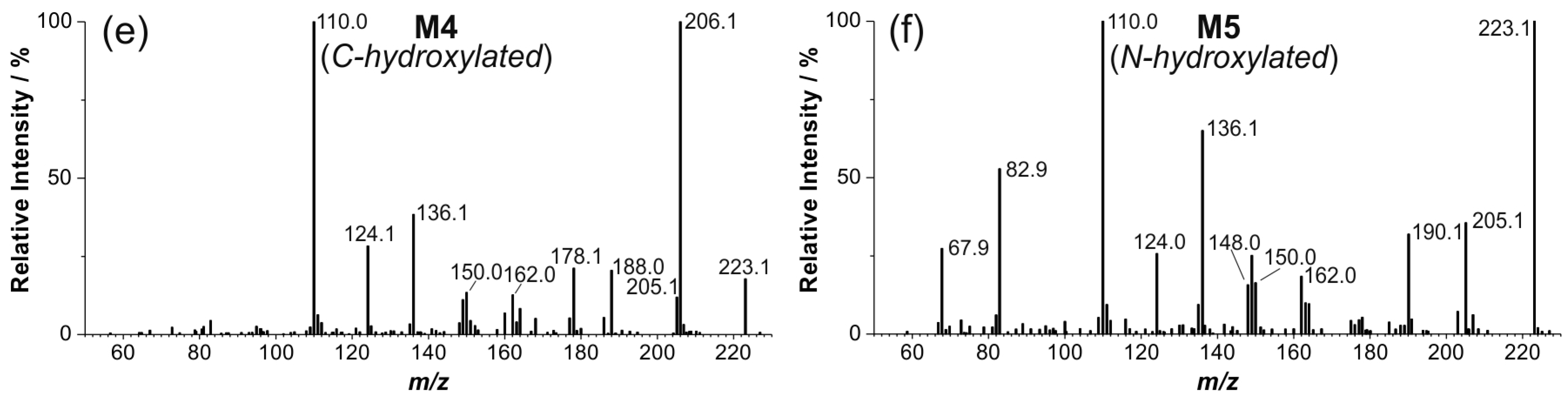
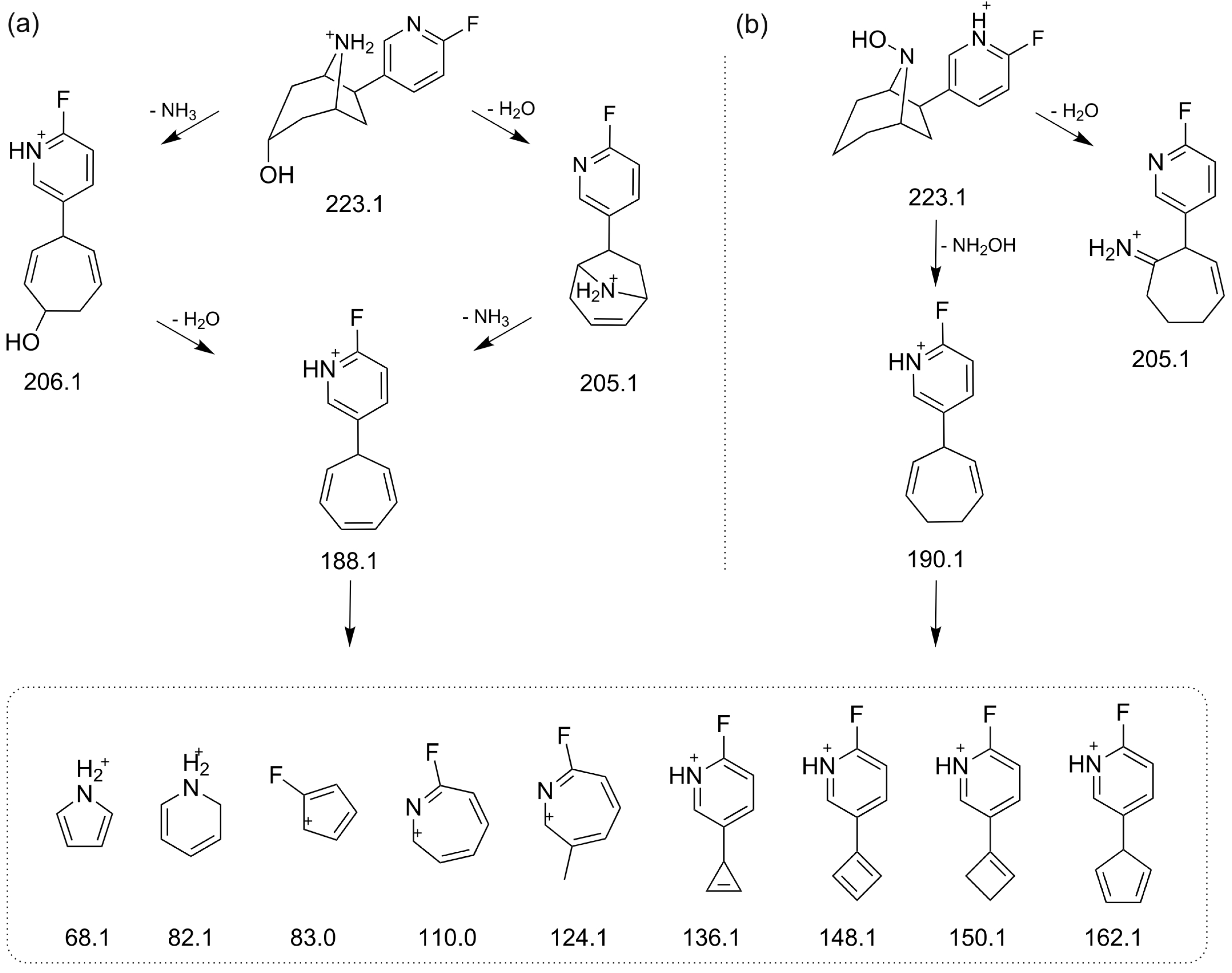
2.4. Structure Elucidation of Monohydroxylated in Vitro Metabolites by MS/MS
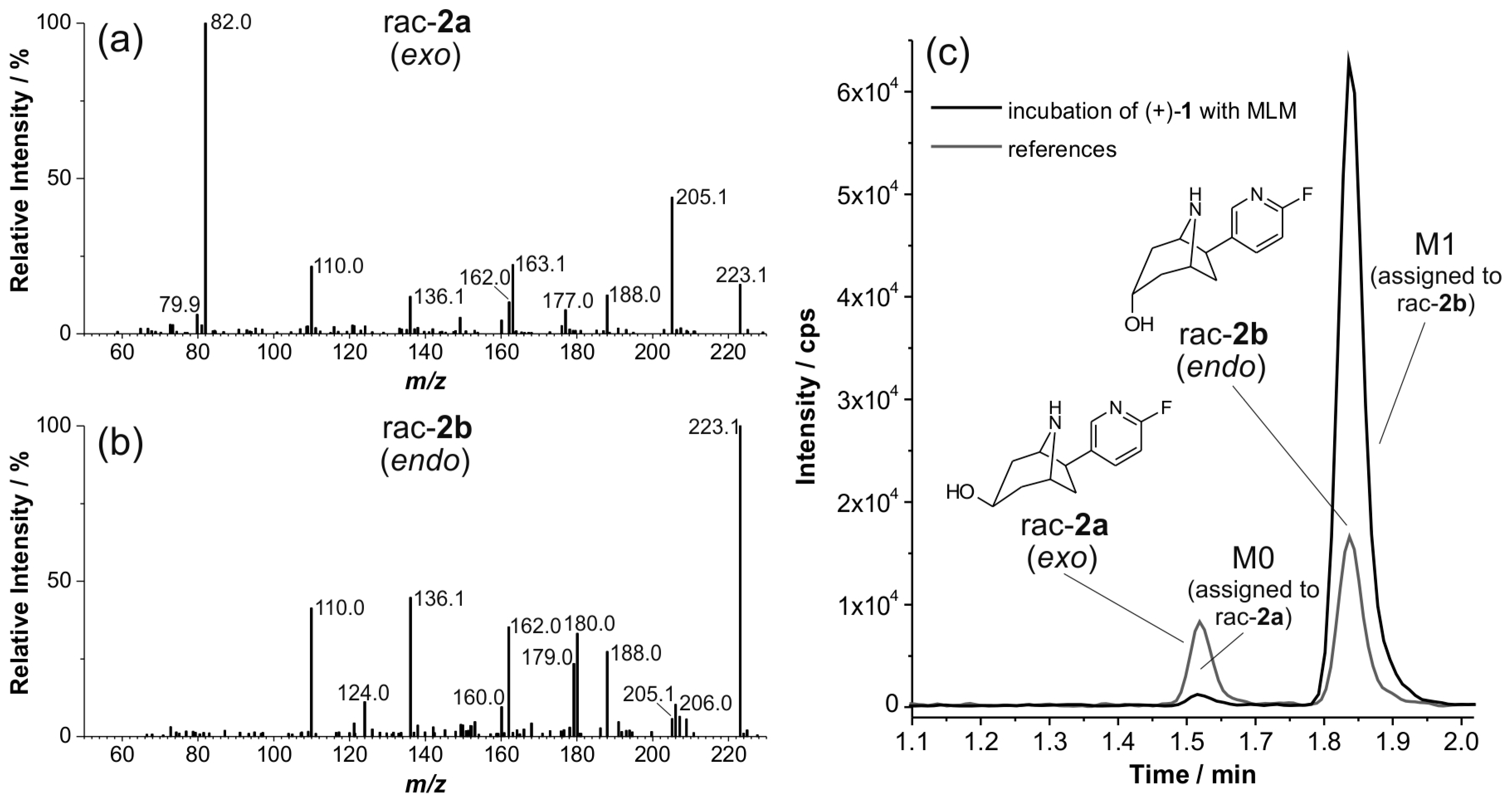
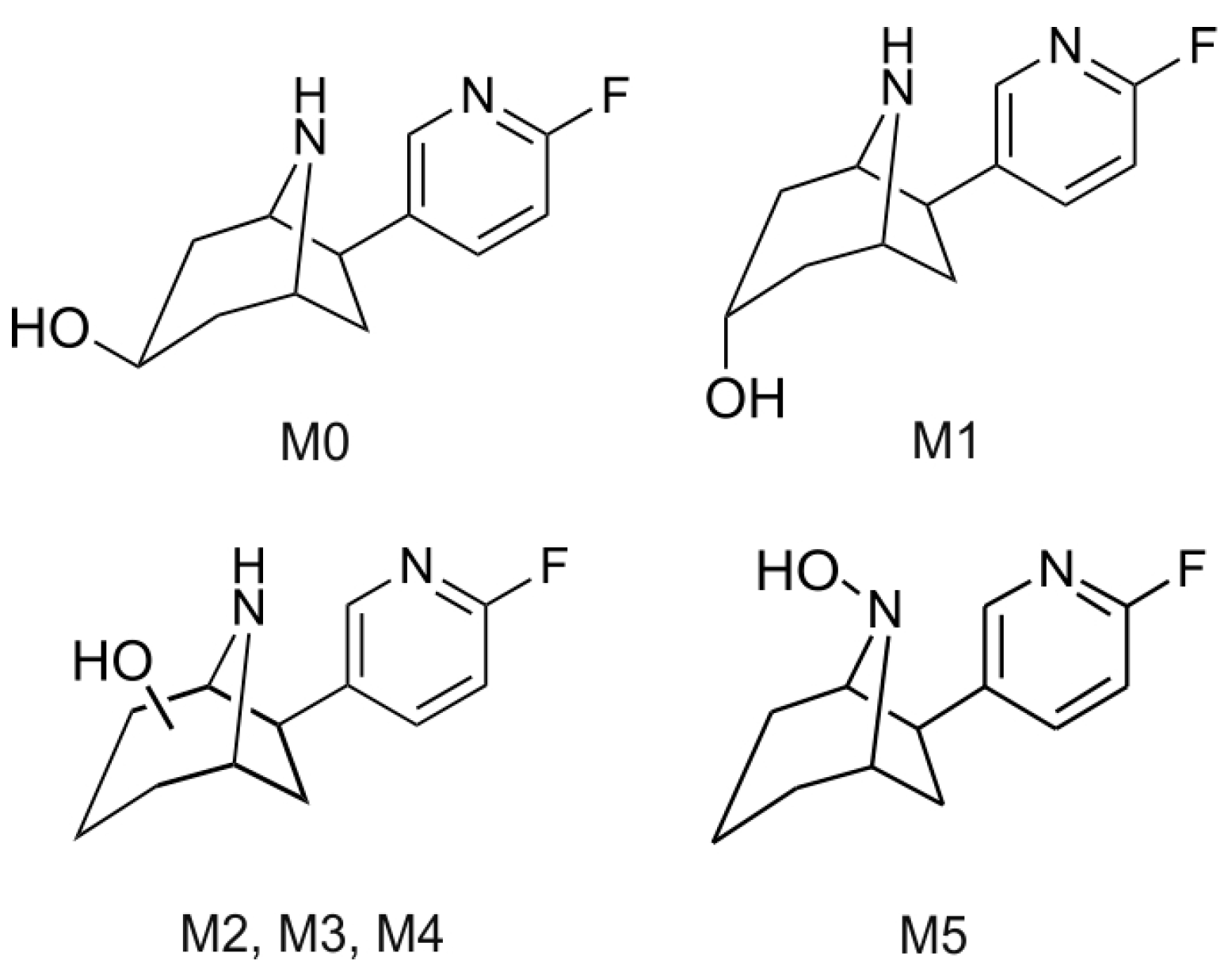
2.5. Structural Identification of Monohydroxylated in Vitro Metabolites by Aid of Reference Compounds
2.6. Dihydroxylated Products and Ketones Formed in Vitro
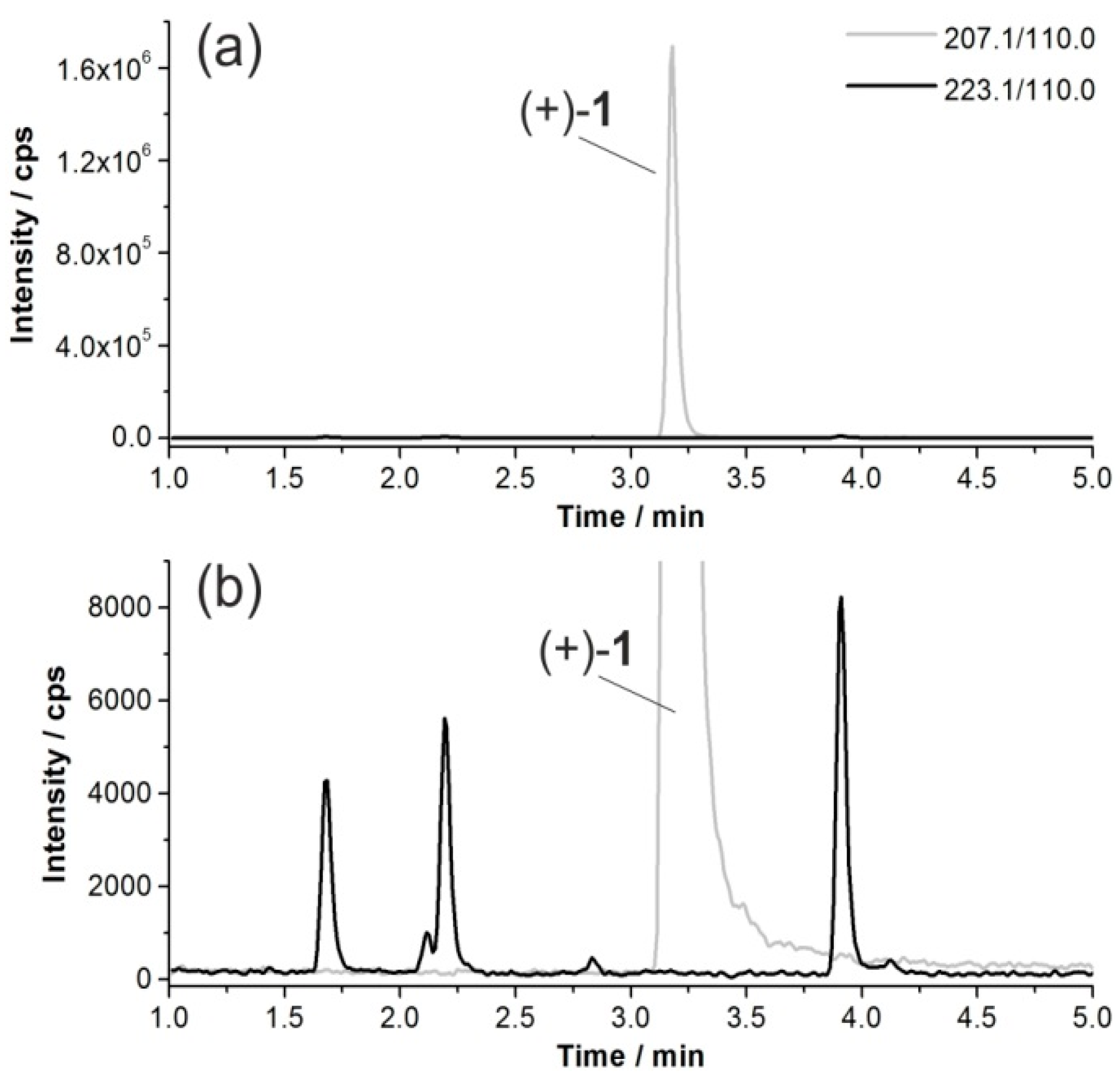
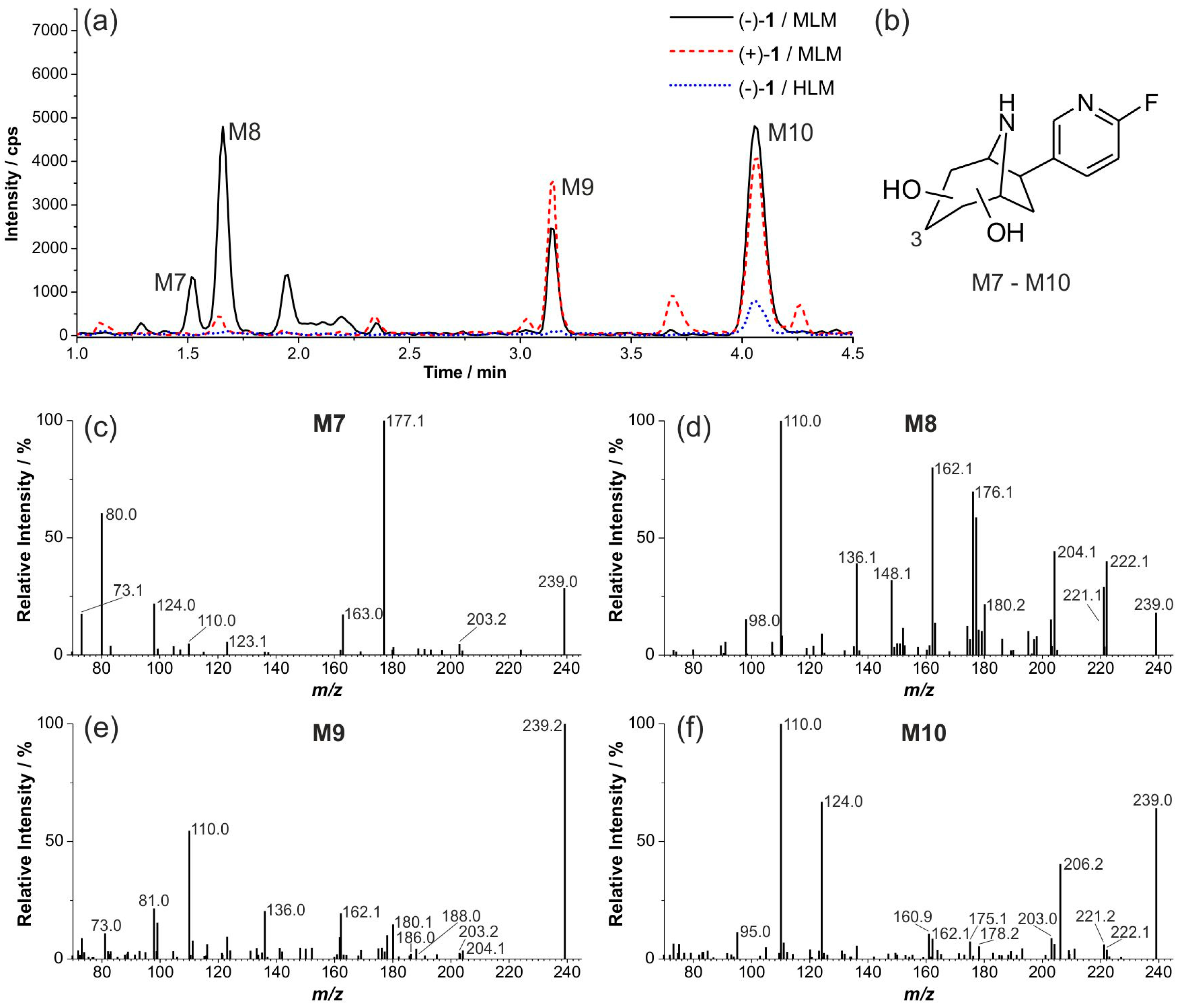
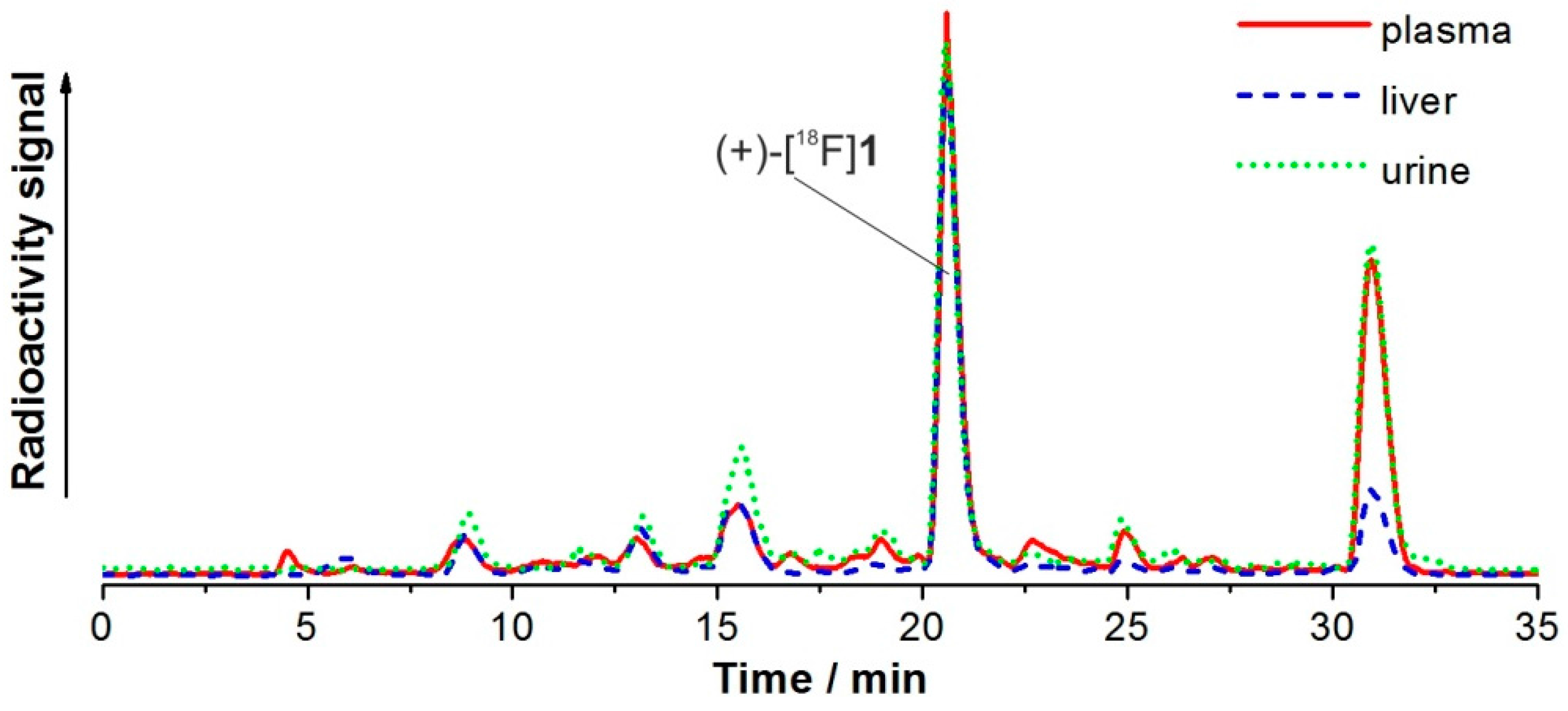
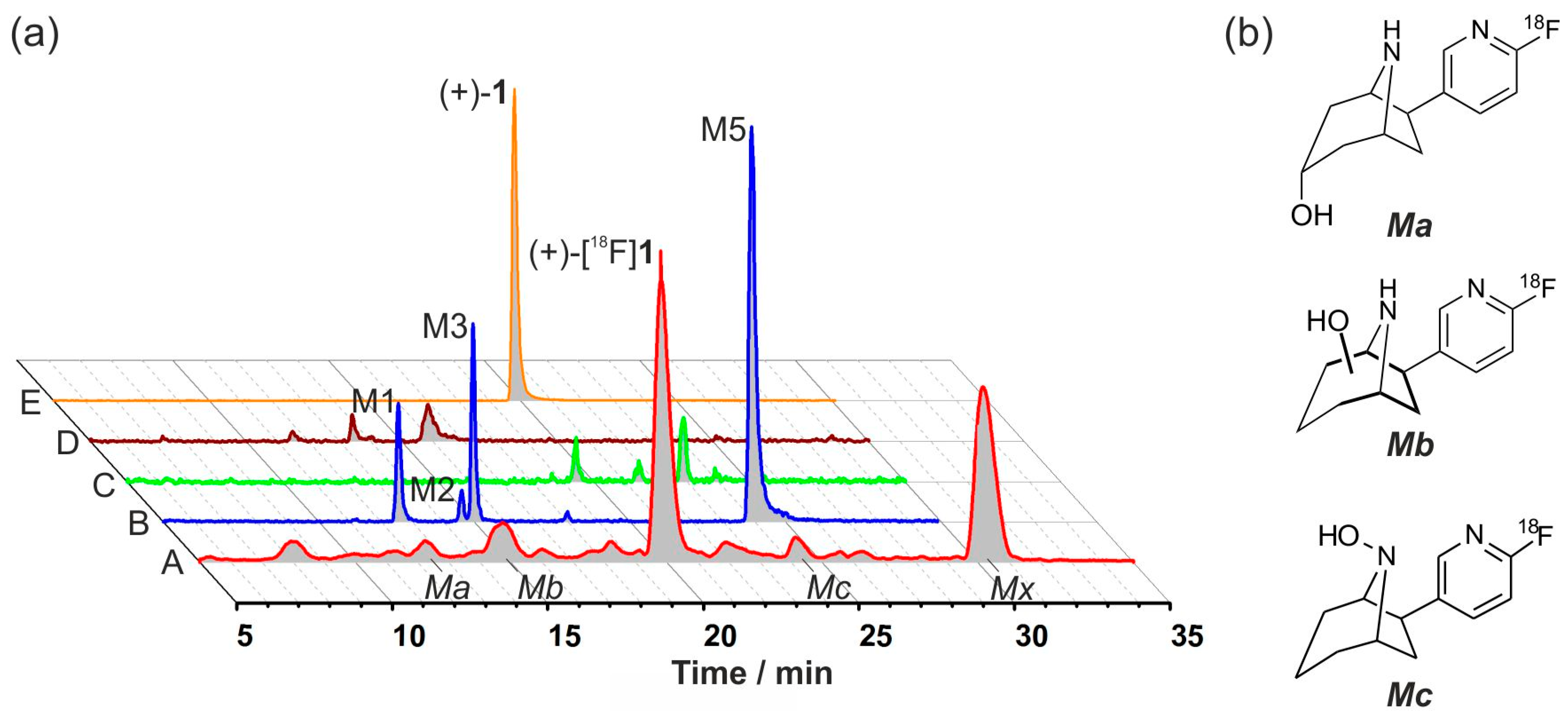
2.7. Investigation of the in Vivo Metabolism of (+)-[18F]1
2.8. Studies on Identification of Radiometabolites of (+)-[18F]1
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Synthesis of Reference Compounds
3.2.1. (±)-exo-6-(6-Fluoro-pyridin-3-yl)-8-aza-bicyclo[3.2.1]octan-3-one ((±)-flubatine-3-one, rac-3)
3.2.2. (±)-3-exo,6-exo-6-(6-Fluoro-pyridin-3-yl)-8-aza-bicyclo[3.2.1]octan-3-ol ((±)-exo-3-hydroxy-flubatine, rac-2a) and (±)-3-endo,6-exo-6-(6-Fluoro-pyridin-3-yl)-8-aza-bicyclo[3.2.1]octan-3-ol ((±)-endo-3-hydroxy-flubatine, rac-2b)
3.2.3. (±)-3-endo,6-exo-6-(6-Fluoro-pyridin-3-yl)-3-hydroxy-8-aza-bicyclo[3.2.1]octane-8-carboxylic Acid Benzyl Ester (rac-5b)
3.3. Radiosynthesis of (+)-[18F]1
3.4. Microsomal Incubations
3.5. In Vivo Study of (+)-[18F]1 in Mouse
3.6. LC-MS Analyses
- Multiple reaction monitoring (MRM) scan type: Substance-specific parameters were optimized for rac-1 delivered by syringe injection. Measurements were performed for the MRM transitions 207.1/110.0, 221.1/110.0, 223.1/110.0 and 239.1/110.0, applying the following parameters for method I: CAD high, scan time 80 ms, declustering potential (DP) 126, entrance potential (EP) 10, collision energy (CE) 37, collision cell exit potential (CXP) 10. For method II: CUR 30, CAD medium, IS 5500, TEM 500, GS1 50, GS2 50, scan time 100 ms, DP 136, EP 10, CE 44, CXP 14.
- Q3 Multiple ion (MI) scan type: Measurements were performed for m/z of 223.1, applying the following parameters: scan time 250 ms, DP 120, EP 10, CXP 37. For presentation purposes, chromatograms of blank samples were subtracted from chromatograms obtained initially, completed by final curve fitting.
- Enhanced product ion (EPI) scan type: By measuring solutions of rac-2a and rac-2b, supplied by a syringe pump, substance-specific parameters, allowing the detection of particular fragments of interest in EPI scan mode, were optimized. For measurements of the prepared incubation samples the following parameters were used: CAD high, IS 5500, TEM 650, GS1 60, GS2 60, products of m/z 221.1, 223.1, or 239.1 (respectively), dynamic fill time, DP 110, EP 10, CE 33, collision energy spread (CES) 0. In ion chromatograms obtained, a range of background was selected manually and subtracted from ranges of interest to result in EPI spectra as shown.
3.7. Radio-HPLC
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schubiger, P.A.; Lehmann, L.; Friebe, M. PET Chemistry—The Driving Force in Molecular Imaging; Springer: Berlin, Germany; Heidelberg, Germany, 2007. [Google Scholar]
- Brust, P.; van den Hoff, J.; Steinbach, J. Development of 18F-labeled radiotracers for neuroreceptor imaging with positron emission tomography. Neurosci. Bull. 2014, 30, 777–811. [Google Scholar] [CrossRef] [PubMed]
- Badio, B.; Garraffo, H.M.; Spande, T.F.; Daly, J.W. Epibatidine: Discovery and definition as a potent analgesic and nicotinic agonist. Med. Chem. Res. 1994, 4, 440–448. [Google Scholar]
- Molina, P.E.; Ding, Y.-S.; Carroll, F.I.; Liang, F.; Volkow, N.D.; Pappas, N.; Kuhar, M.; Abumrad, N.; Gatley, S.J.; Fowler, J.S. Fluoro-norchloroepibatidine: Preclinical assessment of acute toxicity. Nucl. Med. Biol. 1997, 24, 743–747. [Google Scholar] [CrossRef]
- Ding, Y.-S.; Molina, P.E.; Fowler, J.S.; Logan, J.; Volkow, N.D.; Kuhar, M.J.; Carroll, F.I. Comparative studies of epibatidine derivatives [18F]NFEP and [18F]N-Methyl-NFEP: Kinetics, nicotine effect, and toxicity. Nucl. Med. Biol. 1999, 26, 139–148. [Google Scholar] [CrossRef]
- Patt, J.T.; Spang, J.E.; Westera, G.; Buck, A.; Schubiger, P.A. Synthesis and in vivo studies of [C-11]N-methylepibatidine: Comparison of the stereoisomers. Nucl. Med. Biol. 1999, 26, 165–173. [Google Scholar] [CrossRef]
- Smits, R.; Fischer, S.; Hiller, A.; Deuther-Conrad, W.; Wenzel, B.; Patt, M.; Cumming, P.; Steinbach, J.; Sabri, O.; Brust, P.; et al. Synthesis and biological evaluation of both enantiomers of [18F]flubatine, promising radiotracers with fast kinetics for the imaging of α4β2-nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2014, 22, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Deuther-Conrad, W.; Patt, J.T.; Feuerbach, D.; Wegner, F.; Brust, P.; Steinbach, J. Norchloro-fluoro-homoepibatidine: Specificity to neuronal nicotinic acetylcholine receptor subtypes in vitro. Farmaco 2004, 59, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Martin-Ruiz, C.M.; Court, J.A. Nicotinic receptor subtypes in human brain related to aging and dementia. Alcohol 2001, 24, 63–68. [Google Scholar] [CrossRef]
- Nordberg, A. Nicotinic receptor abnormalities of Alzheimer’s disease: Therapeutic implications. Biol. Psychiatry 2001, 49, 200–210. [Google Scholar] [CrossRef]
- Patt, J.T.; Deuther-Conrad, W.; Wohlfarth, K.; Feuerbach, D.; Brust, P.; Steinbach, J. Norchloro-fluoro-homoepibatidine: 18F-labelling and evaluation of affinity and selectivity at neuronal nicotinic acetylcholine receptors. J. Label. Compd. Radiopharm. 2003, 46, S168. [Google Scholar]
- Smits, R.; Hoepping, A.; Fischer, S.; Hiller, A.; Deuther-Conrad, W.; Brust, P.; Steinbach, J.; Patt, M.; Patt, J.T.; Sabri, O. Precursor Compounds for the Radiosynthesis of [18F]norchloro-Fluorohomoepibatidine. CA2841800 A1, 7 February 2015. [Google Scholar]
- Patt, M.; Schildan, A.; Habermann, B.; Fischer, S.; Hiller, A.; Deuther-Conrad, W.; Wilke, S.; Smits, R.; Hoepping, A.; Wagenknecht, G.; et al. Fully automated radiosynthesis of both enantiomers of [18F]Flubatine under GMP conditions for human application. Appl. Radiat. Isot. 2013, 80, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Hiller, A.; Smits, R.; Hoepping, A.; Funke, U.; Wenzel, B.; Cumming, P.; Sabri, O.; Steinbach, J.; Brust, P. Radiosynthesis of racemic and enantiomerically pure (−)-[18F]flubatine—A promising PET radiotracer for neuroimaging of α4β2 nicotinic acetylcholine receptors. Appl. Radiat. Isot. 2013, 74, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hockley, B.G.; Stewart, M.N.; Sherman, P.; Quesada, C.; Kilbourn, M.R.; Albin, R.L.; Scott, P.J.H. (−)-[18F]Flubatine: Evaluation in rhesus monkeys and a report of the first fully automated radiosynthesis validated for clinical use. J. Label. Compd. Radiopharm. 2013, 56, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Gallezot, J.-D.; Esterlis, I.; Bois, F.; Zheng, M.-Q.; Lin, S.-F.; Kloczynski, T.; Krystal, J.H.; Huang, Y.; Sabri, O.; Carson, R.E.; et al. Evaluation of the sensitivity of the novel α4β2* nicotinic acetylcholine receptor PET radioligand 18F-(−)-NCFHEB to increases in synaptic acetylcholine levels in rhesus monkeys. Synapse 2014, 68, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Bois, F.; Gallezot, J.-D.; Zheng, M.-Q.; Lin, S.-F.; Esterlis, I.; Cosgrove, K.P.; Carson, R.E.; Huang, Y. Evaluation of [18F]-(−)-norchlorofluorohomoepibatidine ([18F]-(−)-NCFHEB) as a PET radioligand to image the nicotinic acetylcholine receptors in non-human primates. Nucl. Med. Biol. 2015, 42, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Sattler, B.; Kranz, M.; Starke, A.; Wilke, S.; Donat, C.K.; Deuther-Conrad, W.; Patt, M.; Schildan, A.; Patt, J.; Smits, R.; et al. Internal dose assessment of (−)-18F-flubatine, comparing animal model datasets of mice and piglets with first-in-human results. J. Nucl. Med. 2014, 55, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Patt, M.; Becker, G.A.; Grossmann, U.; Habermann, B.; Schildan, A.; Wilke, S.; Deuther-Conrad, W.; Graef, S.; Fischer, S.; Smits, R.; et al. Evaluation of metabolism, plasma protein binding and other biological parameters after administration of (−)-[18F]Flubatine in humans. Nucl. Med. Biol. 2014, 41, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Sabri, O.; Becker, G.-A.; Meyer, P.M.; Hesse, S.; Wilke, S.; Graef, S.; Patt, M.; Luthardt, J.; Wagenknecht, G.; Hoepping, A.; et al. First-in-human PET quantification study of cerebral α4β2* nicotinic acetylcholine receptors using the novel specific radioligand (−)-[18F]Flubatine. NeuroImage 2015, 118, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hillmer, A.T.; Esterlis, I.; Gallezot, J.D.; Bois, F.; Zheng, M.Q.; Nabulsi, N.; Lin, S.F.; Papke, R.L.; Huang, Y.; Sabri, O.; et al. Imaging of cerebral α4β2* nicotinic acetylcholine receptors with (−)-[18F]Flubatine PET: Implementation of bolus plus constant infusion and sensitivity to acetylcholine in human brain. NeuroImage 2016, 141, 71–80. [Google Scholar] [CrossRef] [PubMed]
- German Clinical Trials Register/Deutsches Register Klinischer Studien (DRKS). Validation of the New Positron Emissions Tomography (PET) Radioligand (+)-[18F]-Flubatine for Imaging of Nicotinic Acetylcholine Receptors (nAChR) in Alzheimer’s Dementia. DRKS-ID: DRKS00005819. Available online: https://drks-neu.uniklinik-freiburg.de/drks_web/ (accessed on 7 September 2016).
- Pawelke, B. Metabolite analysis in positron emission tomography studies: Examples from food sciences. Amino Acids 2005, 29, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Bier, D.; Holschbach, M.H.; Wutz, W.; Olsson, R.A.; Coenen, H.H. Metabolism of the A1 adenosine receptor positron emission tomography ligand [18F]8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine ([18F]CPFPX) in rodents and humans. Drug Metab. Dispos. 2006, 34, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Peyronneau, M.-A.; Saba, W.; Dollé, F.; Goutal, S.; Coulon, C.; Bottlaender, M.; Valette, H. Difficulties in dopamine transporter radioligand PET analysis: The example of LBT-999 using [18F] and [11C] labelling: Part II: Metabolism studies. Nucl. Med. Biol. 2012, 39, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Peyronneau, M.-A.; Saba, W.; Goutal, S.; Damont, A.; Dollé, F.; Kassiou, M.; Bottlaender, M.; Valette, H. Metabolism and quantification of [18F]DPA-714, a new TSPO positron emission tomography radioligand. Drug Metab. Dispos. 2013, 41, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Amini, N.; Nakao, R.; Schou, M.; Halldin, C. Identification of PET radiometabolites by cytochrome P450, UHPLC/Q-ToF-MS and fast radio-LC: Applied to the PET radioligands [11C]flumazenil, [18F]FE-PE2I, and [11C]PBR28. Anal. Bioanal. Chem. 2013, 405, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kiesewetter, D.O.; Lang, L.; Gu, D.; Chen, X. Applications of LC-MS in PET radioligand development and metabolic elucidation. Curr. Drug Metab. 2010, 11, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Watt, A.P.; Hitzel, L.; Morrison, D.; Locker, K.L. Determination of the in vitro metabolism of (+)- and (−)-epibatidine. J. Chromatogr. A 2000, 896, 229–238. [Google Scholar] [CrossRef]
- Spande, T.F.; Garraffo, H.M.; Edwards, M.W.; Yeh, H.J.C.; Pannell, L.; Daly, J.W. Epibatidine: A novel (chloropyridyl)azabicycloheptane with potent analgesic activity from an Ecuadoran poison frog. J. Am. Chem. Soc. 1992, 114, 3475–3478. [Google Scholar] [CrossRef]
- Stan’kov, I.N.; Kondrat’ev, V.B.; Derevyagina, I.D.; Kazakov, P.V.; Lukashov, O.I.; Morozova, O.T.; Semchenko, F.M.; Sosnov, A.V. Gas chromatographic determination of trace amounts of epibatidine and its biomarker in blood plasma. J. Anal. Chem. 2015, 70, 213–219. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Oh, C.-Y.; Lee, K.-Y.; Lee, Y.-S.; Jung, Y.-H.; Park, H.-J.; Ham, W.-H. Syntheses of (±)-homoepibatidine analogues. Arch. Pharm. Res. 2002, 25, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Seger, S.T.; Rydberg, P.; Olsen, L. Mechanism of the N-hydroxylation of primary and secondary amines by cytochrome P450. Chem. Res. Toxicol. 2015, 28, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.












© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludwig, F.-A.; Smits, R.; Fischer, S.; Donat, C.K.; Hoepping, A.; Brust, P.; Steinbach, J. LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in Vivo Metabolism of (+)-[18F]Flubatine—A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors. Molecules 2016, 21, 1200. https://doi.org/10.3390/molecules21091200
Ludwig F-A, Smits R, Fischer S, Donat CK, Hoepping A, Brust P, Steinbach J. LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in Vivo Metabolism of (+)-[18F]Flubatine—A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors. Molecules. 2016; 21(9):1200. https://doi.org/10.3390/molecules21091200
Chicago/Turabian StyleLudwig, Friedrich-Alexander, René Smits, Steffen Fischer, Cornelius K. Donat, Alexander Hoepping, Peter Brust, and Jörg Steinbach. 2016. "LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in Vivo Metabolism of (+)-[18F]Flubatine—A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors" Molecules 21, no. 9: 1200. https://doi.org/10.3390/molecules21091200
APA StyleLudwig, F.-A., Smits, R., Fischer, S., Donat, C. K., Hoepping, A., Brust, P., & Steinbach, J. (2016). LC-MS Supported Studies on the in Vitro Metabolism of both Enantiomers of Flubatine and the in Vivo Metabolism of (+)-[18F]Flubatine—A Positron Emission Tomography Radioligand for Imaging α4β2 Nicotinic Acetylcholine Receptors. Molecules, 21(9), 1200. https://doi.org/10.3390/molecules21091200